1. Introduction
With increasing demands on the properties of composite materials, the use of new materials has gained much interest. Graphene is of particular importance due to its outstanding mechanical, thermal and electrical properties, which are a result of the special structure of its carbon atoms [
1,
2,
3]. In addition, graphene and its derivatives exhibit a very large surface area, which enables a significant impact on the characteristics of the composite material using very low weight fractions of the used additive [
4,
5].
There is a vast number of manufacturing methods to produce graphene and its related materials. At the same time, the production of large quantities at comparatively low costs that is of interest for the composite industry is currently only possible via the Hummers’ method and its associated varieties [
6,
7]. With the Hummers’ method, graphite is first oxidized using different chemicals, such as sulfuric acid, potassium permanganate and hydrogen peroxide, which leads to graphene oxide (GO) as the reaction product [
8]. By a subsequent chemical or thermal reduction, the GO is transformed to create reduced graphene oxide [
9]. As this procedure is associated with relatively harsh conditions, the quality of the resulting material is significantly lower than the characteristics that may be achieved with other methods, such as mechanical exfoliation or chemical vapor deposition [
6,
10]. However, the functional groups that remain on the surface of the GO and reduced graphene oxide particles usually allow better coupling actions with polymeric matrices and, hence, these nanocomposites mostly demonstrate a better performance than nanocomposites with single-layer graphene inclusions [
11,
12].
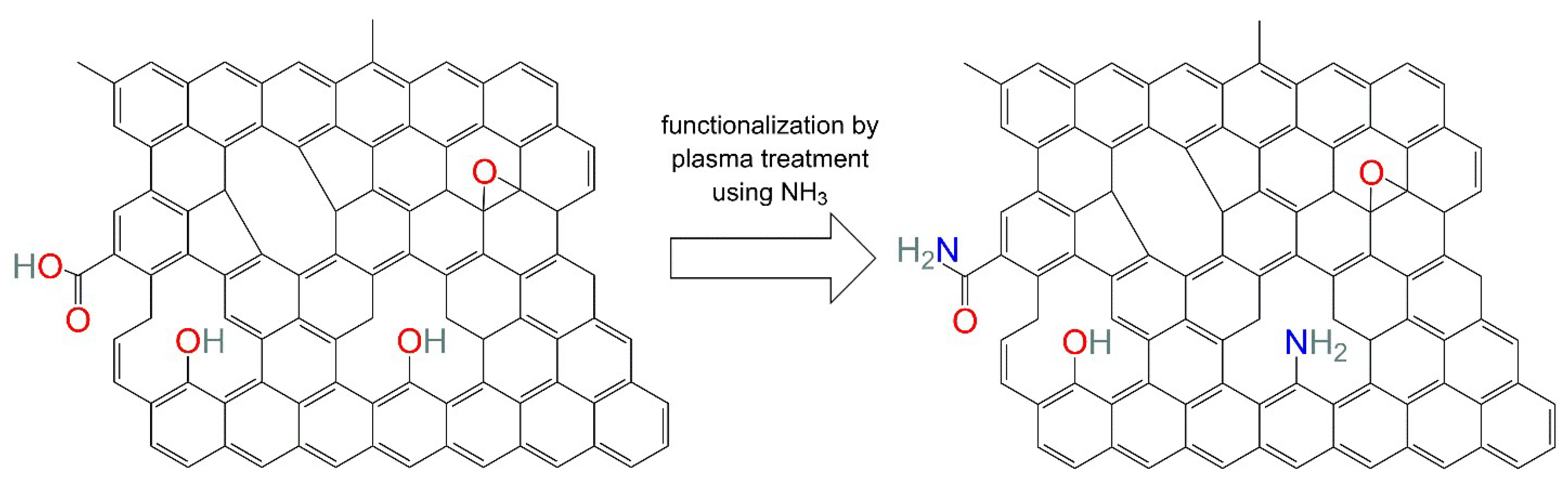
To further enhance the bonding of the graphene materials with the polymeric matrix and to improve the dispersibility, a tailored functionalization may be applied to the particles [
13,
14]. In general, it is possible to differentiate between two types of functionalization, non-covalent and covalent functionalization, where the latter enables a larger spectrum of properties and a stronger bonding of the functional groups to the nanoparticle [
13]. Furthermore, a covalent bonding between the particles and matrix allows an improved transfer of mechanical loads, phonons and electrons across the interface, which results in a more homogeneous dispersion, as well as superior mechanical, thermal and electrical properties [
15,
16,
17,
18]. When using epoxies as the matrix material, a surface functionalization of the particles with amines is of particular interest, since these groups can participate in the polymerization process, especially when an epoxy resin is cured with amine-based hardeners that are commonly used for most composite applications [
19,
20].
The covalent functionalization of graphene and its derivatives is usually conducted using tedious chemical processes, in which large quantities of hazardous reagents and solvents are used in a number of different non-robust processing steps that currently cannot be scaled to larger volumes [
21,
22]. An alternative is the use of plasma treatment, which uses the ionized forms of gases, such as NH
3 or O
2, to create the respective functional groups on the particle’s surface [
13]. Even though specialized equipment is required, it is easier to scale by, e.g., increasing the number or size of the reactors. Furthermore, it is a very quick method with typical process times of a few seconds to minutes and does not require further processing steps, such as purification, which is mandatory in chemical functionalization processes [
23]. In addition, the plasma typically only impairs the particle’s surface up to depths of a few nanometers, while the bulk composition of the inner layers of few-layered graphene derivatives remain unaffected [
24,
25,
26]. This might result in an enlarged surface roughness, which can be associated with an enhanced interlocking with the matrix and, hence, improved shear modulus and strength [
27]. However, the performance of nanocomposites with plasma-functionalized graphene and its derivatives is currently poorly understood.
In this study, we report the influence of increasing the weight fraction from 0.00 to 1.50 wt% of two forms of reduced graphene oxide on the mechanical, thermal and electrical characteristics of nanocomposite samples with a matrix consisting of a bisphenol-A-(epichlorohydrin) epoxy resin and an amine-based hardener. A non-functionalized grade of reduced graphene oxide (rGO) is compared to an amine-functionalized form of reduced graphene oxide (frGO), for which the functional groups are realized via plasma treatment. First, the impact of the plasma processing on the morphology and elemental composition of the rGO and frGO powders is evaluated. Second, a thorough characterization of the mechanical, thermal and electrical properties is carried out to classify the effects of both rGO and frGO. We believe that this information will help to predict the properties of nanocomposites with inclusions of rGO as well as frGO, and will also facilitate the choice of materials with respect to the use of functionalized additives. It will also enable further areas of applications, such as the use in fiber-reinforced polymers [
28], thermal interface materials [
29,
30], electromagnetic shielding [
31] or sensory materials [
32,
33].
2. Materials and Methods
2.1. Materials
Two varieties of reduced graphene oxide were supplied by Graphit Kropfmühl (Hauzenberg, Germany). Both particles were produced by the manufacturer by treating natural graphite flakes (RFL 99,5 O) with a modified Hummers’ method. EXG 98 300 R was used as the non-functionalized form of reduced graphene oxide (rGO). The material EXG 98 300 R FNH was exposed to a cold, low-pressure gas plasma process using NH3 as the source for the plasma treatment and, hence, leading to an amine-functionalized form of reduced graphene oxide (frGO). The two powder materials were used as received.
The bisphenol-A-(epichlorohydrin) epoxy resin Biresin® CR83 was used in conjunction with the amine-based hardener Biresin® CH83-10 from Sika Deutschland (Stuttgart, Germany) to create the matrix material. This two-part system has a low-mixed viscosity (155 mPa s at 25 °C), which facilitates the manufacturing of nanocomposite samples. Furthermore, the amine-based hardener matches the applied functionalization of the frGO particles.
2.2. Sample Preparation
In order to achieve a homogeneous dispersion of the particles in the polymeric matrix, the rGO or frGO particles were pre-dispersed in the neat epoxy resin by manual mixing. This suspension was subsequently led through the three-roll mill 80S PLUS from EXAKT Advanced Technologies (Norderstedt, Germany) using eight cycles with different gap widths, as stated in
Table 1. During the dispersion process, the three-roll mill was running at a speed ratio of 1:3:9, where the velocity of the fastest roller was set to 200 rpm. The hereby-created masterbatches allowed the production of specimens with a maximum concentration of 1.50 wt% rGO or frGO. For the manufacturing of the nanocomposite samples with concentrations of 0.25 wt%, 0.50 wt%, 0,75 wt%, 1.00 wt% and 1.50 wt%, the hardener was added to the respective amount of masterbatch and, if required, more resin was added to dilute the masterbatch to lower concentrations. A constant resin/hardener mixing ratio of 100:30 parts by weight was applied for all nanocomposite and neat polymer samples. The respective suspension was subsequently stirred by hand and degassed in a vacuum chamber. Later, the uncured materials were casted into molds and left to cure for 48 h at room temperature. Next, the various cured material configurations underwent a thermal treatment, in which they were heated from room temperature to 70 °C at a rate of 0.2 °C min
−1, kept at 70 °C for 12 h and subsequently cooled to room temperature at a rate of 0.5 °C min
−1.
2.3. Sample Characterization
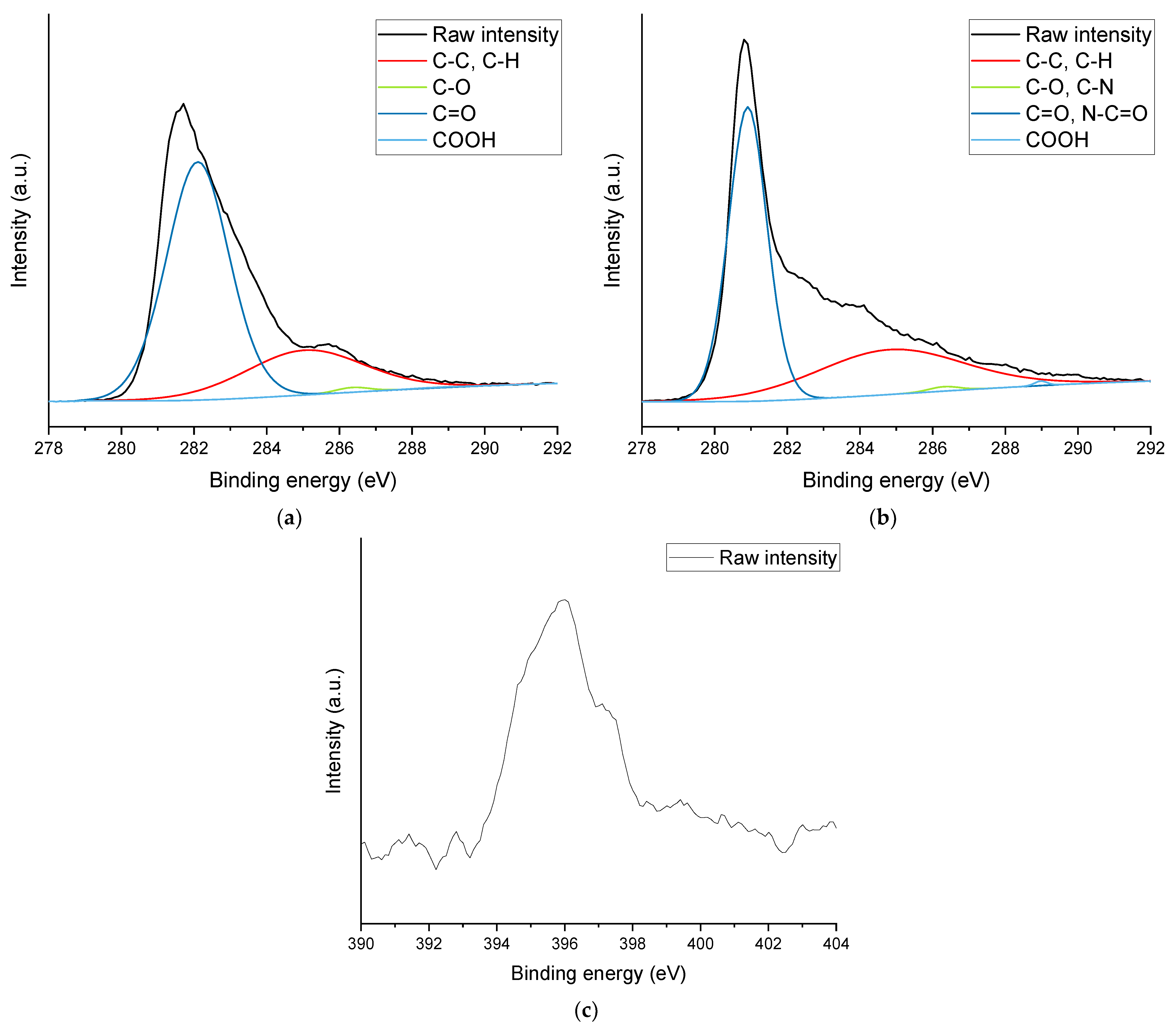
The morphology and elemental composition of the powder materials was examined by scanning electron microscopy (SEM), as well as energy-dispersive X-ray spectroscopy (EDX) with an FEI (Hillsboro, OR, USA) Helios NanoLab 600 equipped with an X-Max (50 mm2) detector from Oxford Instruments (High Wycombe, UK). The elemental analysis was obtained using an operating voltage and current of 10 kV and 11 nA, respectively, for the area visible at a magnification of 200×, and the powders were deposited on carbon tape.
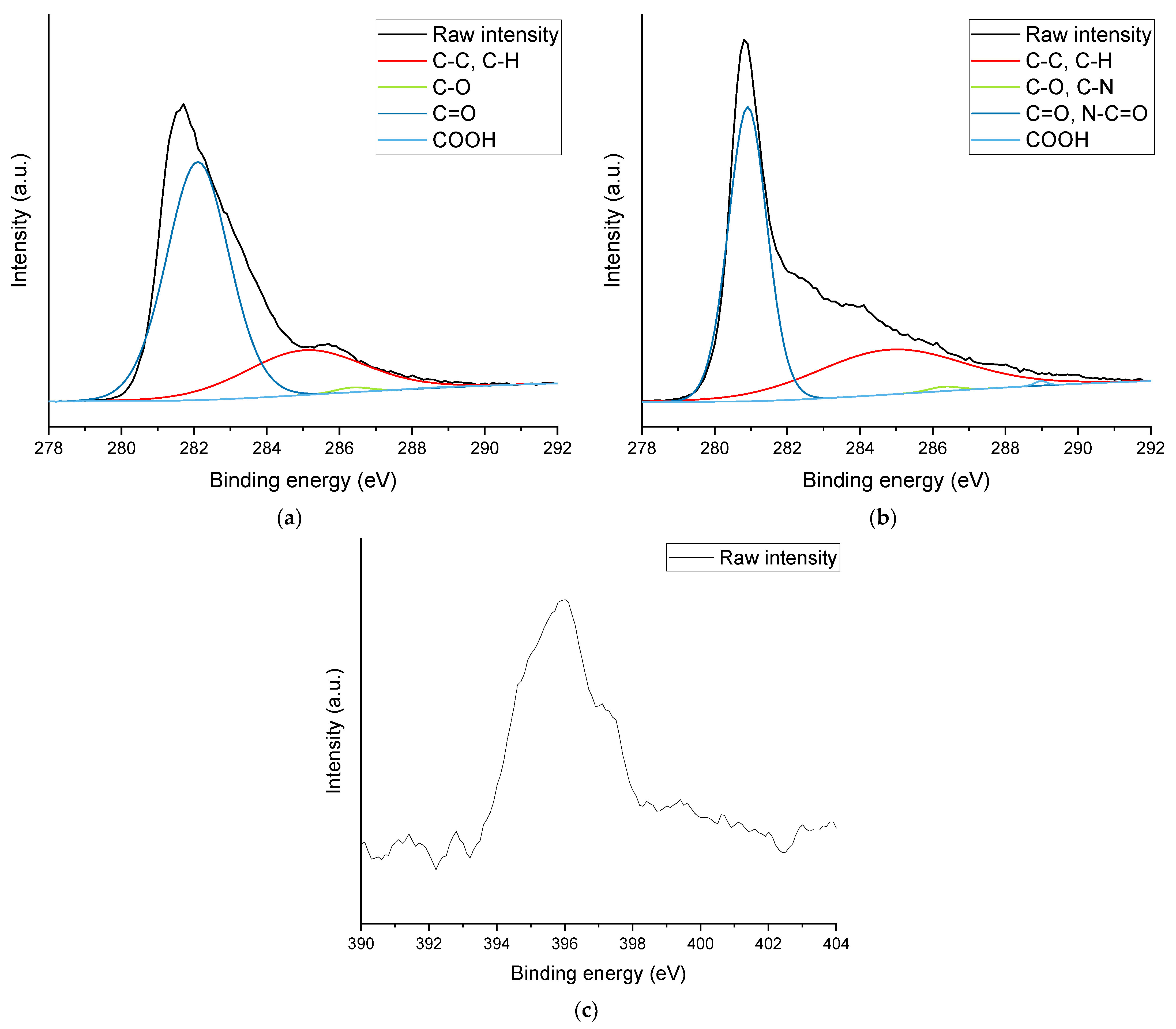
X-ray photoelectron spectroscopy (XPS) was used to confirm the presence of nitrogen and, hence, amine functional groups and the elemental composition of the powder materials. It was performed using a Kratos (Manchester, UK) AXIS Nova using a monochromatic Al kα X-ray source operating at a power of 225 W. Survey and detailed spectra were obtained at 160 eV and 20 eV, respectively. Three spots with an elliptical area of 0.3 × 0.7 mm were examined for each powder material.
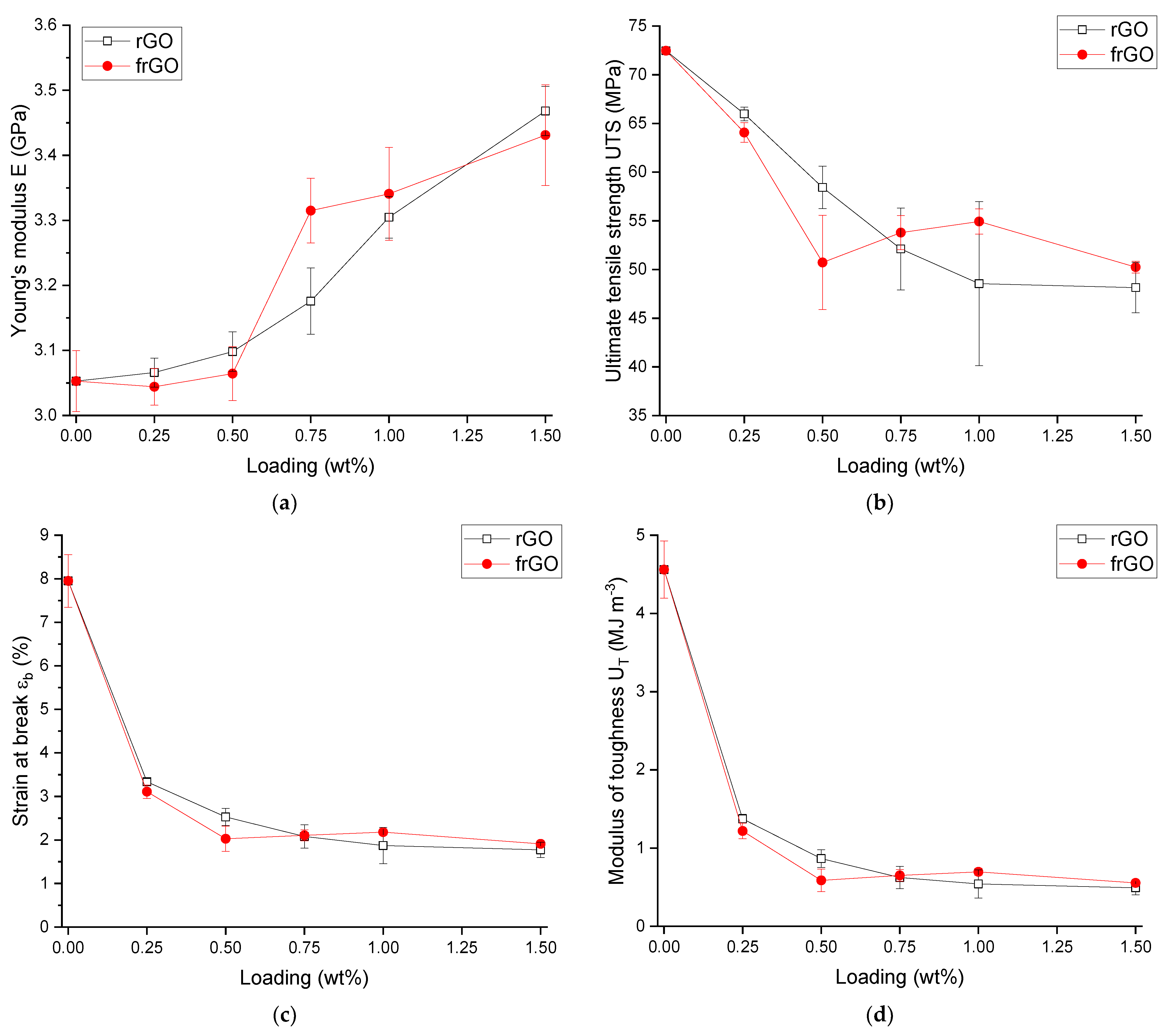
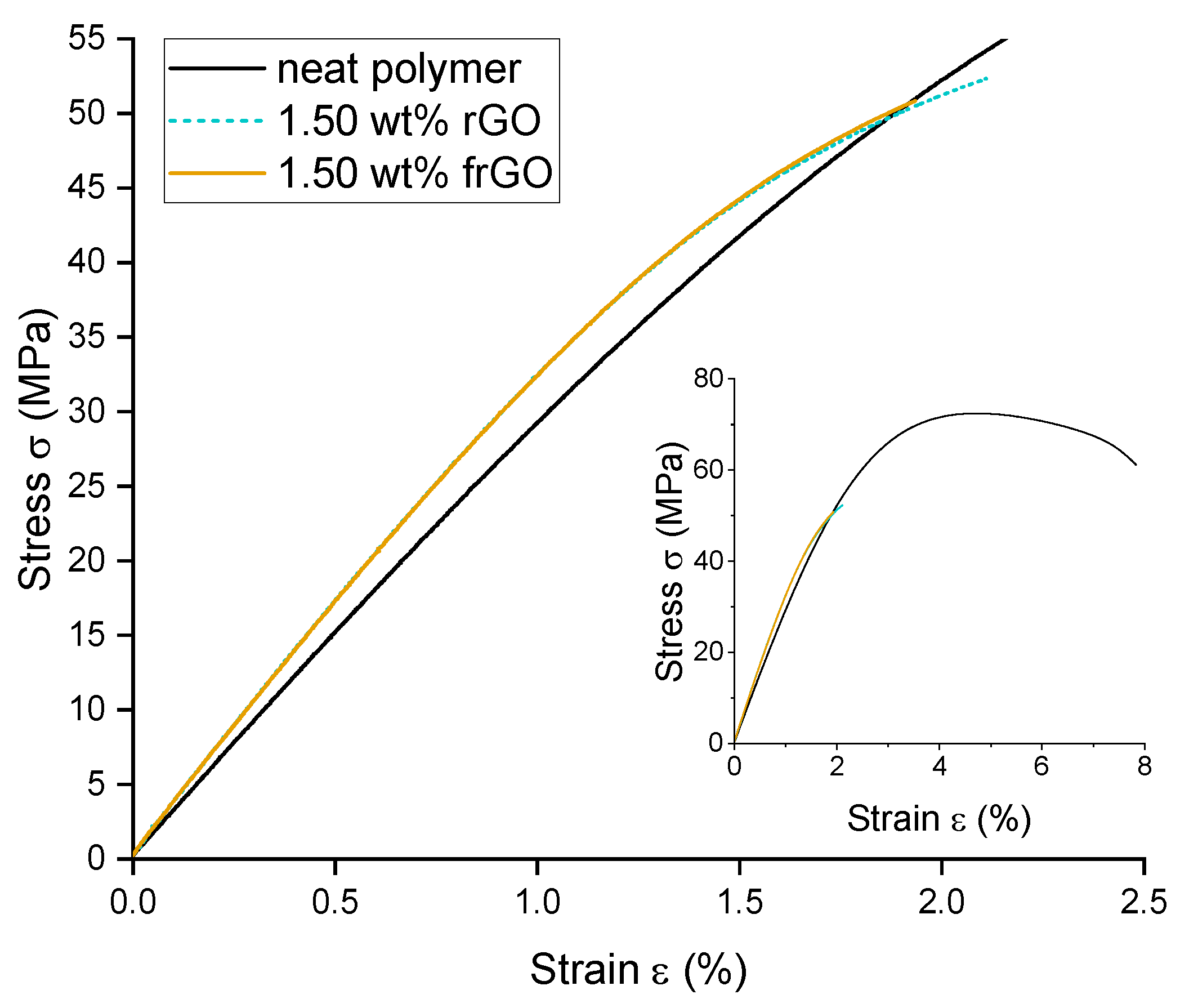
The tensile properties of the various material configurations were evaluated according to DIN EN ISO 527-2:2012 [
34] and using specimen-type 1B. The tensile tests were performed at room temperature using the universal testing machine Inspekt Table 20 kN from Hegewald and Peschke (Nossen, Germany). A total of 5 samples were measured per material configuration with a pre-force of 10 N and a testing speed of 1 mm min
−1. The morphology of the respective fracture surfaces were subsequently analyzed by scanning electron microscopy (SEM) using a Zeiss (Oberkochen, Germany) DSM 940.
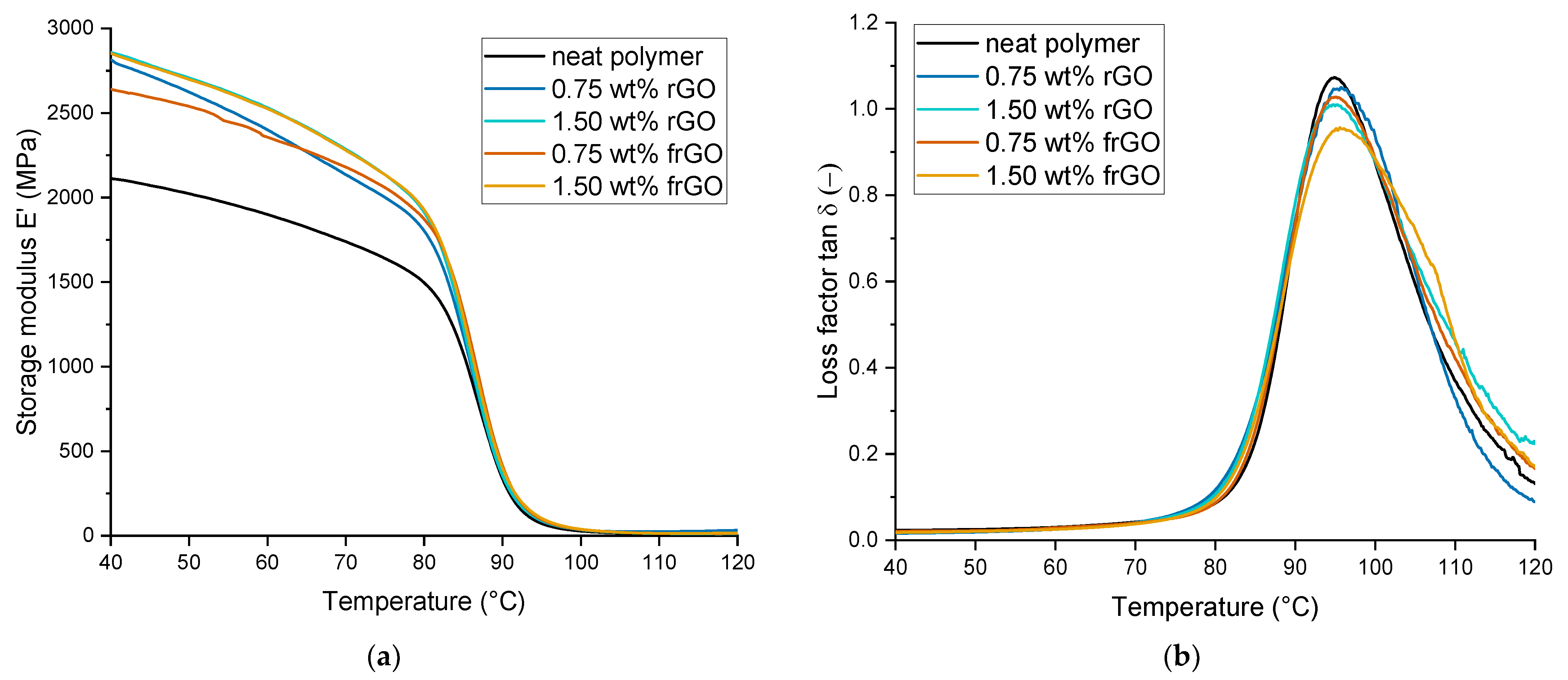
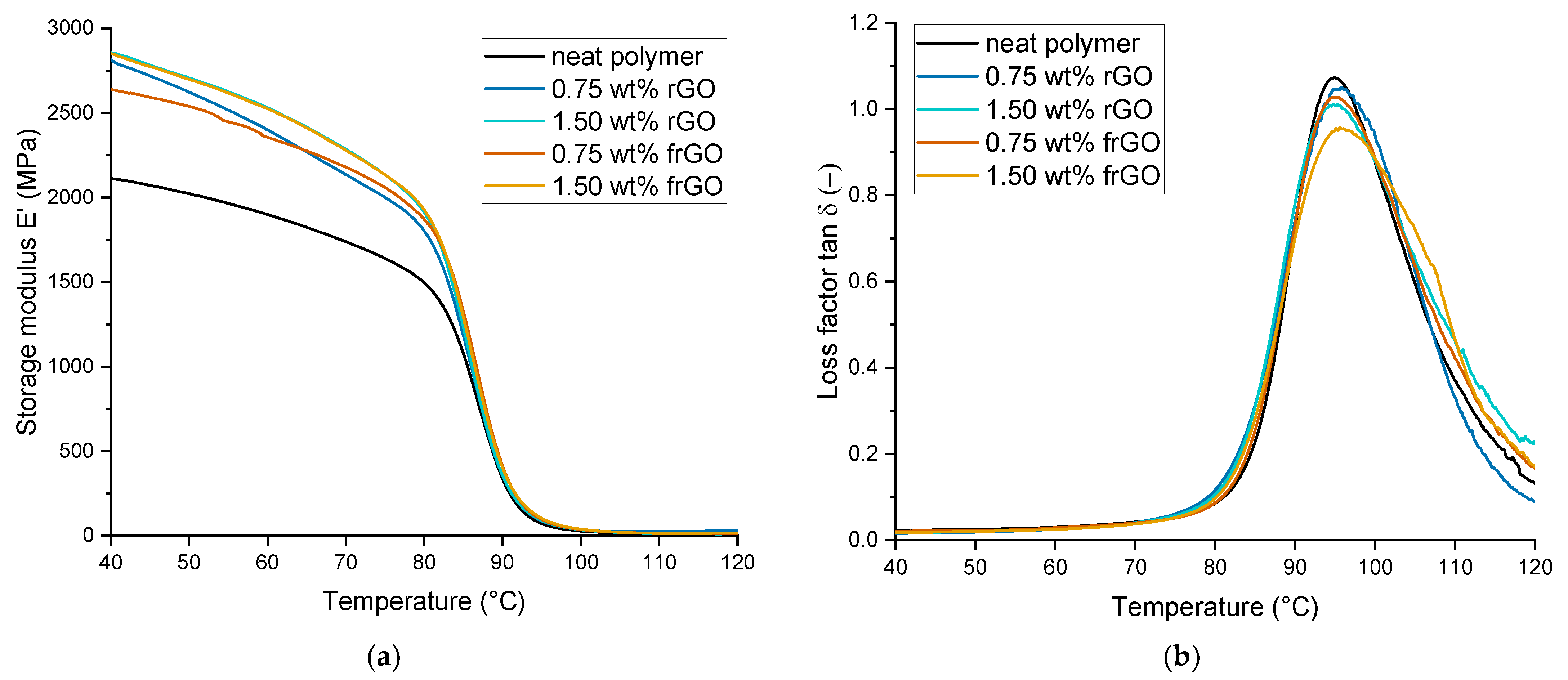
The thermomechanical as well as viscoelastic behavior was evaluated by dynamic mechanical analysis (DMA) and using a DMA 2980 from TA Instruments (New Castle, DE). In accordance to DIN 65583:1999 [
35], the tests were executed in dual-cantilever mode at a frequency of 1 Hz within a temperature range of room temperature to 130 °C and at a heating rate of 3 °C min
−1. A total of three specimens per material configuration was evaluated.
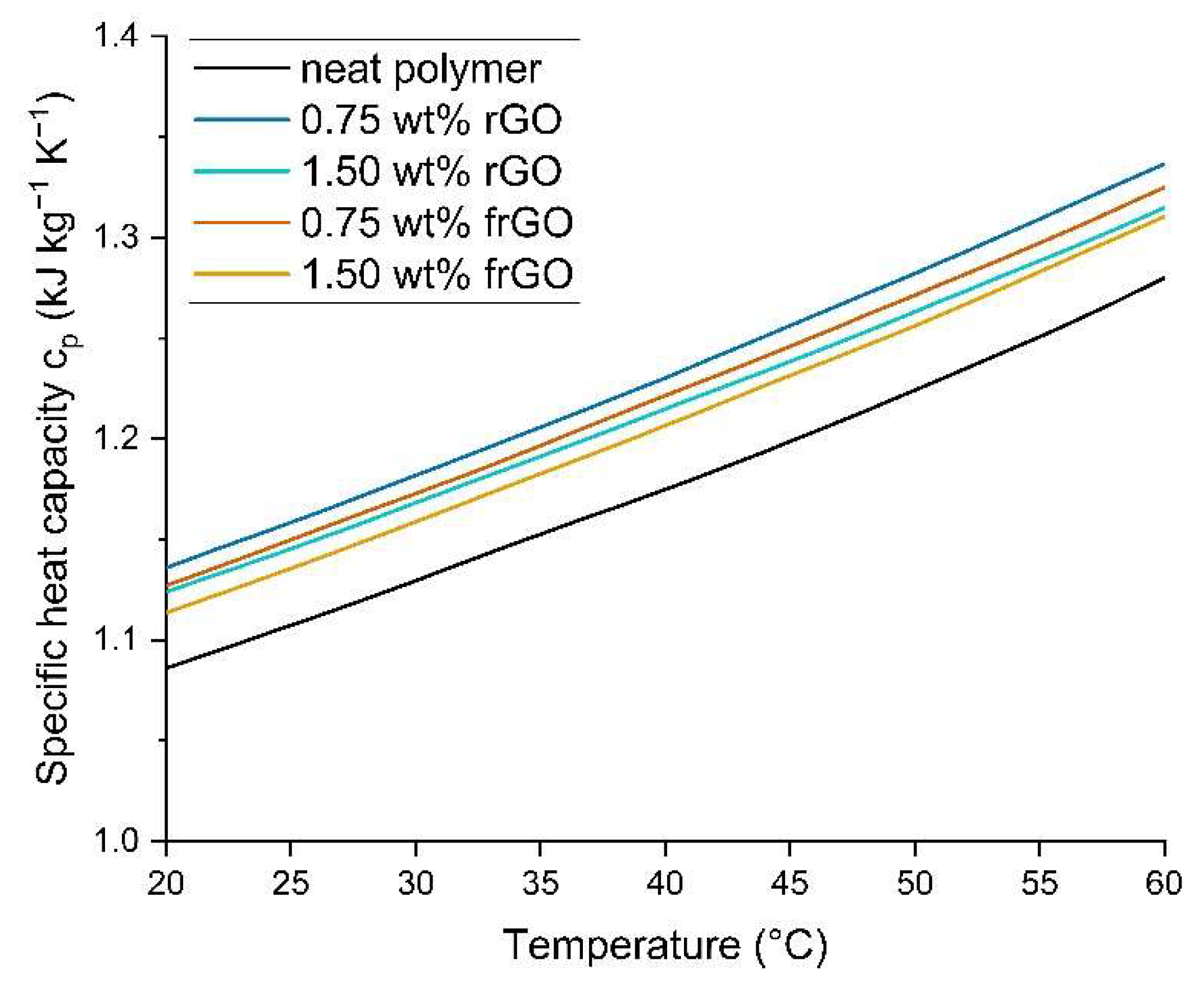
Differential scanning calorimetry (DSC) was performed using a DSC 2920 from TA Instruments and according to DIN EN ISO 11357-4:2021 [
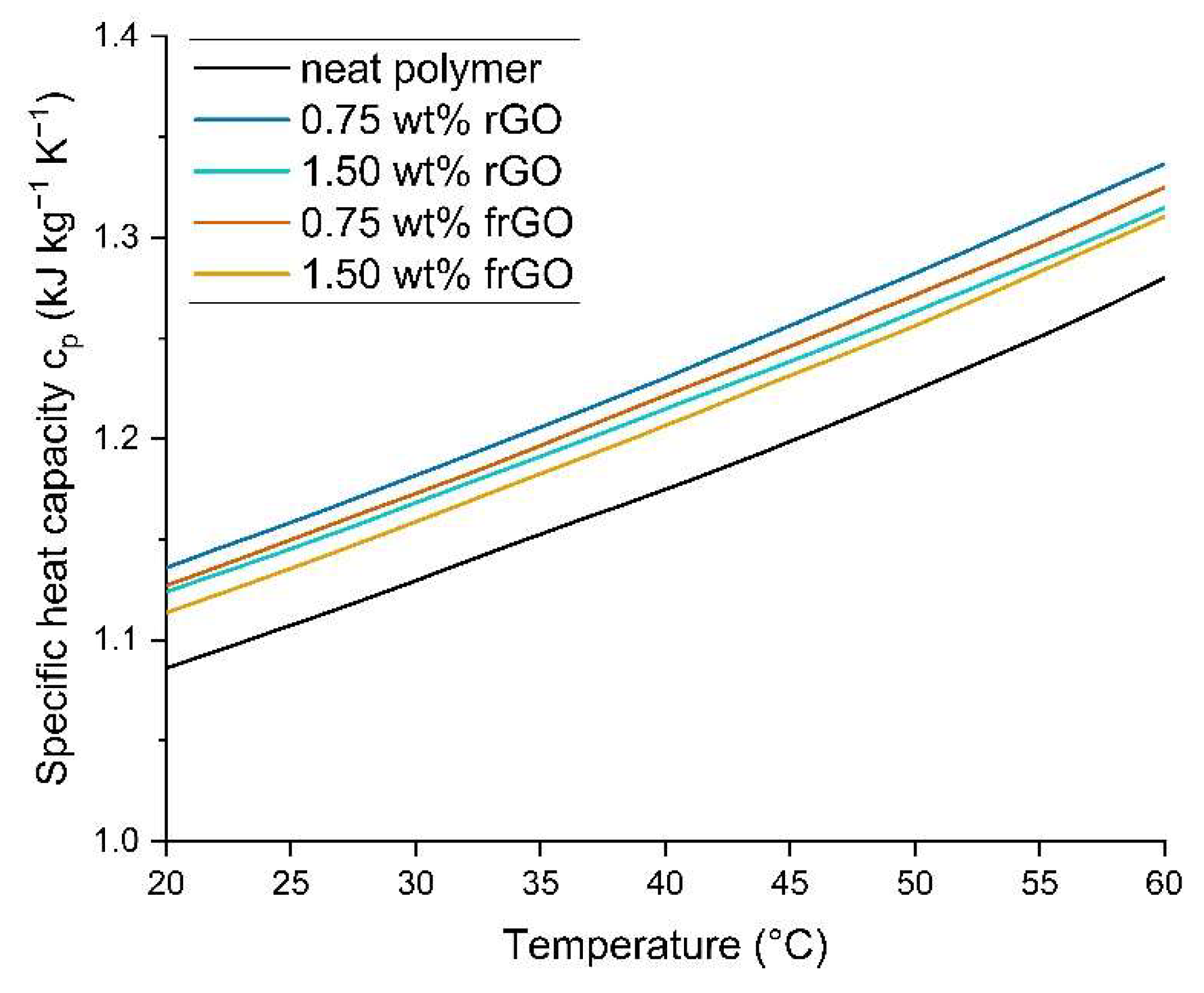
36] to determine the specific heat capacity c
p of the neat polymer and nanocomposite samples. The respective sample was initially kept at −20 °C for 2 min and then heated using a modulated DSC from −20 °C to 70 °C at a heating rate of 5 °C min
−1. Each measurement was acquired in a nitrogen atmosphere and measurements were obtained from three samples of the respective material.
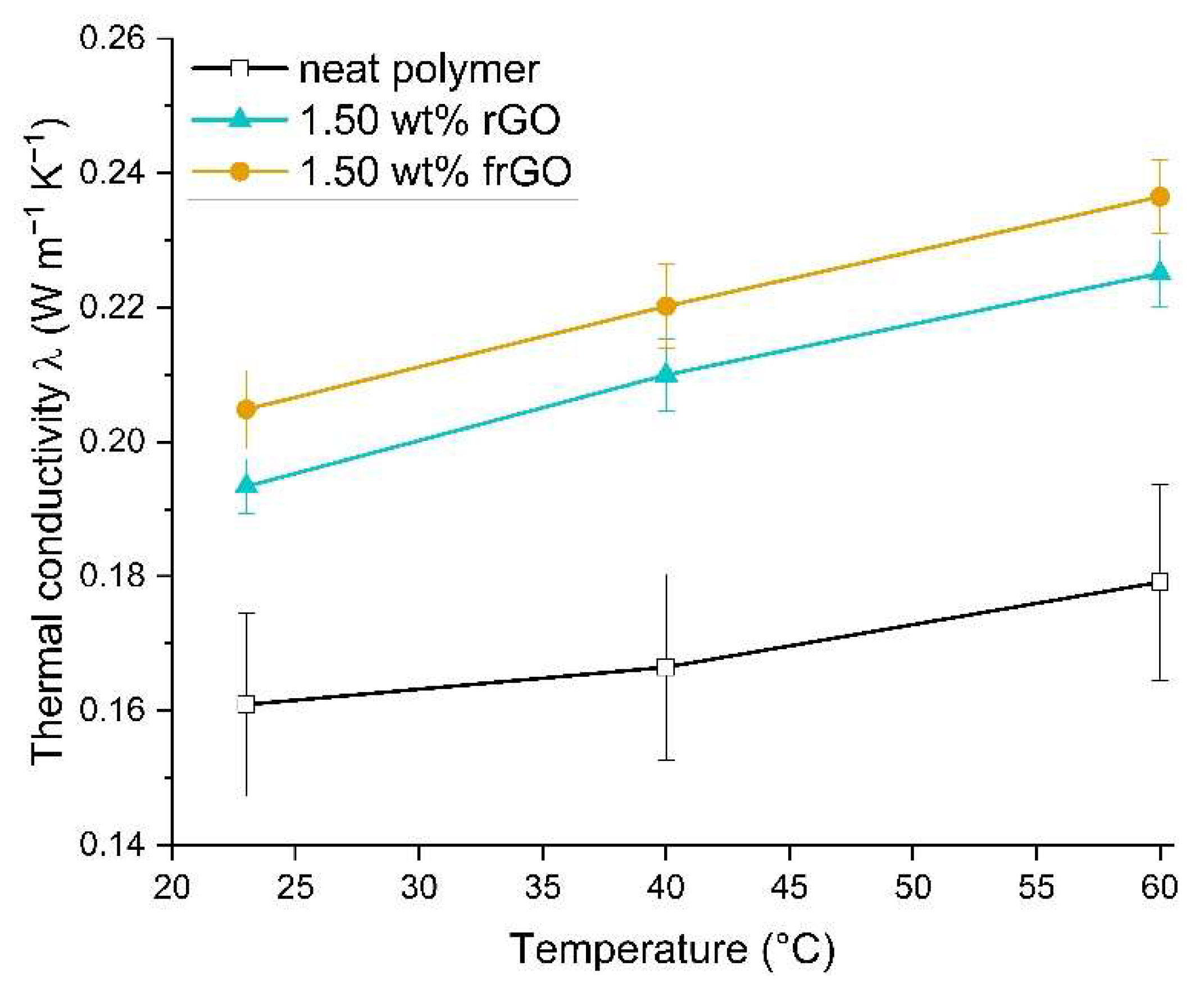
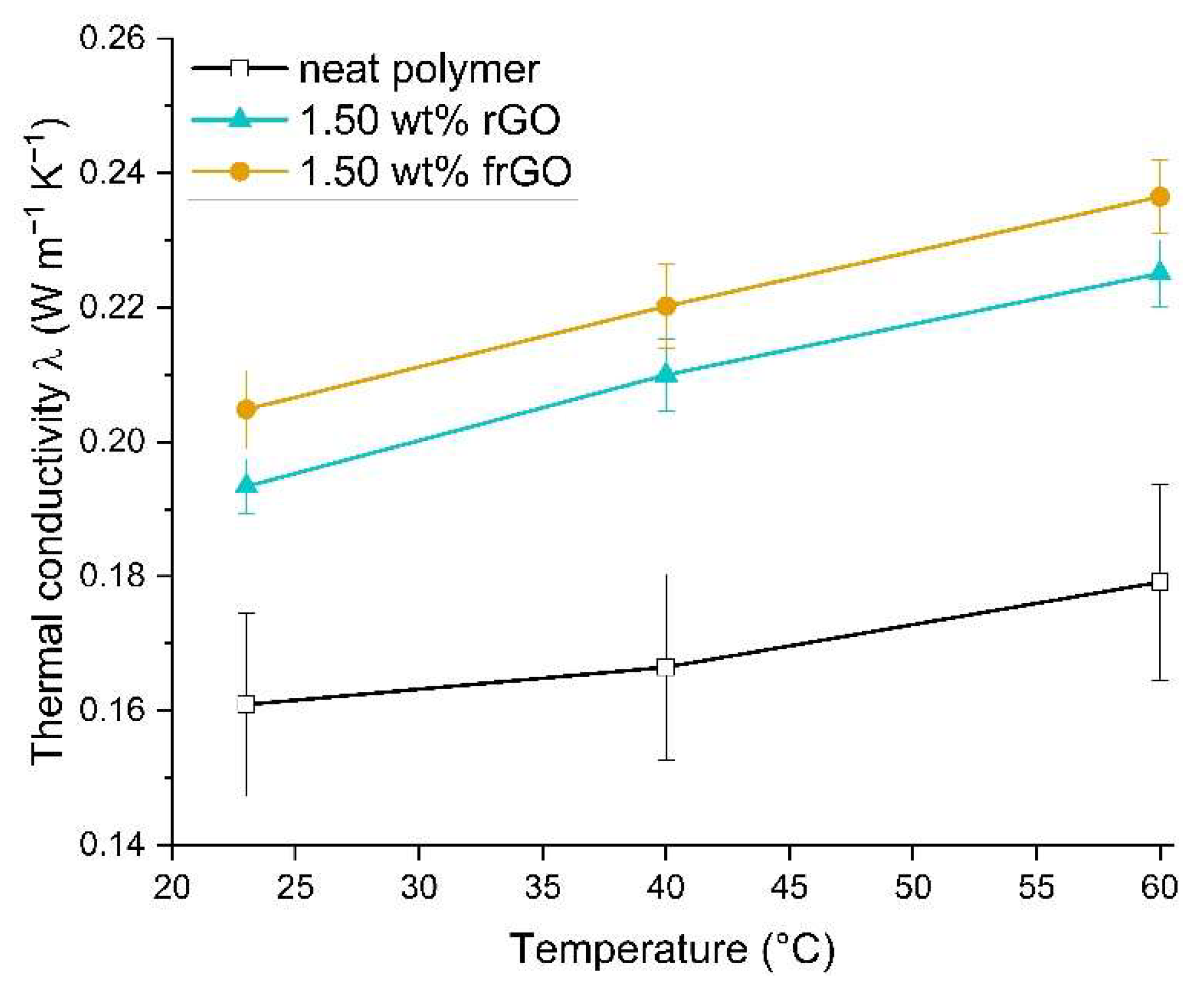
Laser flash analysis (LFA) was conducted in accordance to DIN EN ISO 22007-4:2017 [
37] and using a Netzsch (Selb, Germany) Nanoflash LFA 447. The thermal diffusivity a of each specimen was evaluated four times in series at each isothermal testing temperature of room temperature, 40 °C and 60 °C. A total of three specimens was analyzed for each material configuration. This was used to evaluate the thermal conductivity λ using
where c
p is the specific heat capacity and ρ is the density of the material.
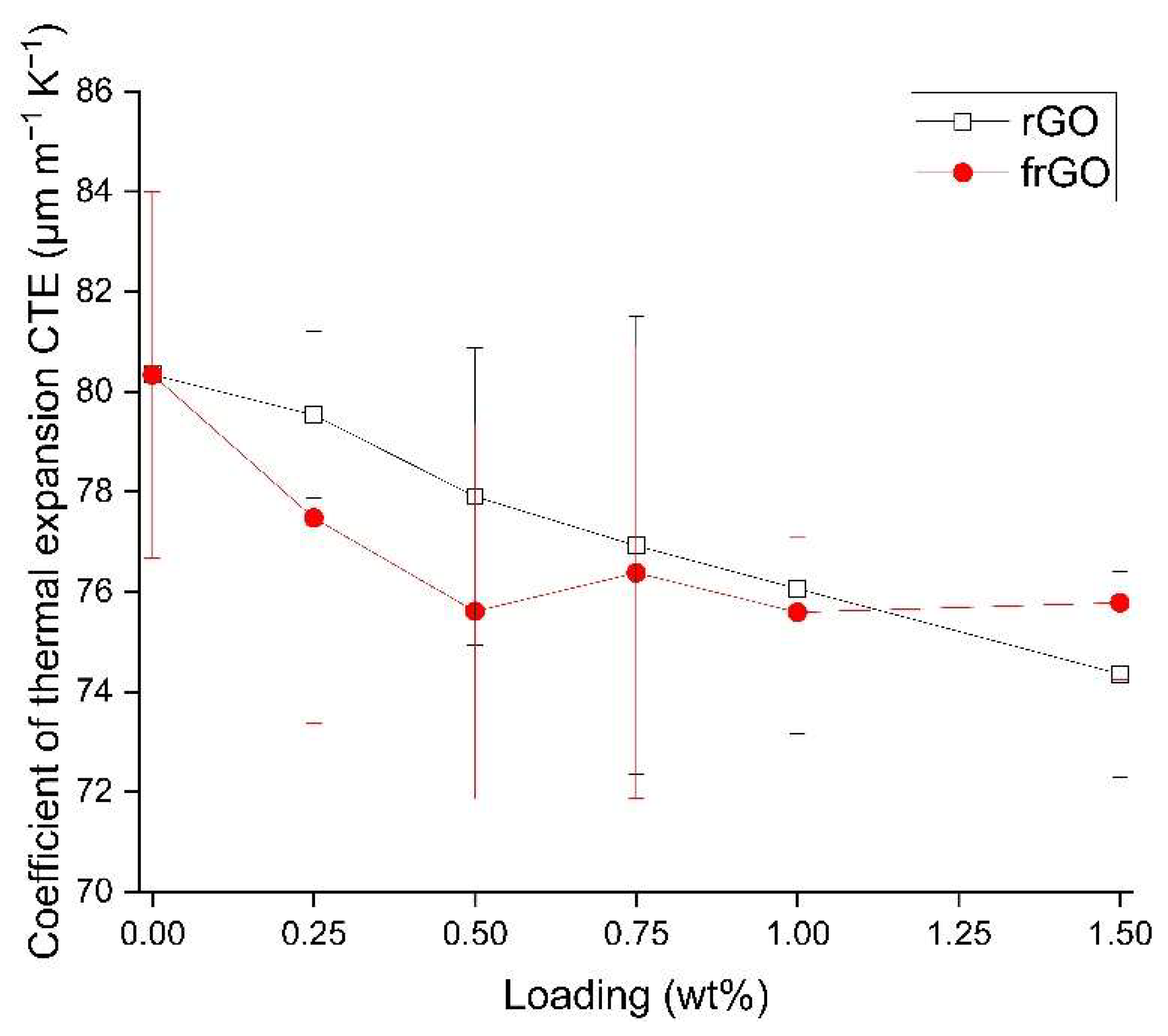
The coefficient of thermal expansion (CTE) of the examined materials was evaluated based on thermomechanical analysis (TMA) and using a TA Instruments TMA 2940. In accordance to DIN 53752:1980 [
38], a total of three samples was assessed for each material configuration. Each specimen was heated 3 times in succession from room temperature to 70 °C at a heating rate of 3 °C min
−1.
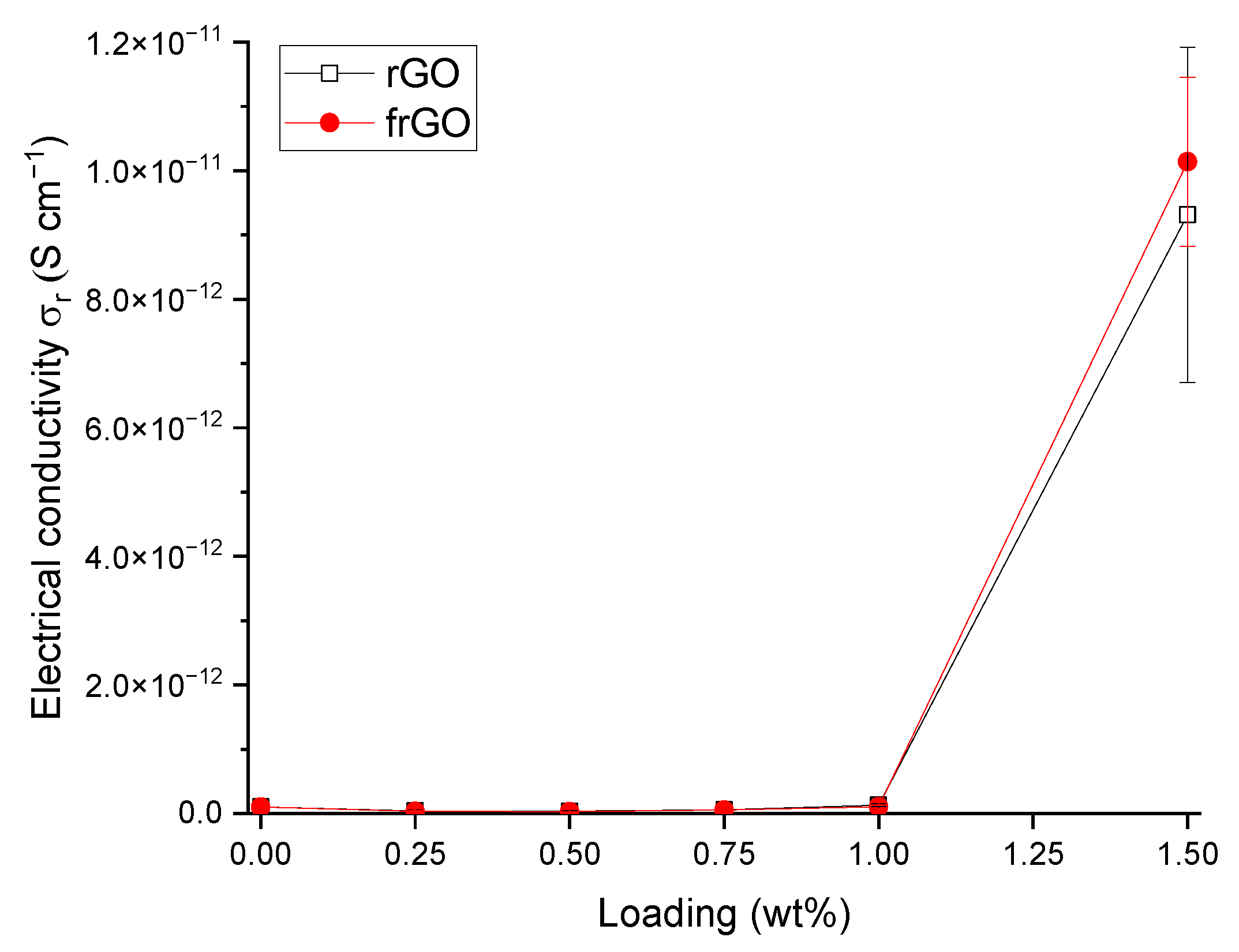
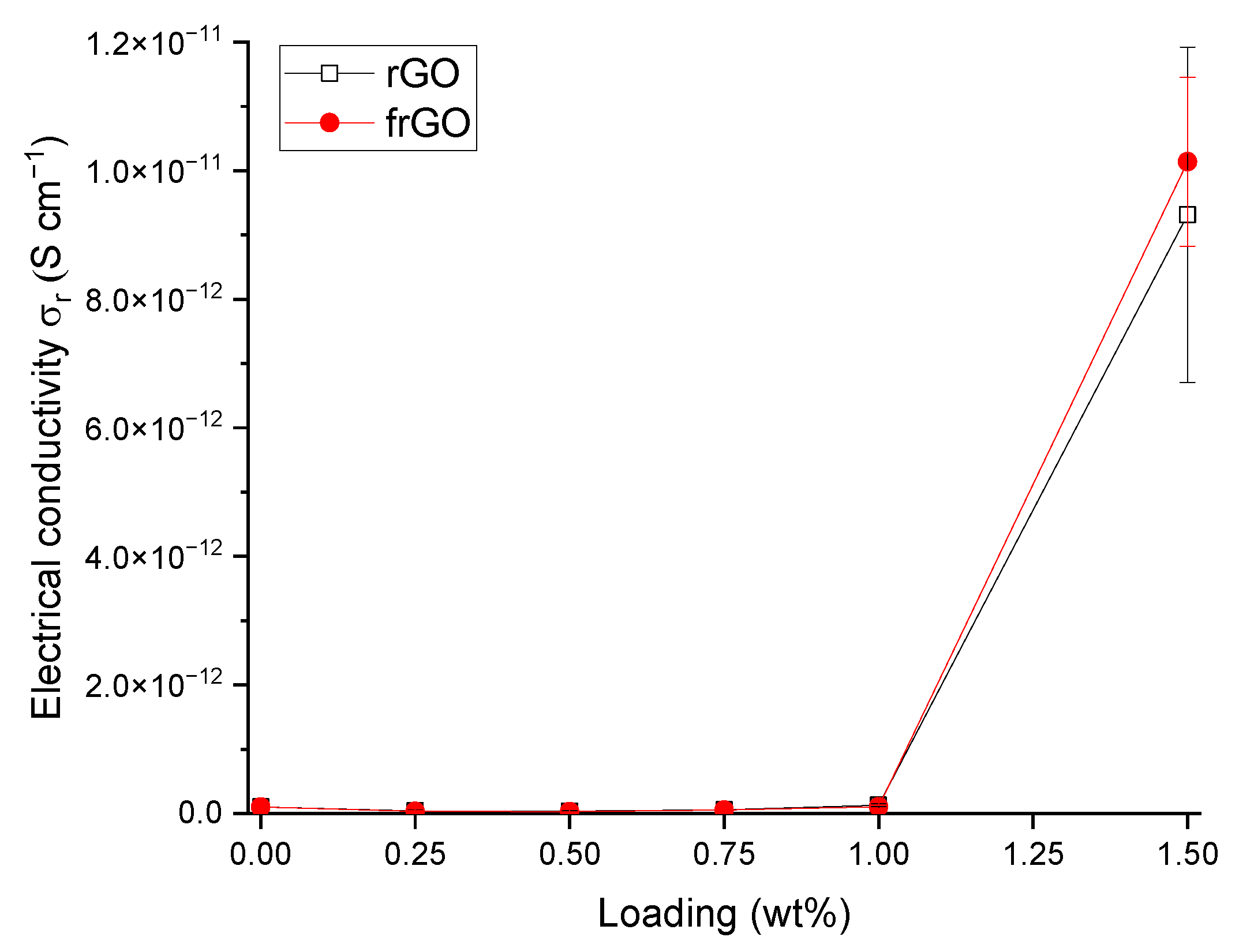
In order to determine the electrical conductivity of the neat polymer and nanocomposite samples, the volume resistance of these materials was measured based on DIN EN 62631-3-1 (VDE 0307-3-1):2017 [
39] and using a Keysight (Santa Rosa, CA, USA) N1424 resistivity cell connected to a Keysight N1424 volume/surface selector box, which was attached to a Keysight B2987A electrometer/high resistance meter. The respective specimen was clamped with a force of 5 kg using a circular electrode with a diameter of 88 mm. The volume resistance R
x was noted after each specimen was exposed to the voltage source for 1 min to allow a settling of the measured resistance. The volume resistance was used to obtain the sample’s electrical conductivity
by
where
is the effective area of the electrode and h is the specimen’s mean height. The electrical conductivity was assessed based on three specimens for each material configuration, where each sample was measured three times. As the measurement procedure might lead to the charging of the sample, it was ensured that repetitive measurements of the respective sample were only repeated after 24 h.
4. Conclusions
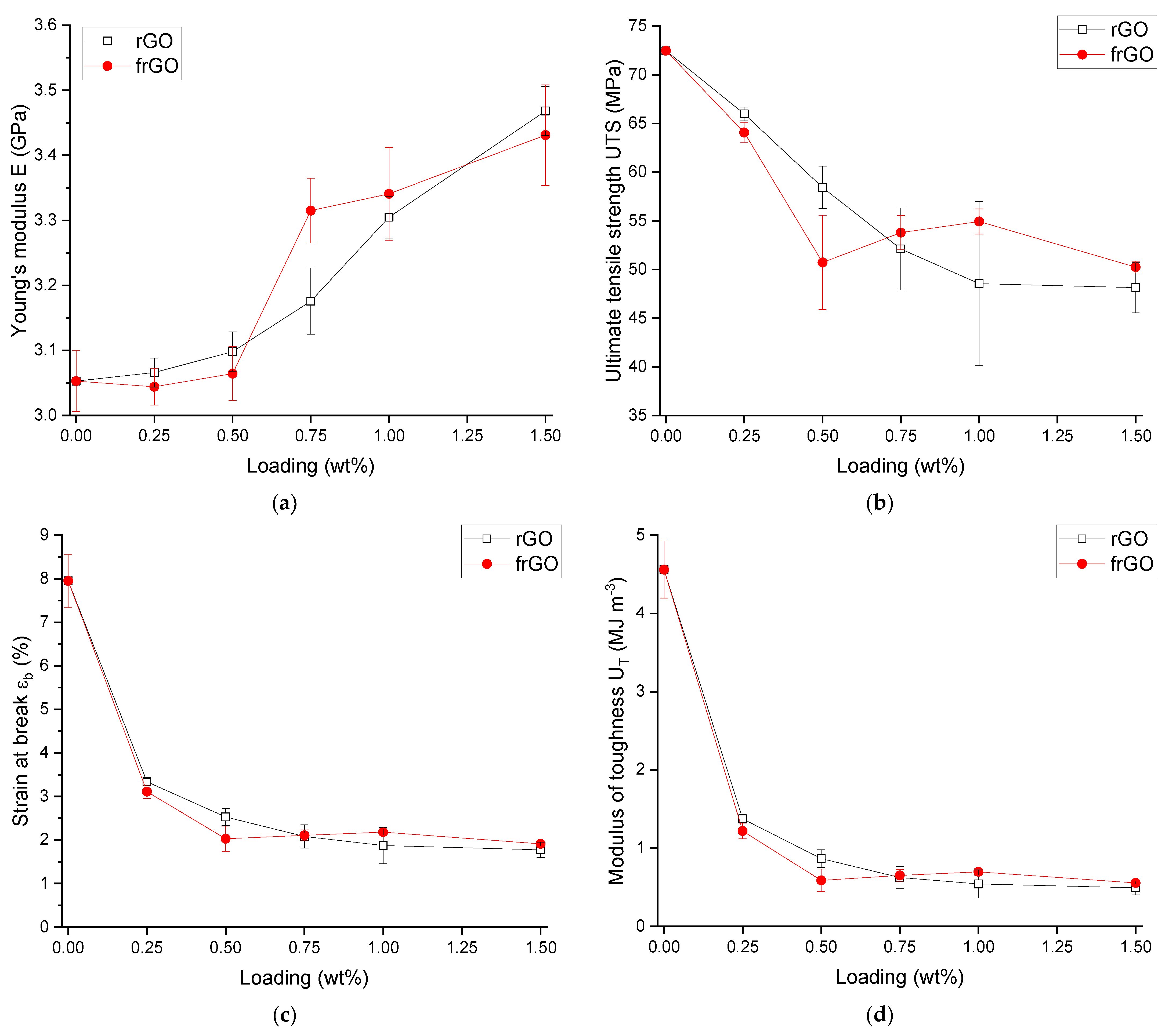
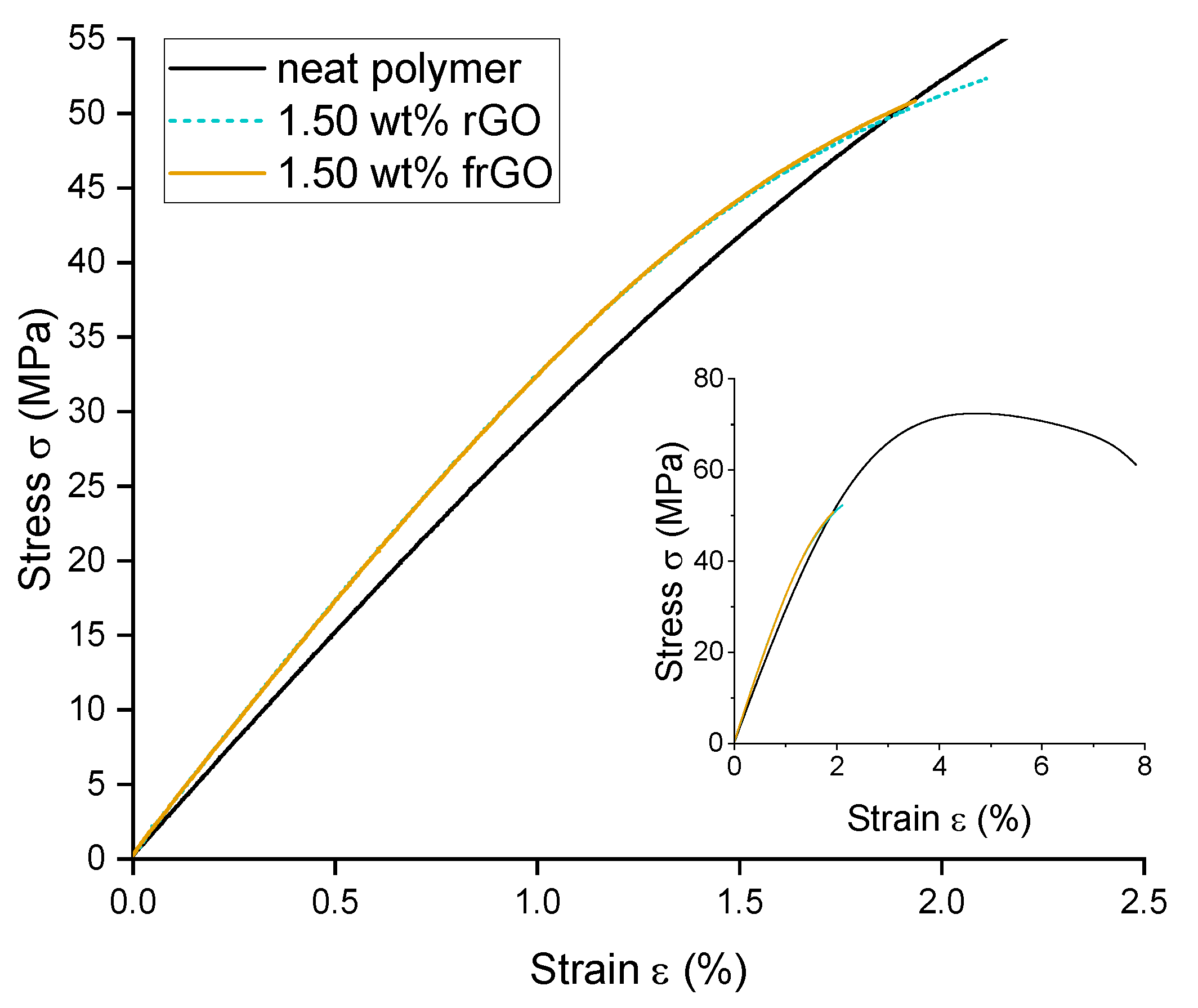
This work evaluated the influence of an amine-functionalization of reduced graphene oxide on the mechanical, thermal and electrical properties of epoxy/amine-based hardener nanocomposites. The morphology as examined by the use of SEM images of the rGO and frGO particles showed that no major visual differences were observed between the used additives. The elemental composition, however, demonstrated a marginally higher C/O ratio and the presence of nitrogen functionalities in the case of the frGO particles. The mechanical characteristics of the produced nanocomposites were examined by the use of tensile tests, the evaluation of the resulting fracture surfaces and DMA. As proven by both the tensile tests and DMA, an increased stiffness with increasing weight fractions of (f)rGO was determined for all material configurations, but this comes at the cost of a deterioration of the strength and toughness, as well as a more brittle fracture behavior. Furthermore, the more rugged fracture surfaces and the DMA measurements of the frGO-including nanocomposites indicated an improved interfacial bonding caused by the amine-functionalities on the frGO particles. Hence, the bonding, as achieved by the used plasma functionalization process, seems to be too weak to result in a significant enhancement of the mechanical properties. A similar behavior with no significant difference between the rGO- and frGO-including nanocomposites was observed in the case of the coefficient of thermal expansion, specific heat capacity and electrical conductivity of the prepared nanocomposites. However, the frGO-including nanocomposite demonstrated significantly higher values of the thermal diffusivity and thermal conductivity than the nanocomposites with rGO as the used additive. This confirms the effectiveness of the used functionalization by the applied plasma processing with respect to the thermal conductivity. Furthermore, an approximately linear increase in the thermal conductivity was observed with both increasing weight fractions of the used additives and rising temperatures. These findings suggest the potential of rGO and especially frGO to further increase the thermal performance in composite materials at room temperature, but also at elevated temperatures. This will be useful to enable applications, such as the use in thermal interface materials or fiber-reinforced polymers.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}