Polydopamine-Coated Paraffin Microcapsules as a Multifunctional Filler Enhancing Thermal and Mechanical Performance of a Flexible Epoxy Resin





Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Sample Preparation
2.2.1. Modification of Microcapsules with PDA
2.2.2. Preparation of Epoxy-Based Composites
2.3. Experimental Techniques
3. Results and Discussion
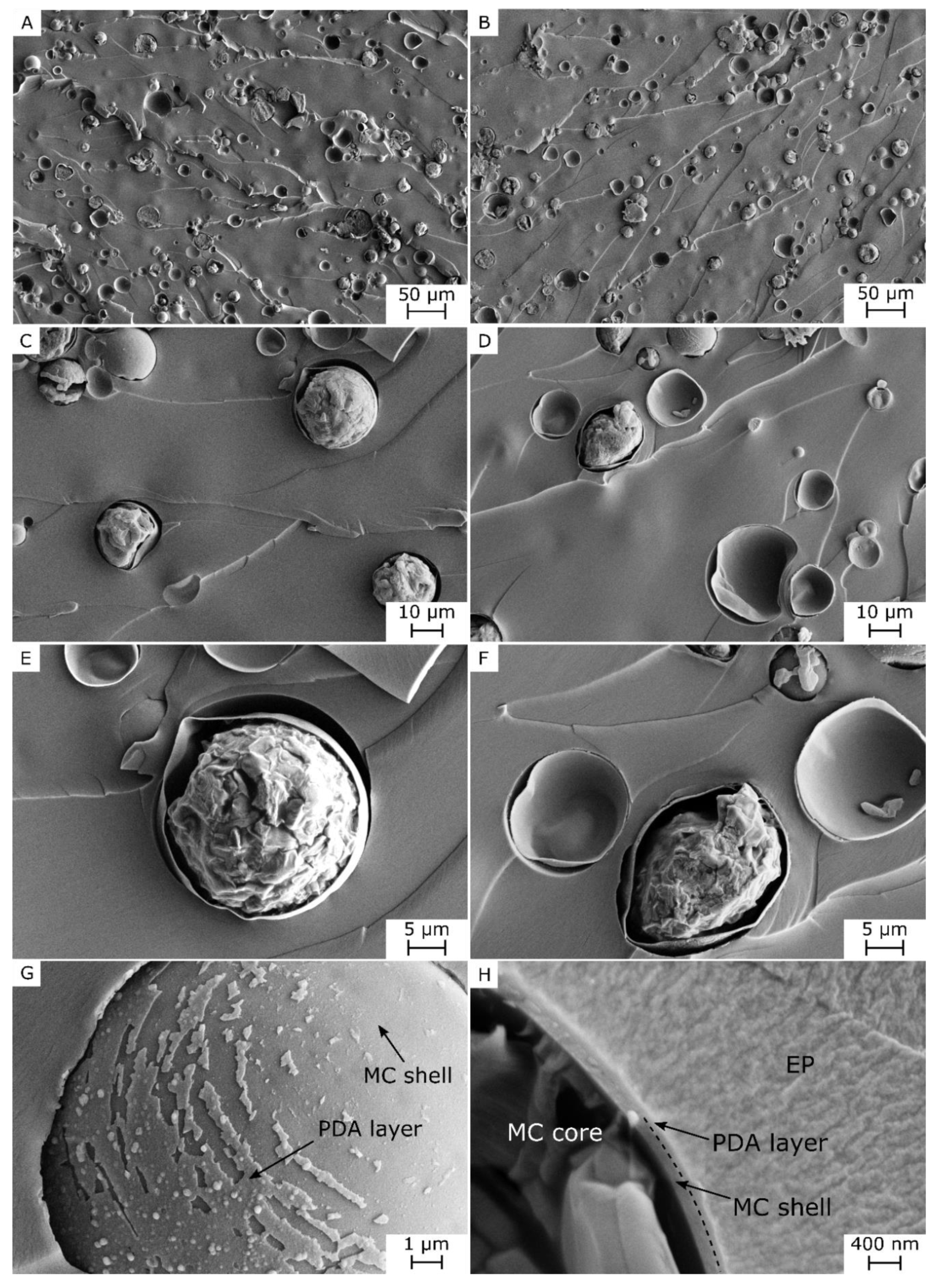
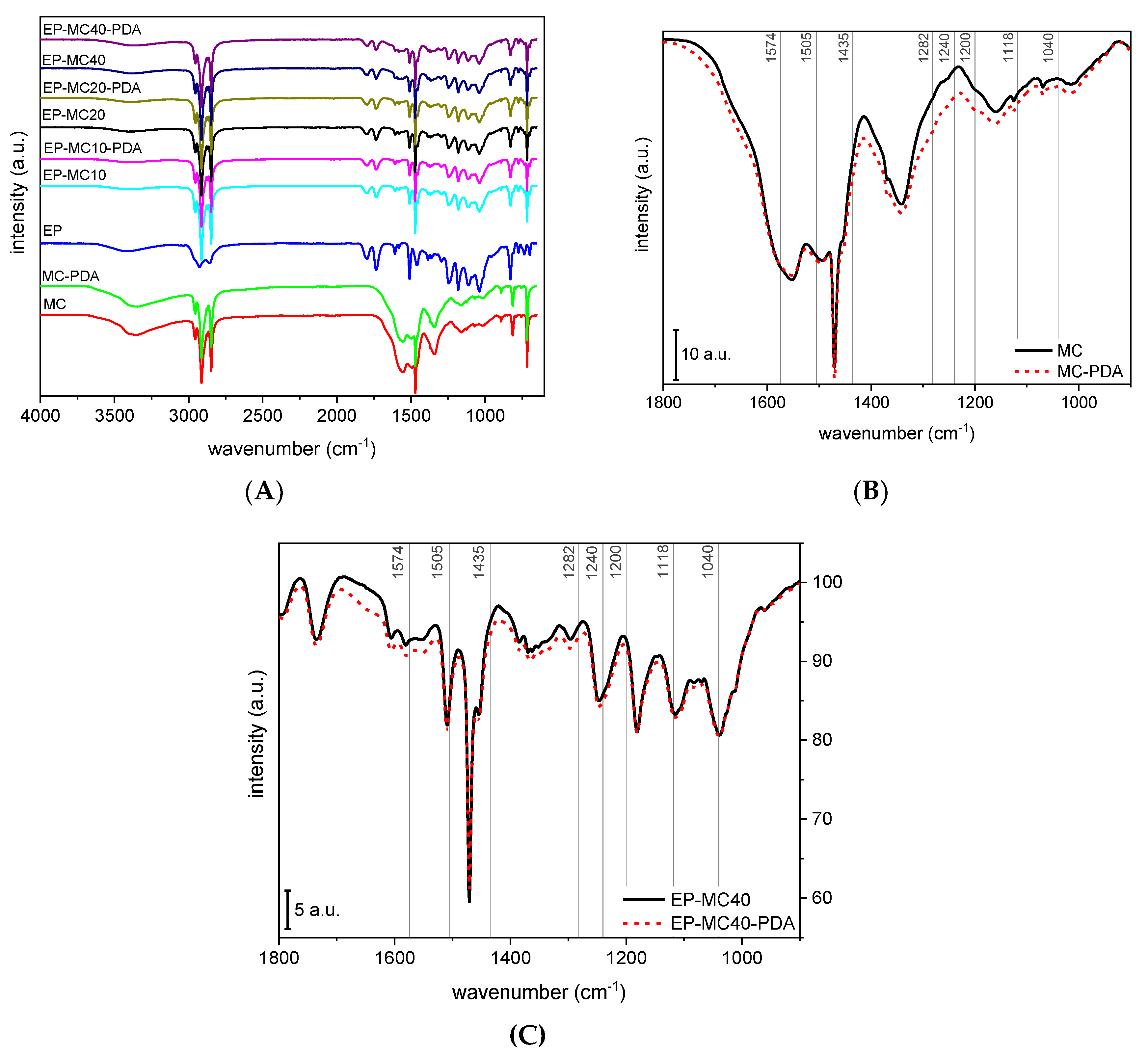
3.1. Microstructural and Spectroscopic Properties
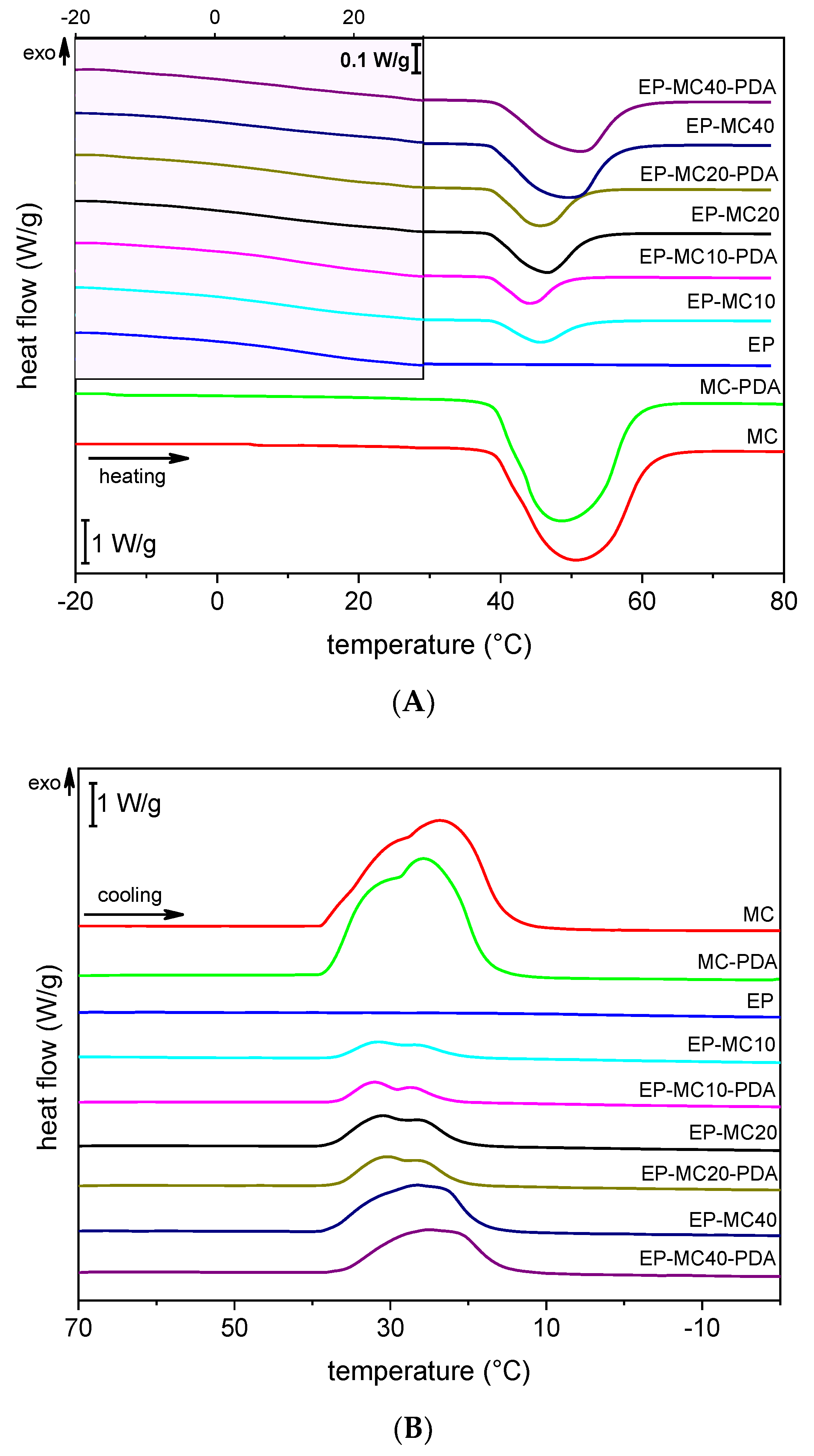
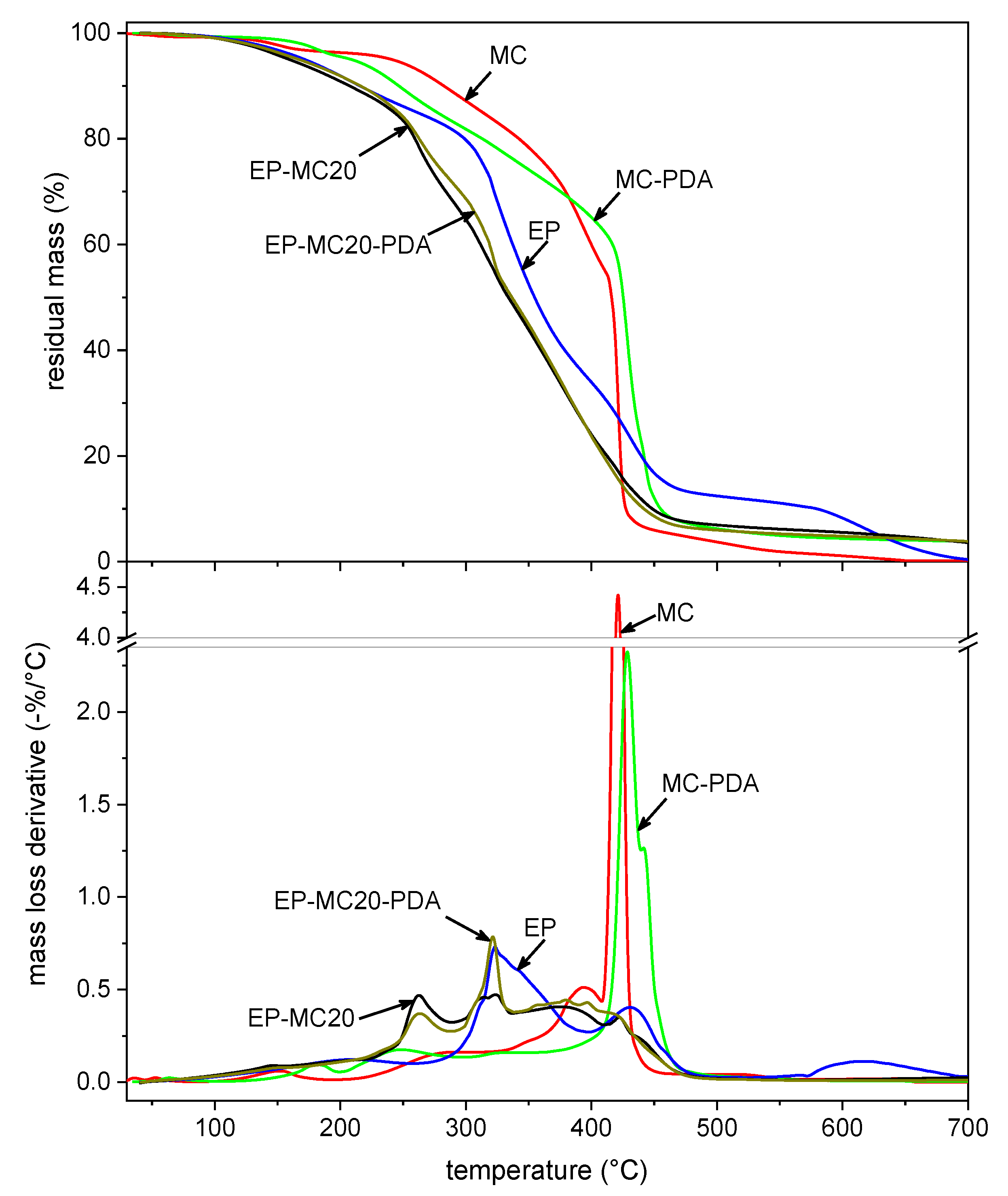
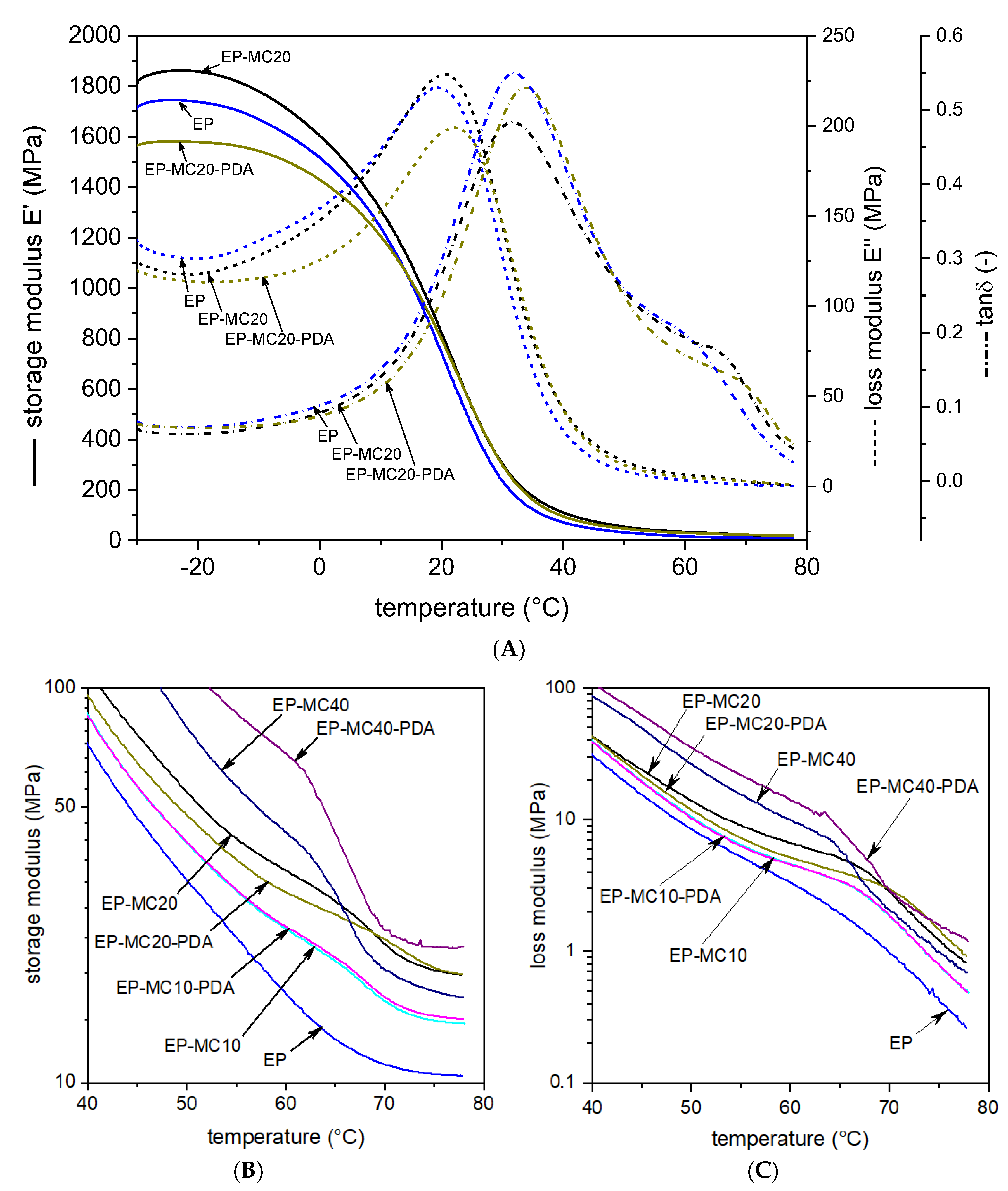
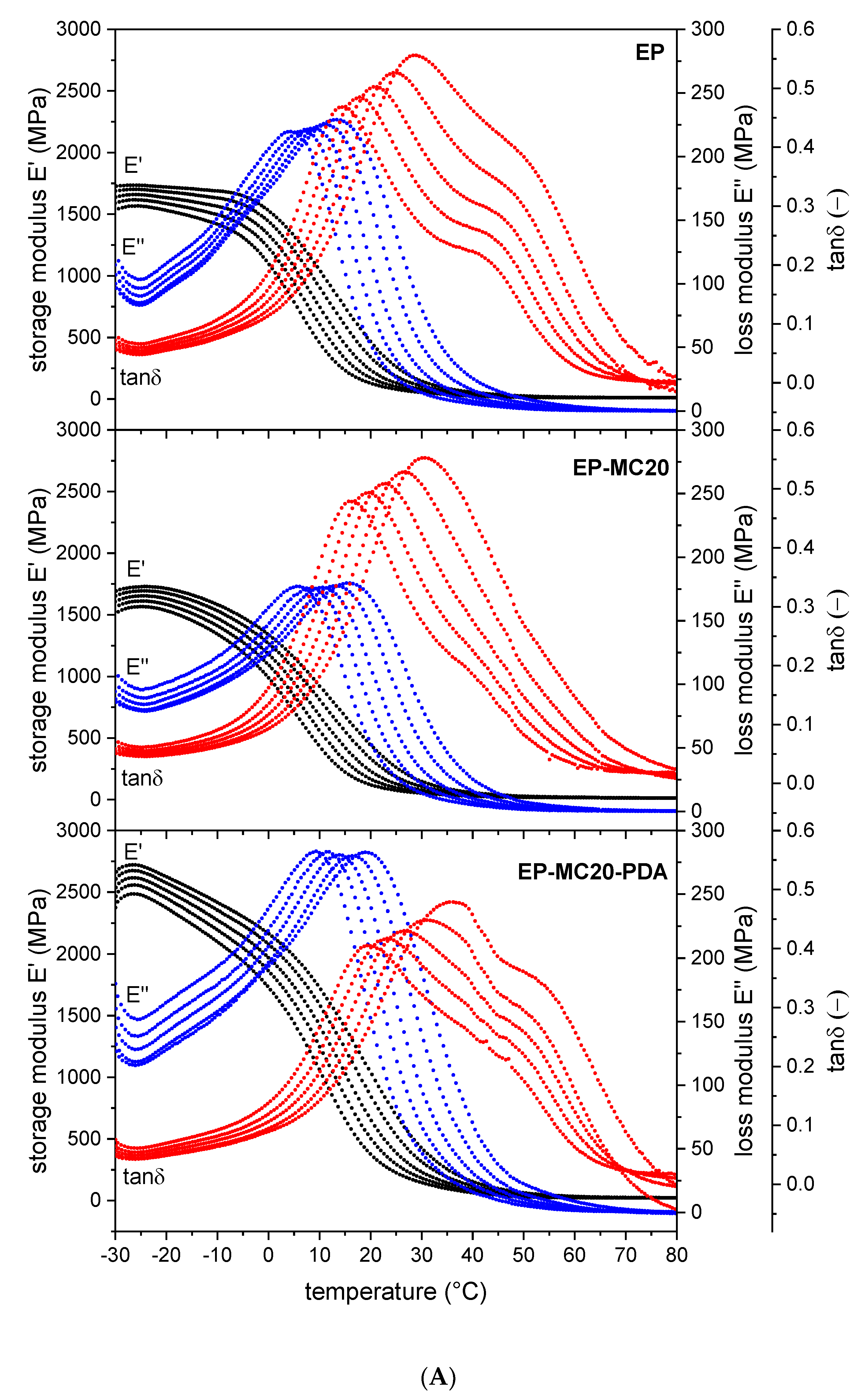
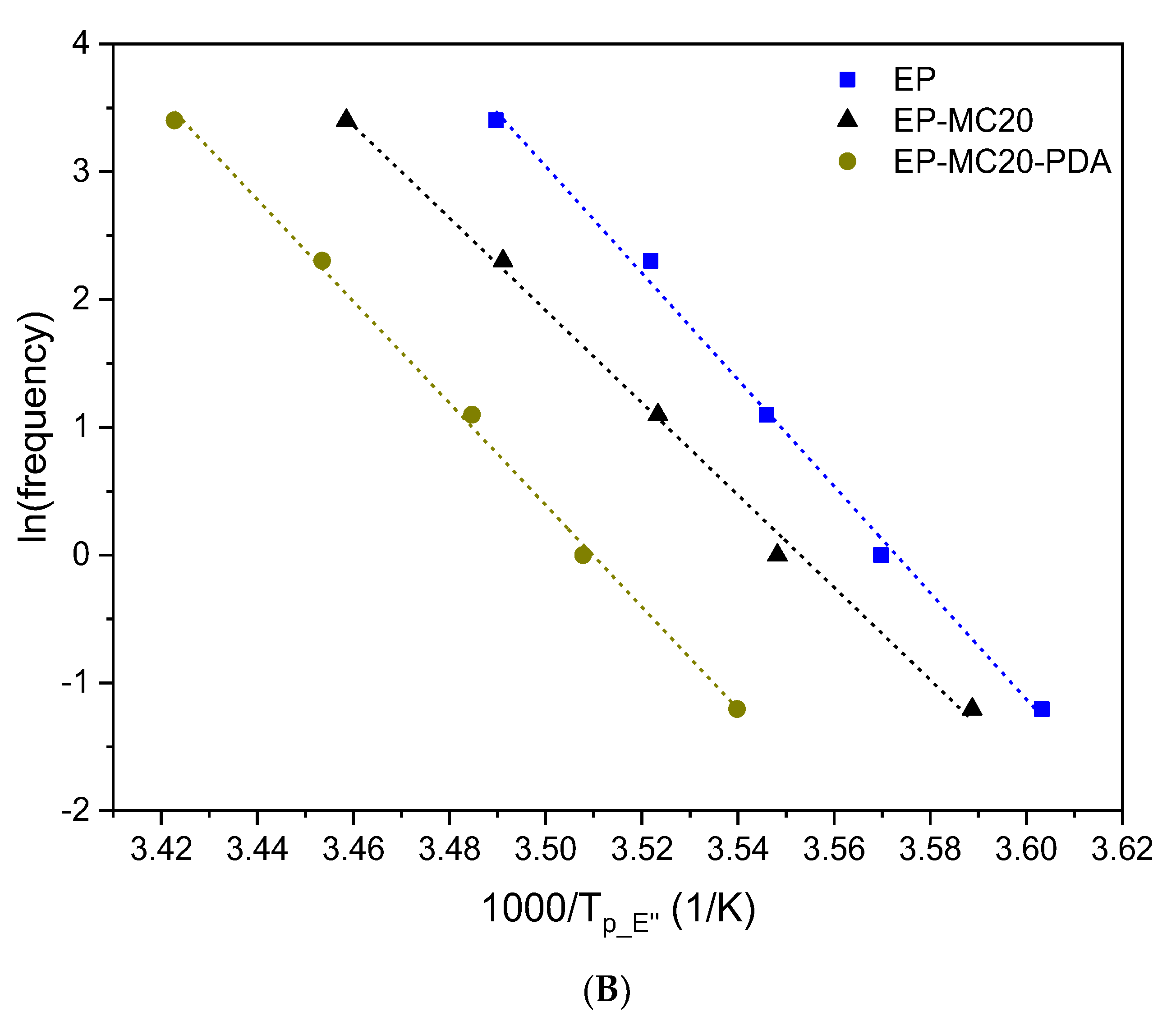
3.2. Thermal and Dynamic Mechanical Properties
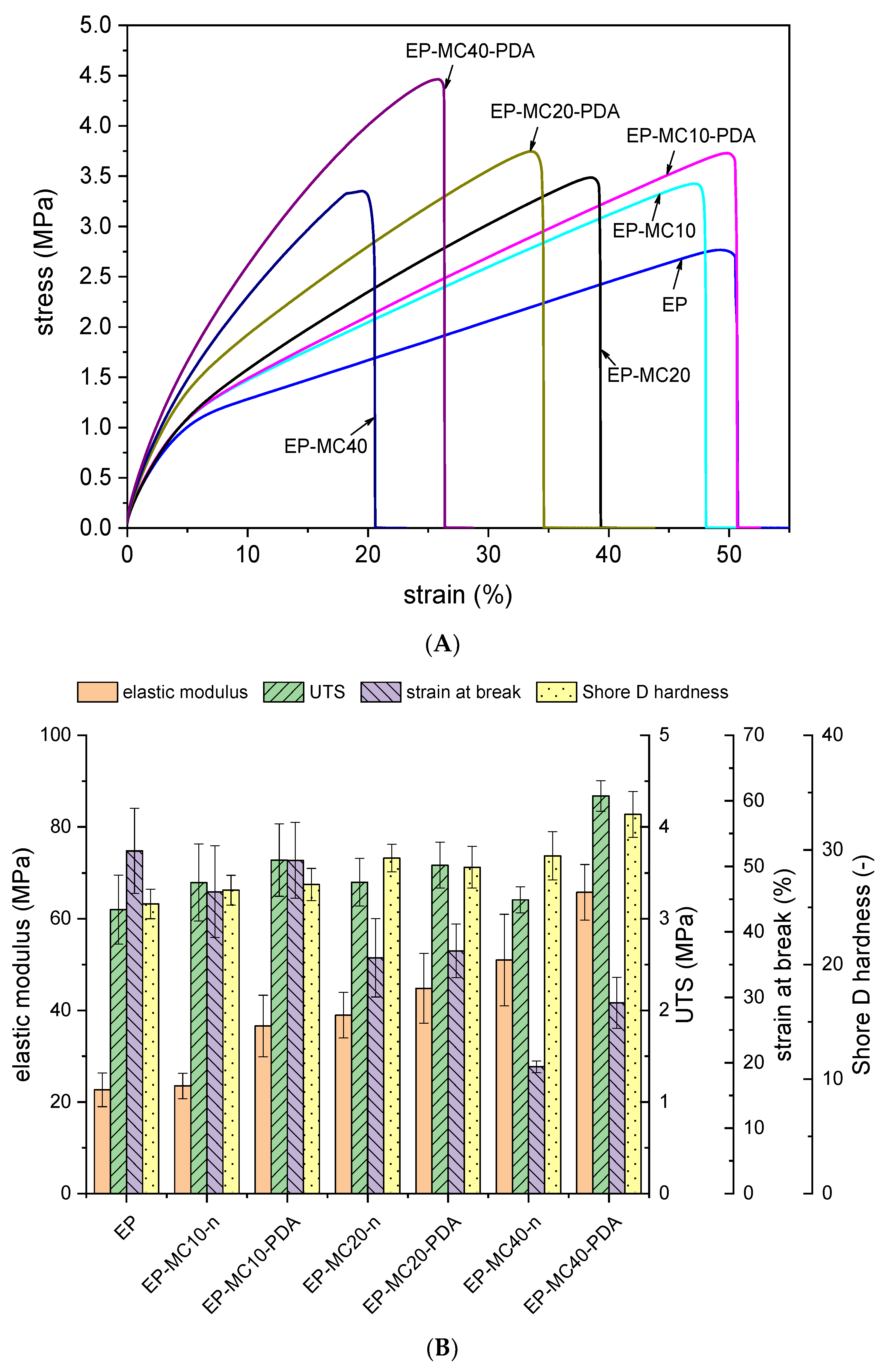
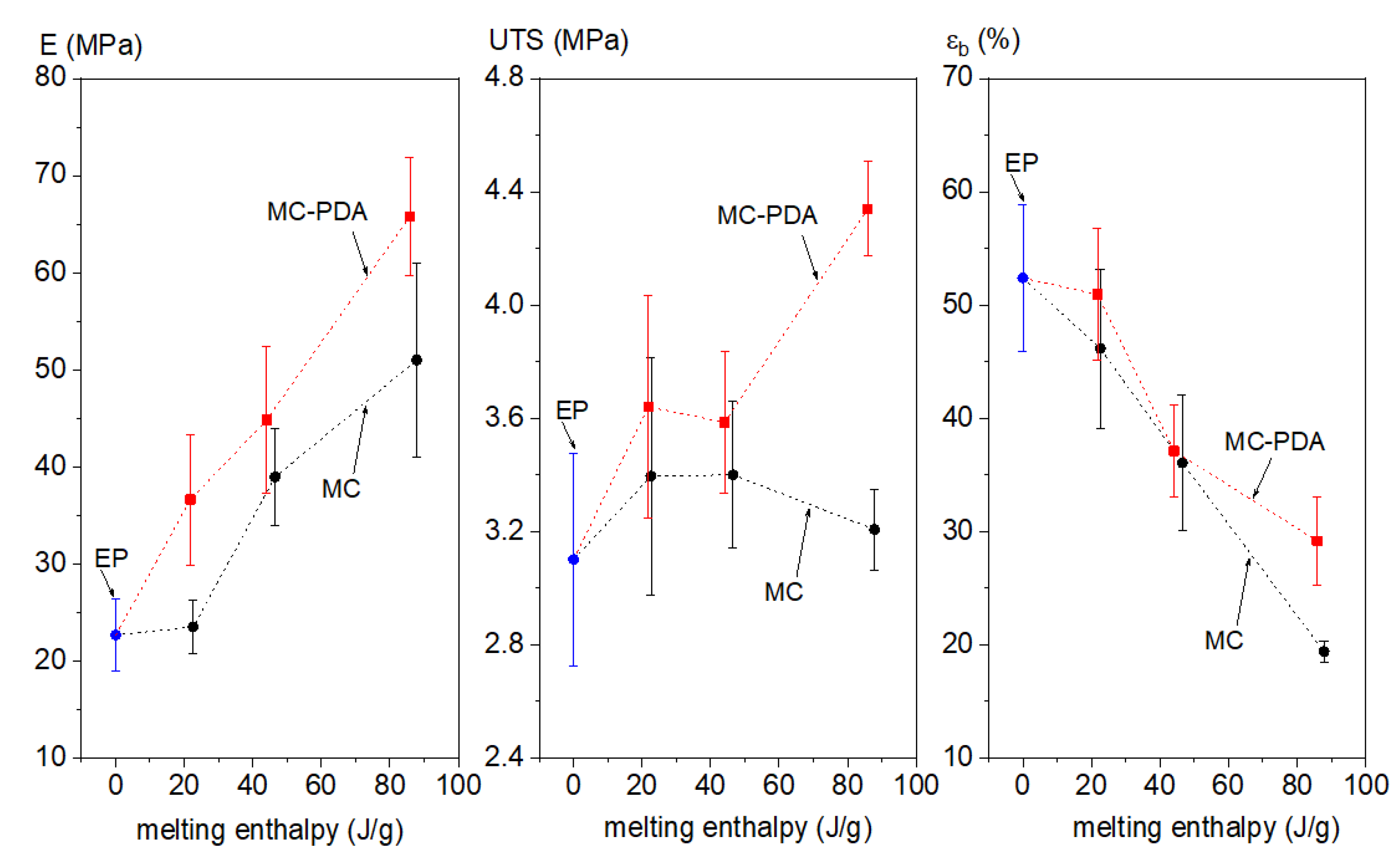
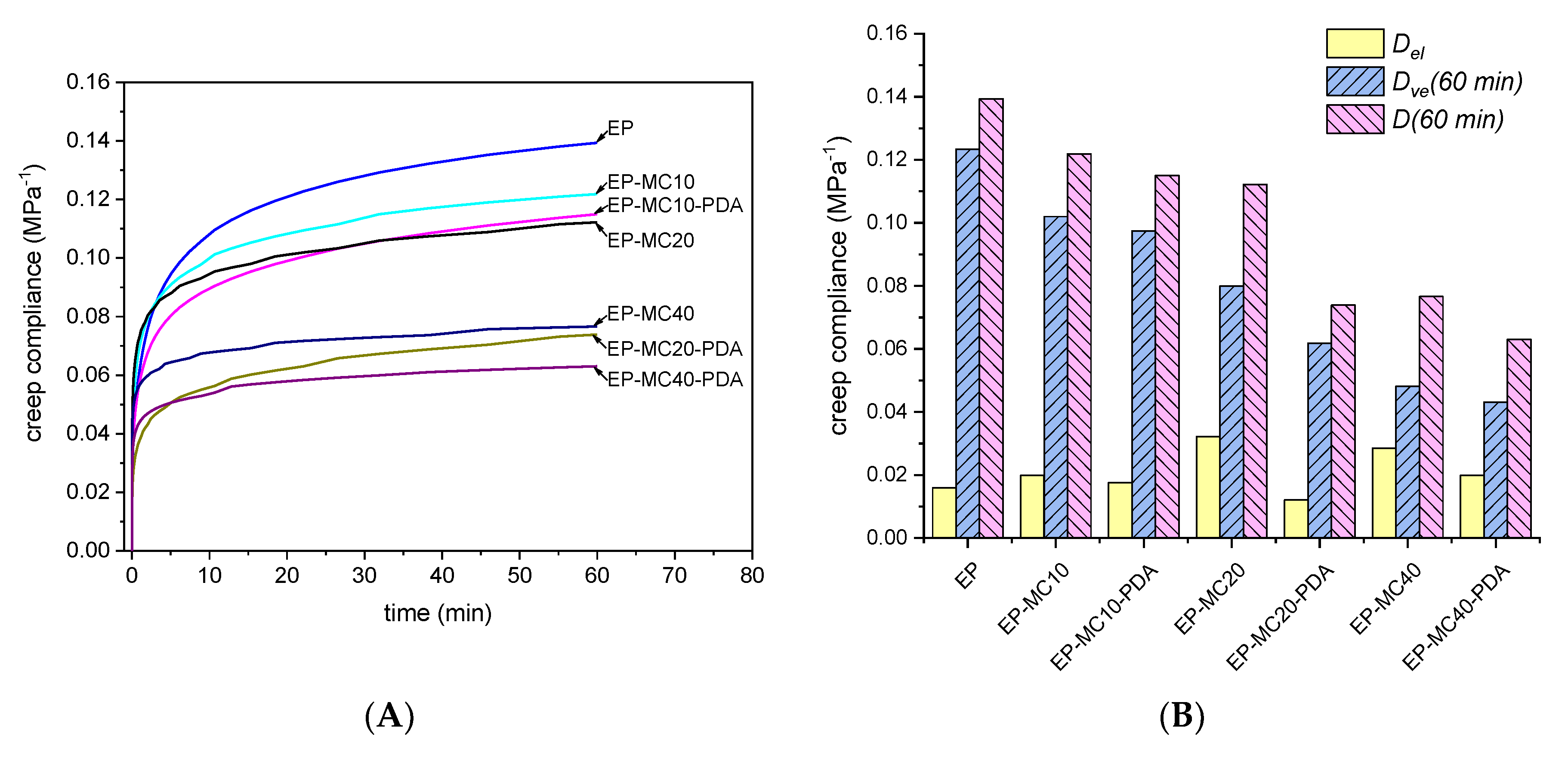
3.3. Mechanical Properties
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pielichowska, K.; Pielichowski, K. Phase change materials for thermal energy storage. Prog. Mater. Sci. 2014, 65, 67–123. [Google Scholar] [CrossRef]
- Fleischer, A.S. Thermal Energy Storage Using Phase Change Materials—Fundamentals and Applications; Series Springer Briefs in Thermal Engineering and Applied Science; Springer: Heidelberg, Germany, 2015. [Google Scholar]
- Khadiran, T.; Hussein, M.Z.; Zainal, Z.; Rusli, R. Encapsulation techniques for organic phase change materials as thermal energy storage medium: A review. Sol. Energy Mater. Sol. Cells 2015, 143, 78–98. [Google Scholar] [CrossRef]
- Zhang, P.; Xiao, X.; Ma, Z.W. A review of the composite phase change materials: Fabrication, characterization, mathematical modeling and application to performance enhancement. Appl. Energy 2016, 165, 472–510. [Google Scholar] [CrossRef]
- Abhat, A. Low temperature latent heat thermal energy storage: Heat storage materials. Sol. Energy 1983, 30, 313–332. [Google Scholar] [CrossRef]
- Agyenim, F.; Hewitt, N.; Eames, P.; Smyth, M. A review of materials, heat transfer and phase change problem formulation for latent heat thermal energy storage systems (lhtess). Renew. Sustain. Energy Rev. 2010, 14, 615–628. [Google Scholar] [CrossRef]
- Baştürk, E.; Kahraman, M.V. Preparation and performances of uv-cured methacrylated polyacrylic acid-based core-shell hybrid phase change materials. Polym. Eng. Sci. 2018, 58, 2166–2174. [Google Scholar] [CrossRef]
- Bayés-García, L.; Ventolà, L.; Cordobilla, R.; Benages, R.; Calvet, T.; Cuevas-Diarte, M.A. Phase change materials (pcm) microcapsules with different shell compositions: Preparation, characterization and thermal stability. Sol. Energy Mater. Sol. Cells 2010, 94, 1235–1240. [Google Scholar] [CrossRef]
- Zhou, J.; Cui, Y.; Yao, H.; Ma, J.; Ren, H. Nanocapsules containing binary phase change material obtained via miniemulsion polymerization with reactive emulsifier: Synthesis, characterization, and application in fabric finishing. Polym. Eng. Sci. 2019, 59, E42–E51. [Google Scholar] [CrossRef]
- Fredi, G.; Dirè, S.; Callone, E.; Ceccato, R.; Mondadori, F.; Pegoretti, A. Docosane-organosilica microcapsules for structural composites with thermal energy storage/release capability. Materials 2019, 12, 1286. [Google Scholar] [CrossRef]
- Cui, Y.; Xie, J.; Liu, J.; Pan, S. Review of phase change materials integrated in building walls for energy saving. Procedia Eng. 2015, 121, 763–770. [Google Scholar] [CrossRef]
- Kastiukas, G.; Zhou, X.M.; Castro-Gomes, J. Development and optimisation of phase change material-impregnated lightweight aggregates for geopolymer composites made from aluminosilicate rich mud and milled glass powder. Constr. Build. Mater. 2016, 110, 201–210. [Google Scholar] [CrossRef]
- Biswas, K.; Lu, J.; Soroushian, P.; Shrestha, S. Combined experimental and numerical evaluation of a prototype nano-pcm enhanced wallboard. Appl. Energy 2014, 131, 517–529. [Google Scholar] [CrossRef]
- Zhu, K.; Li, X.; Su, J.; Li, H.; Zhao, Y.; Yuan, X. Improvement of anti-icing properties of low surface energy coatings by introducing phase-change microcapsules. Polym. Eng. Sci. 2018, 58, 973–979. [Google Scholar] [CrossRef]
- Fok, S.C.; Shen, W.; Tan, F.L. Cooling of portable hand-held electronic devices using phase change materials in finned heat sinks. Int. J. Sci. 2010, 49, 109–117. [Google Scholar] [CrossRef]
- Sahoo, S.K.; Das, M.K.; Rath, P. Application of tce-pcm based heat sinks for cooling of electron components: A review. Renew. Sustain. Energy Rev. 2016, 59, 550–582. [Google Scholar] [CrossRef]
- Tomizawa, Y.; Sasaki, K.; Kuroda, A.; Takeda, R.; Kaito, Y. Experimental and numerical study on phase change material (pcm) for thermal management of mobile devices. Appl. Therm. Eng. 2016, 98, 320–329. [Google Scholar] [CrossRef]
- Ianniciello, L.; Biwolé, P.H.; Achard, P. Electric vehicles batteries thermal management systems employing phase change materials. J. Power Sources 2018, 378, 383–403. [Google Scholar] [CrossRef]
- Lazrak, A.; Fourmigué, J.-F.; Robin, J.-F. An innovative practical battery thermal management system based on phase change materials: Numerical and experimental investigations. Appl. Therm. Eng. 2018, 128, 20–32. [Google Scholar] [CrossRef]
- Ji, C.; Wang, Y.; Ye, Z.; Tan, L.; Mao, D.; Zhao, W.; Zeng, X.; Yan, C.; Sun, R.; Kang, D.J.; et al. Ice-templated mxene/ag-epoxy nanocomposites as high-performance thermal management materials. ACS Appl. Mater. Interfaces 2020, 12, 24298–24307. [Google Scholar] [CrossRef]
- Xiao, C.; Guo, Y.; Tang, Y.; Ding, J.; Zhang, X.; Zheng, K.; Tian, X. Epoxy composite with significantly improved thermal conductivity by constructing a vertically aligned three-dimensional network of silicon carbide nanowires/ boron nitride nanosheets. Compos. Part B Eng. 2020, 187, 107855. [Google Scholar] [CrossRef]
- Kandasamy, R.; Wang, X.-Q.; Mujumdar, A.S. Application of phase change materials in thermal management of electronics. Appl. Therm. Eng. 2007, 27, 2822–2832. [Google Scholar] [CrossRef]
- Cui, Y.; Li, M.; Hu, Y. Emerging interface materials for electronics thermal management: Experiments, modeling, and new opportunities. J. Mater. Chem. C 2020, 8, 10568–10586. [Google Scholar] [CrossRef]
- Dorigato, A.; Ciampolillo, M.V.; Cataldi, A.; Bersani, M.; Pegoretti, A. Polyethylene wax/epdm blends as shape-stabilized phase change materials for thermal energy storage. Rubber Chem. Technol. 2017, 90, 575–584. [Google Scholar] [CrossRef]
- Rigotti, D.; Dorigato, A.; Pegoretti, A. 3d printable thermoplastic polyurethane blends with thermal energy storage/release capabilities. Mater. Today Commun. 2018, 15, 228–235. [Google Scholar] [CrossRef]
- Resch-Fauster, K.; Hengstberger, F.; Zauner, C.; Holper, S. Overheating protection of solar thermal façades with latent heat storages based on paraffin-polymer compounds. Energy Build. 2018, 169, 254–259. [Google Scholar] [CrossRef]
- Resch-Fauster, K.; Feuchter, M. Thermo-physical characteristics, mechanical performance and long-term stability of high temperature latent heat storages based on paraffin-polymer compounds. Thermochim. Acta 2018, 663, 34–45. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, C.; Jia, Y.; Wu, J.; Huang, Y.; Wang, L. Review on electrospun ultrafine phase change fibers (pcfs) for thermal energy storage. Appl. Energy 2018, 210, 167–181. [Google Scholar] [CrossRef]
- Jeong, S.-G.; Kim, S.; Huh, W. Preparation of epoxy resin using n-hexadecane based shape stabilized pcm for applying wood-based flooring. J. Adhes. Sci. Technol. 2014, 28, 711–721. [Google Scholar] [CrossRef]
- Fredi, G.; Dorigato, A.; Pegoretti, A. Multifunctional glass fiber/polyamide composites with thermal energy storage/release capability. Express Polym. Lett. 2018, 12, 349–364. [Google Scholar] [CrossRef]
- Dorigato, A.; Fredi, G.; Pegoretti, A. Novel Phase Change Materials Using Thermoplastic Composites. In Proceedings of the AIP Conference—9th International Conference “Times of Polymers and Composites” (TOP), Ischia, Italy, 17–21 June 2018. [Google Scholar] [CrossRef]
- Dorigato, A.; Fredi, G.; Meneghini, T.; Pegoretti, A. Thermo-mechanical behaviour of thermoplastic composite laminates with thermal energy storage/release capability. In Proceedings of the ECCM 2018—18th European Conference on Composite Materials, Megaron Athens International Conference Centre (MAICC), Athens, Greece, 24–28 June 2018. [Google Scholar]
- Fredi, G.; Dorigato, A.; Unterberger, S.; Artuso, N.; Pegoretti, A. Discontinuous carbon fiber/polyamide composites with microencapsulated paraffin for thermal energy storage. J. Appl. Polym. Sci. 2019, 136, 47401–47408. [Google Scholar] [CrossRef]
- Fredi, G.; Dorigato, A.; Fambri, L.; Pegoretti, A. Multifunctional polymer composites reinforced with discontinuous carbon fibers for thermal energy storage. In Proceedings of the ECCM 2018—18th European Conference on Composite Materials, Megaron Athens International Conference Centre (MAICC), Athens, Greece, 24–28 June 2018. [Google Scholar]
- Fredi, G.; Dorigato, A.; Fambri, L.; Pegoretti, A. Wax confinement with carbon nanotubes for phase changing epoxy blends. Polymers 2017, 9, 405. [Google Scholar] [CrossRef] [PubMed]
- Fredi, G.; Dorigato, A.; Fambri, L.; Pegoretti, A. Multifunctional epoxy/carbon fiber laminates for thermal energy storage and release. Compos. Sci. Technol. 2018, 158, 101–111. [Google Scholar] [CrossRef]
- Fredi, G.; Dorigato, A.; Fambri, L.; Pegoretti, A. Thermal Energy Storage with Polymer Composites. In Proceedings of the American Society for Composites—34th Technical Conference, Atlanta, GA, USA, 23–25 September 2019. [Google Scholar]
- Fredi, G.; Dorigato, A.; Fambri, L.; Pegoretti, A. Detailed experimental and theoretical investigation of the thermo-mechanical properties of epoxy composites containing paraffin microcapsules for thermal management. Polym. Eng. Sci. 2020, 60, 1202–1220. [Google Scholar] [CrossRef]
- Fredi, G.; Dorigato, A.; Fambri, L.; Unterberger, S.H.; Pegoretti, A. Effect of phase change microcapsules on the thermo-mechanical, fracture and heat storage properties of unidirectional carbon/epoxy laminates. Polym. Test. 2020, 91, 106741–106747. [Google Scholar] [CrossRef]
- Fredi, G.; Dirè, S.; Dorigato, A.; Fambri, L.; Pegoretti, A. Multifunctional carbon fiber/epoxy laminates for thermal energy storage and release. In Proceedings of the ECCM 2018—18th European Conference on Composite Materials, Megaron Athens International Conference Centre (MAICC), Athens, Greece, 24–28 June 2018. [Google Scholar]
- Fredi, G.; Dorigato, A.; Pegoretti, A. Novel reactive thermoplastic resin as a matrix for laminates containing phase change microcapsules. Polym. Compos. 2019, 40, 3711–3724. [Google Scholar] [CrossRef]
- Fredi, G.; Dorigato, A.; Pegoretti, A. Dynamic-mechanical response of carbon fiber laminates with a reactive thermoplastic resin containing phase change microcapsules. Mech. Time-Depend. Mater. 2020, 24, 395–418. [Google Scholar] [CrossRef]
- Fredi, G.; Brünig, H.; Vogel, R.; Scheffler, C. Melt-spun polypropylene filaments containing paraffin microcapsules for multifunctional hybrid yarns and smart thermoregulating thermoplastic composites. Express Polym. Lett. 2019, 13, 1071–1087. [Google Scholar] [CrossRef]
- Dorigato, A.; Fredi, G.; Negri, M.; Pegoretti, A. Thermo-mechanical behaviour of novel wood laminae-thermoplastic starch biodegradable composites with thermal energy storage/release capability. Front. Mater. 2019, 6, 1–12. [Google Scholar] [CrossRef]
- Su, J.-F.; Wang, X.-Y.; Wang, S.-B.; Zhao, Y.-H.; Zhu, K.-Y.; Yuan, X.-Y. Interface stability behaviors of methanol-melamine-formaldehyde shell micropcms/epoxy matrix composites. Polym. Compos. 2011, 32, 810–820. [Google Scholar] [CrossRef]
- Su, J.-F.; Zhao, Y.-H.; Wang, X.-Y.; Dong, H.; Wang, S.B. Effect of interface debonding on the thermal conductivity of microencapsulated-paraffin filled epoxy matrix composites. Compos. Pt. A Appl. Sci. Manuf. 2012, 43, 325–332. [Google Scholar] [CrossRef]
- Dorigato, A.; Fredi, G.; Pegoretti, A. Application of the thermal energy storage concept to novel epoxy/short carbon fiber composites. J. Appl. Polym. Sci. 2019, 136, 47431–47434. [Google Scholar] [CrossRef]
- Lynge, M.E.; Van Der Westen, R.; Postmab, A.; Stadler, B. Polydopamine—A nature-inspired polymer coating for biomedical science. Nanoscale 2011, 3, 4916–4928. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.K.; Park, S.H.; Choi, J.W. Mussel-inspired coating and adhesion for rechargeable batteries: A review. Acs Appl Mater. Interfaces 2018, 10, 7562–7573. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Dong, W.; Tang, L.; Zhang, Y.; Kong, J.; Gu, J. Fabrication and investigations on the polydopamine/kh-560 functionalized pbo fibers/cyanate ester wave-transparent composites. Compos. Commun. 2018, 8, 36–41. [Google Scholar] [CrossRef]
- Shanmugam, L.; Feng, X.; Yang, J. Enhanced interphase between thermoplastic matrix and uhmwpe fiber sized with cnt-modified polydopamine coating. Compos. Sci. Technol. 2019, 174, 212–220. [Google Scholar] [CrossRef]
- Fredi, G.; Simon, F.; Sychev, D.; Melnyk, I.; Janke, A.; Scheffler, C.; Zimmerer, C. Bioinspired polydopamine coating as an adhesion enhancer between paraffin microcapsules and an epoxy matrix. ACS Omega 2020, 5, 19639–19653. [Google Scholar] [CrossRef]
- Nishizawa, N.; Kawamura, A.; Kohri, M.; Nakamura, Y.; Fujii, S. Polydopamine particle as a particulate emulsifier. Polymers 2016, 8, 62. [Google Scholar] [CrossRef]
- Roldán, M.L.; Centeno, S.A.; Rizzo, A. An improved methodology for the characterization and identification of sepia in works of art by normal raman and sers, complemented by ftir, py-gc/ms, and xrf. J. Raman Spectrosc. 2014, 45, 1160–1171. [Google Scholar] [CrossRef]
- Alyamaç, E. Self-Stratifying Coatings. PhD Thesis, The Graduate Faculty of The University of Akron, Akron, OH, USA, 2009. [Google Scholar]
- Ding, R.; Torres, S.W.; Messman, J.; Bowen, D.E.; Bowler, N. Dynamics of model polycyclic aromatic hydrocarbon compound-epoxy composites: A dielectric study. Polymer 2018, 136, 6–16. [Google Scholar] [CrossRef]
- Vryonis, O.; Andritsch, T.; Vaughan, A.S.; Lewin, P.L. Effect of surfactant molecular structure on the electrical and thermal performance of epoxy/functionalized-graphene nanocomposites. Polym. Compos. 2020, 41, 2753–2767. [Google Scholar] [CrossRef]
- Ferry, J.D. Viscoelastic Properties of Polymers, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 1980. [Google Scholar]
- Bohn, M.A. The connection between the parameters of wlf equation and of arrhenius equation. Propellants Explos. Pyrotech. 2019, 44, 696–705. [Google Scholar] [CrossRef]
- Kolařík, J.; Pegoretti, A. Proposal of the boltzmann-like superposition principle for nonlinear tensile creep of thermoplastics. Polym. Test. 2008, 27, 596–606. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Epoxy Resin (Base + Hardener) (wt %) | Neat or PDA-Modified Microcapsules (wt %) |
|---|---|---|
| EP | 100 | 0 |
| EP-MC10 | 90 | 10 |
| EP-MC10-PDA | 90 | 10 |
| EP-MC20 | 80 | 20 |
| EP-MC20-PDA | 80 | 20 |
| EP-MC40 | 60 | 40 |
| EP-MC40-PDA | 60 | 40 |
| Sample | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| MC | - | 44.7 | 227.7 | 100 | 29.0 | 224.2 | - | 44.4 | 226.1 |
| MC-PDA | - | 45.7 | 221.1 | 100 | 29.3 | 220.8 | - | 45.5 | 220.9 |
| EP | 11.1 | - | - | - | - | - | 9.3 | - | - |
| EP-MC10 | 11.3 | 44.0 | 22.6 | 99.2 | 32.8 | 22.9 | 7.8 | 44.0 | 23.4 |
| EP-MC10-PDA | 10.7 | 43.1 | 21.8 | 98.6 | 32.9 | 21.7 | 8.8 | 43.1 | 21.7 |
| EP-MC20 | 7.5 | 44.6 | 46.6 | 102.2 | 32.6 | 46.5 | 8.2 | 44.5 | 46.1 |
| EP-MC20-PDA | 11.1 | 44.2 | 44.0 | 99.6 | 31.8 | 43.8 | 9.9 | 44.0 | 43.8 |
| EP-MC40 | 8.6 | 47.0 | 87.8 | 96.4 | 28.9 | 88.8 | 9.1 | 46.5 | 87.3 |
| EP-MC40-PDA | 10.7 | 49.6 | 85.8 | 97.1 | 26.6 | 85.4 | 9.9 | 49.5 | 85.6 |
| Sample | |||||
|---|---|---|---|---|---|
| MC | 118.4 | 164.0 | 241.2 | 421.2 | 0.12 |
| MC-PDA | 140.4 | 180.9 | 209.0 | 428.7 | 3.81 |
| EP | 107.7 | 147.0 | 172.0 | 323.3 | 0.39 |
| EP-MC10 | 105.9 | 140.2 | 164.7 | 321.2 | 1.35 |
| EP-MC10-PDA | 116.0 | 153.9 | 179.2 | 319.5 | 4.89 |
| EP-MC20 | 101.4 | 136.9 | 159.6 | 323.4 | 3.60 |
| EP-MC20-PDA | 101.9 | 141.5 | 167.1 | 321.4 | 3.85 |
| EP-MC40 | 100.8 | 139.5 | 158.5 | 330.2 | 3.10 |
| EP-MC40-PDA | 112.1 | 150.7 | 176.9 | 337.1 | 4.31 |
| Sample | |||||
|---|---|---|---|---|---|
| EP | 1737 | 454.5 | 16.8 | 20.1 | 31.8 |
| EP-MC10 | 1720 | 535.3 | 24.5 | 21.8 | 33.5 |
| EP-MC10-PDA | 1490 | 514.2 | 24.8 | 22.0 | 33.5 |
| EP-MC20 | 1859 | 527.7 | 34.5 | 20.8 | 31.4 |
| EP-MC20-PDA | 1579 | 530.1 | 30.4 | 22.4 | 34.0 |
| EP-MC40 | 2045 | 841.9 | 43.0 | 24.4 | 35.2 |
| EP-MC40-PDA | 3300 | 1137.6 | 68.1 | 22.8 | 37.8 |
| Sample | ||
|---|---|---|
| EP | 346 ± 13 | 0.996 |
| EP-MC20 | 300 ± 10 | 0.997 |
| EP-MC20-PDA | 331 ± 8 | 0.998 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fredi, G.; Zimmerer, C.; Scheffler, C.; Pegoretti, A. Polydopamine-Coated Paraffin Microcapsules as a Multifunctional Filler Enhancing Thermal and Mechanical Performance of a Flexible Epoxy Resin. J. Compos. Sci. 2020, 4, 174. https://doi.org/10.3390/jcs4040174
Fredi G, Zimmerer C, Scheffler C, Pegoretti A. Polydopamine-Coated Paraffin Microcapsules as a Multifunctional Filler Enhancing Thermal and Mechanical Performance of a Flexible Epoxy Resin. Journal of Composites Science. 2020; 4(4):174. https://doi.org/10.3390/jcs4040174
Chicago/Turabian StyleFredi, Giulia, Cordelia Zimmerer, Christina Scheffler, and Alessandro Pegoretti. 2020. "Polydopamine-Coated Paraffin Microcapsules as a Multifunctional Filler Enhancing Thermal and Mechanical Performance of a Flexible Epoxy Resin" Journal of Composites Science 4, no. 4: 174. https://doi.org/10.3390/jcs4040174
APA StyleFredi, G., Zimmerer, C., Scheffler, C., & Pegoretti, A. (2020). Polydopamine-Coated Paraffin Microcapsules as a Multifunctional Filler Enhancing Thermal and Mechanical Performance of a Flexible Epoxy Resin. Journal of Composites Science, 4(4), 174. https://doi.org/10.3390/jcs4040174