Abstract
A novel byphenyl hydrazone ligand developed as a chemosensor for the detection of Cu2+ was studied using a theoretical analysis based on the density functional theory (DFT) and time-dependent DFT (TD-DFT). The geometries of the ligand (L) and the Cu2+-ligand complex were optimized at the CAM-B3LYP/631+G(d,p) level of theory in dimethyl sulfoxide, using the conductor-like polarizable continuum model. The adsorption spectra of these molecular systems were analyzed and compared with the experimental data. Theoretical study of the structural, electronics and optical properties allowed us to understand the chemical changes that the ligand undergoes in the complexation process with the Cu+2 ion.
Published: 14 November 2018
1. Introduction
The design and development of selective chemosensors of cations that are biologically important have gained considerable attention. In particular, copper is the third most abundant essential trace element in the human body and is commonly found as Cu2+ in natural water. However, excessive intake of Cu2+ in the body can cause serious health effects such as respiratory ailments, gastrointestinal problems, kidney damage, Alzheimer’s disease or Wilson’s disease [1]. Moreover, copper is also widely used in industry and agriculture, and consequently could cause significant environmental pollution. Therefore, exploring new detection methods for Cu2+ that allow us simple, fast and selective determination is very important.
Some analytical methods for Cu2+ detection such as inductively coupled plasma-optical emission spectrometry, atomic absorption spectrometry, and atomic emission spectrometry have been widely used. However, these methods often require expensive instrumentation and consume a lot of time. Therefore, in recent years, the identification of various heavy metal ions by fluorogenic and colorimetric chemosensors has drawn much attention due to their selectivity, sensitivity, low cost, operability, and velocity of detection [2,3].
Fluorogenic chemosensors are attractive and versatile tools for analytical sensing because of their high sensibility, fast response time, and technical simplicity [3]. In this context, we recently reported the design and synthesis of a novel chemosensor derived from 5,5′-bis-vanillin for the determination of Cu2+ [4].
In this work, we undertook a theoretical study of this chemosensor using Density Functional Theory (DFT) and time-dependent DFT (TD-DFT) to understand the chemical changes that occur in the ligand in the complexation process with the Cu+2 ion. We used the experimental data from UV-Vis spectroscopy and compared them with the theoretical data obtained by simulations to study the chelation phenomenon between the ligand and Cu2+.
2. Computational Methods
The ground state geometry optimizations of the ligand and complex Cu-ligand were carried out at the DFT level of theory using the CAM-B3LYP functional and the 6-31G(d,p) basis set. The excited states of these compounds were calculated with the time-dependent DFT (TD-DFT) method using the same functional and basis set as in the ground state calculations (TD CAM-B3LYP/6-31G(d,p)). These calculations were performed with solvent models for dimethyl sulfoxide (DMSO) using the conductor-like polarizable continuum model (CPCM) [5]. Frequency calculations allowed us to verify that the currently located geometries were minimal.
Molecular orbital calculations and natural orbital (NBO) analysis were performed to characterize the exited-state properties. All of these calculations were developed using the Gaussian 09 program package [6].
3. Results and Discussion
3.1. Ground State Geometry
The optimized structures of ligand (L) and L-Cu2+ complex molecules obtained with CAM-B3LYP/6-31G(d,p) level are presented in Figure 1. It is important to consider the solvent effect on theoretical calculations when seeking to compare the experimental data; in this respect, the CPCM is an effective tool for the treatment of bulk solvent effects. Also, the CAM-B3LYP is recommended to obtain a good prediction of the excitation states of the aromatic system.
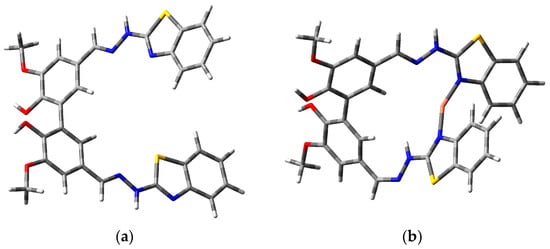
Figure 1.
Optimized geometries of (a) ligand L and (b) L-Cu2+ complex.
From these geometry optimizations, the values of the dihedral angles of the biphenyl structures were obtained. The dihedral angles for L and L-Cu2+ were 62.65° and 53.05°, respectively. These results demonstrate that the dihedral angle decreases (~10°) because of L chelation with Cu2+, increasing the conformational rigidity of the ligand. This effect will be responsible for the bathochromic shift of L-Cu2+ in the UV-vis spectra and could also be responsible for the enhanced fluorescence intensity of this complex [7].
It was found that Cu2+ was coordinated with both -N sites of benzothiazoles, as shown in Figure 1b, forming two coordinate bonds. The Cu2+-N bond length values were 1.93 Å and 1.92 Å for each -N. The total energy of the optimized structures of L and L-Cu2+ were −2582.60 and −4222.71 Hartrees, respectively. The optimization energy values suggest that the total energy of the complex was lower compared to L, indicating higher stability of the L-Cu2+ complex.
The complexation energy (EC) of L-Cu2+ was evaluated according to the following total energy difference: Ec = E(L-Cu2+) − E(L) − E(Cu2+), whose value was −159.13 Kcal/mol. The negative value of EC indicated exothermic processes.
3.2. Electronic Properties and Charge Distribution
To understand the electrical transport properties, the HOMO-LUMO gap of L and L-Cu2+ were also estimated using the same DFT method. The negative energy values for HOMO and LUMO in L and in L-Cu2+ signified that both molecules were stable. The energy gaps between the HOMO and LUMO (ΔEH-L) in L and L-Cu2+ were 146.21 Kcal/mol and 67.44 Kcal/mol, respectively. This shows that the complexation decreases the HOMO-LUMO energy gap, stabilizing the system. It also suggested the process of electron transfer from electron rich L to electron deficient Cu2+.
The NBO charge distributions of L and L-Cu2+ were calculated, and the most important differences in charge were observed in the region of the Cu-N bonds, compared with charges of L free. In L, the N atoms of the benzothiazoles presented the same charge (−0.55), while in L-Cu2+ the charge of these atoms increased (−0.58 and −0.65). The Cu2+ ion charge was 2.00 for free state, while in L-Cu2+ it decreased to 0.67 due to the charge in the complex being delocalized.
3.3. Optical Properties
To understand the electronic transitions from of these compounds, quantum calculations on the electronic absorption spectra in DMSO were performed using the TD CAM-B3LYP/6-31G(d,p) level to obtain the main vertical excitation energies of L and L-Cu2+.
The UV-Vis absorption spectrum of L in DMSO solution exhibited two bands at 257 and 360 nm. After adding Cu2+ to the solution, the band of L at 360 nm gradually decreased and moved to a new wideband at 460–490 nm [3].
According to preliminary TD-DFT calculations, the main vertical excitation energies of L were 254 and 322 nm for excited states. The absorption wavelengths of these states lie relatively closely together and are in agreement with the experimental absorption data.
For L-Cu2+ complex, the main vertical excitation energies calculated were 599, 564, 553 and 475 nm, corresponding to different exited states. As was observed with data analysis of L, the theoretical absorption data were in agreement with the experimental absorption wavelengths.
4. Conclusions
We used the density functional theory method to investigate the geometries and electronic properties of a novel chemosensor derived from 5,5’-bis-vanillin for detection of Cu2+. The energy of the ligand-Cu2+ complex in a 1:1 ratio presented more stability than the ligand free, indicating that the formation of the complex is thermodynamically favored with a complexation energy of −159.13 Kcal/mol.
The coordination of Cu2+ with the ligand occurred throughout the nitrogen atoms of the heterocyclic moiety of the corresponding hydrazone, generating two new Cu-N bonds. The energy gap of the ligand decreased with the complexation with copper, increasing the stability of the system. Due to the chemical changes produced in the system, charge delocalization was observed in the atoms that were involved in the formation of the complex.
The TD-DFT calculations were used to replicate the necessary optical transitions to predict the excited states. The predicted results of the absorption wavelengths for the ligand and L-Cu2+ complex were in agreement with the experimental data.
Acknowledgments
This research was supported by the Agencia Nacional de Ciencia y Tecnología (ANCyT) of Argentina -PICT 2014 No. 1587 and by the Universidad Nacional del Litoral, Santa Fe, Argentina.
References
- Mariappan, K.; Alaparthi, M.; Caple, G.; Balasubramanian, V.; Hoffman, M.M.; Hudspeth, M.; Sykes, A.G. Selective Fluorescence Sensing of Copper(II) and Water via Competing Imine Hydrolysis and Alcohol Oxidation Pathways Sensitive to Water Content in Aqueous Acetonitrile Mixtures. Inorg. Chem. 2014, 53, 2953–2962. [Google Scholar] [CrossRef] [PubMed]
- Udhayakumari, D.; Naha, S.; Velmathi, S. Colorimetric and fluorescent chemosensors for Cu2+. A comprehensive review from the years 2013–15. Anal. Methods 2017, 9, 552–578. [Google Scholar] [CrossRef]
- Costero, A.M.; Gil, S.; Parra, M.; Mancini, P.M.E.; Kneeteman, M.N.; Quindt, M.I. 5,5′-Bis-vanillin derivatives as discriminating sensors for trivalent cations. Tetrahedron Lett. 2015, 56, 3988–3991. [Google Scholar] [CrossRef]
- Quindt, M.I.; Gutierrez, L.G.; Kneeteman, M.N.; Mancini, P.M.E.; Parra, M.; Gil, S.; Costero, A.M. A new highly selective chromogenic and fluorogenic chemosensor for Copper (II), Lett. Org. Chem. 2017, 15, 659–664. [Google Scholar]
- Cossi, M.; Raga, N.; Scalmani, G.; Barone, V. Energies, structures, and electronic properties of molecules in solution with the CPCM solvation model. J. Comput. Chem. 2003, 24, 669–681. [Google Scholar] [CrossRef] [PubMed]
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, Gaussian 09 (Gaussian, Inc., Wallingford CT, 2009). Available online: http://gaussian.com/glossary/g09/ (accessed on 26 March 2019).
- Sanjoy, S.T.; Pradip, C.P.; Pramanik, H.A.R. Fluorescent chemosensor based on sensitive Schiff base for selective detection of Zn2+. Spectrochim. Acta Part A 2014, 121, 520–526. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).