1. Introduction
Catalysis is one of the fundamental pillars of green chemistry. The design and application of new catalysts and catalytic systems simultaneously achieve the dual goals of environmental protection and economic benefit [
1]. Thus, it is paramount that new materials that present catalytic activity are developed, allowing target molecules to be obtained in less time and with greater energy efficiency and hence optimizing the sustainability of chemical synthesis processes. In addition, the environmentally benign nature of the catalysts is another desirable property therein [
2].
Activated carbons (ACs) constitute a group of materials that possess several advantageous properties. Apart from the low environmental impact and the reduced cost, activated carbons possess a high surface area that makes them interesting for heterogeneous catalysis. Moreover, their large pore volume and chemical surface nature can be modified in order to improve their catalytic performance [
3]. These materials are interesting as heterogeneous catalysts because the reaction work-up is not complicated as they can be separated from the reaction products by simple filtration. Their use may save energy and may diminish solvent use in many reactions. They can also be recovered in their active form and recycled in many cases.
Activated carbons can be used not only as catalyst support but also as catalysts themselves in fine-chemical synthesis [
4,
5]. The use of activated carbons as catalysts, replacing the liquid-phase systems, is very promising. Examples of their use as catalysts in fine-chemical synthesis relate mainly to basic carbons [
6,
7,
8]. In this context, activated carbons have attracted much attention and have successfully catalyzed several kinds of organic transformations, such as the synthesis of α,β-unsaturated nitriles [
9], the epoxide ring-opening reaction [
10], synthesis of chalcones [
8],
N-alkylation of imidazole [
11,
12,
13,
14,
15,
16,
17], and acetylation of glycerol [
18].
On the other hand, the acetylation of hydroxyl groups in a variety of substrates, such as alcohols, phenols, and carbohydrates, constitutes a major protection procedure that is extensively applied in synthesis to mask this moiety because of its feasibility and reversibility [
19]. According to the principles of green chemistry, protection/deprotection steps should be avoided [
2], but this is sometimes not possible. If protection steps are required, they should proceed readily, quantitatively, and keeping waste formation and costs to a minimum.
O-acetylation is typically performed with an excess of acetic anhydride with basic or acidic catalysis, although the noxious acyl halides are sometimes used instead. Pyridine is commonly used as a catalyst and as a solvent despite its toxicity [
20]. Because this reaction is so widely used, not only in organic and pharmaceutical syntheses but also in the cosmetic and food industry, many reports on different catalytic procedures have been published [
21]. Pyridine derivative 4-dialkylaminopyridine greatly accelerates the reaction when used as cocatalyst with pyridine [
22,
23,
24]. Other amine bases also show good catalytic activity [
25] as do metal triflates [
26,
27,
28,
29,
30,
31,
32,
33,
34,
35,
36,
37]. Some metal salts, mainly perchlorates, have been used successfully as well [
38,
39,
40]. Different heterogeneous catalysts have been reported [
41,
42,
43,
44,
45]. Salen cobalt complex is a reusable catalyst for this reaction [
46], while iodine has been proven as a powerful catalytic acetylating agent [
47]. A series of bicarbonates and carbonates have also been tested, and good catalytic activity for the acetylation of primary alcohols and phenols has been reported [
48]. Modified zirconia catalyzes the process too [
49], while tributylphosphine has been shown as an efficient catalyst for acylation reactions [
50]. Task-specific ionic liquids have also been found to have very good catalytic performance in the acetylation of alcohols and phenols [
51].
Many of the methods reported achieve good results, but some of them present drawbacks associated with heavy-metal waste production, energy costs, use of noxious compounds, harsh reaction conditions, long reaction times, or complicated work-up procedures. For this reason, there is still a need to explore novel methodologies that allow environmentally friendly catalysts to be used while reducing energy costs and solvent use and simplifying work-up.
In our search for catalytic systems that may prove most adequate in terms of sustainable chemical processes, we obtained and characterized three activated carbons. These ACs were obtained from three commercial carbons, which were tested as well. We assessed the catalytic activity of these ACs in the acetylation reaction of alcohols, phenols, and monosaccharides. We report herein a novel and efficient solvent-free, activated-carbon-catalyzed protocol for the acetylation of alcohols, phenol, β-naphthol, and monosaccharides.
2. Materials and Methods
Solvents of HPLC grade were purchased from Scharlab S.L. Reagents were purchased from Acros Organics and Sigma-Aldrich. Three different commercial carbons were acquired: activated carbon Norit RX 3.0 from Cabot Corporation (formerly Norit Nederland B.V.), labeled carbon N; mesoporous carbon xerogel from Xerolutions S.L., labeled carbon X; and activated carbon (charcoal-activated, extra pure food grade) from Merck KGaA, labeled carbon M.
NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer using CDCl3 as solvent and tetramethylsilane as internal standard. Thin-layer chromatography (TLC) was performed on silica gel plates coated with fluorescent indicator F254 from Merck KGaA.
2.1. Synthesis and Characterization of the Catalysts
The commercial carbon materials M, N, and X were treated with a concentrated H
2SO
4 solution at room temperature for 1.5 h (1 g/20 cm
3) [
52]. The materials were then washed with deionized water in soxhlet until constant pH and dried in oven at 110 °C. The activated carbons obtained from M, N, and X were termed M-S, N-S, and X-S, respectively. All six carbons were tested for catalytic activity.
2.1.1. Adsorption Isotherms and Porosimetry
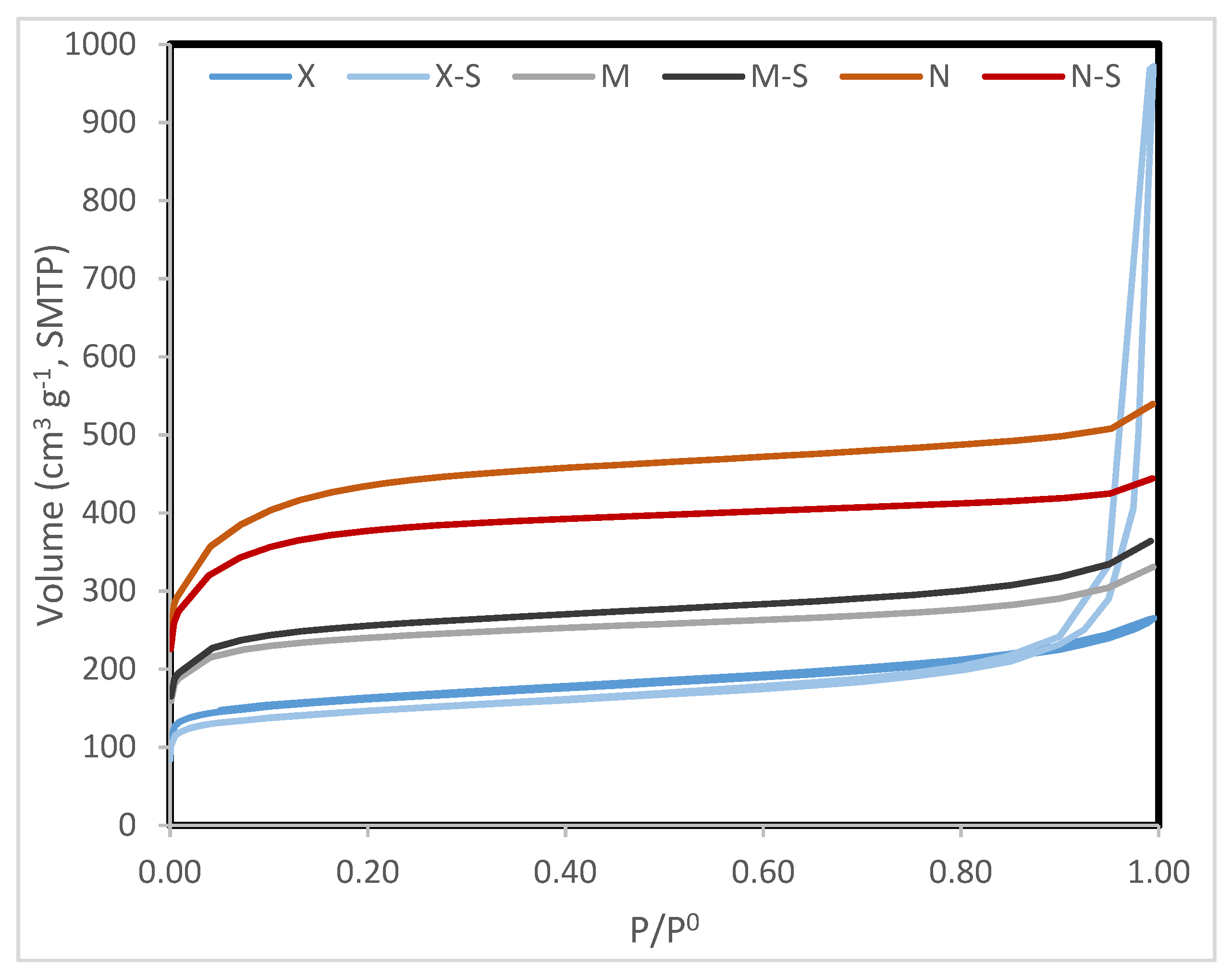
The commercial carbons were mainly microporous, and the nitrogen adsorption capacity was highest for N, followed by M and then finally X (
Figure 1). Treatment with sulfuric acid to produce M-S, N-S, and X-S slightly diminished their N
2 adsorption capacity; however, their porosity remained nearly the same, except for the N-S carbon, which was significantly smaller than that of N (
Table 1) [
53].
The adsorption data was in agreement with those of specific surface obtained by applying the model of Brunauer, Emmett, and Teller (BET) to the data of the previous isotherms (
Table 2).
2.1.2. Elemental Analysis
The organic elemental analysis (C, H, N, and S) was performed in a LECO CHNS 932 equipment. Values are shown in
Table 3.
The treatment of X and M with sulfuric acid lowered the carbon content, oxidized the catalyst, and increased the O and S contents. On the other hand, the treatment of N hardly changed the oxygen and carbon content but increased the sulfur content.
2.1.3. X-ray Photoelectron Spectroscopy (XPS)
The experiments were performed on a K-Alpha equipment from Thermo Scientific with monochromatic Kα Al radiation, 12 KV voltage, and 6 mA current.
The composition, in general, was similar to that of the elemental analysis shown previously (
Table 4). The relative increase in the amount of S in X-S indicated that this atom was mostly on the surface.
The signal of S in these catalysts had peaks close to 168 eV. This corresponded to oxidized forms of sulfur, such as sulfonic groups and sulfates. In untreated M and N carbons, both the 1s orbital peak of N (near 400 eV) and the peak of the 2p orbital of S (near 164 eV) corresponded to reduced forms.
2.1.4. Point of Zero Charge (PZC)
To carry out this measurement, a 0.1 M solution of sodium nitrate was prepared and 7% in weight of the catalyst was added, keeping it stirred for 48 h at 25 °C in a thermostatic bath. Subsequently, it was filtered, and the pH of the filtered solution was measured with a pH meter. This pH value was taken as the point of zero charge (
Table 5).
Catalysts X and N were less alkaline than M. This can be explained by the fact that X and N had a higher oxygen content, which led to the formation of acidic oxygenated groups (alcohols and carboxylic acids). The treatment with sulfuric acid increased the acidity of these materials. This was not only due to oxidation but also due to the formation of sulfonic groups. The effect was similar in the three carbons, but because M was a more alkaline carbon, the final acidity of M-S was lower. In the case of X-S carbon, the acidity most likely originated from mainly the formation of sulfonic groups.
2.2. Acetylation Procedure
According to the best results obtained, the procedure implied that the hydroxylated substrate (1.0 mmol), acetic anhydride (2.5 equiv. per hydroxyl group of the hydroxylated substrate), and 4 mol% of catalyst (percentage referred to acetic anhydride) should be stirred at 60 °C until completion of the reaction, as determined by TLC. Once the reaction was completed, the carbon was filtered off, and the filtrate was treated with distilled water, washed twice with NaHCO3 (10%), and then extracted twice with diethyl ether. The organic phase was dried with anhydrous MgSO4 and evaporated at reduced pressure to yield the pure product. All products obtained were known and were identified by their NMR spectra. In some cases, the acetylated product could be isolated by distillation.
In the case of the monosaccharides, the carbon was filtered off, and the filtrate was treated with distilled water, washed twice with NaHCO3 (10%), then extracted twice with dichloromethane. The organic phase was dried with anhydrous MgSO4 and evaporated at reduced pressure to obtain the desired product.
3. Results and Discussion
To test the catalytic performance of the different activated carbons, we chose the acetylation reaction of benzyl alcohol (BA) with acetic anhydride (AA) in the absence of solvent. Based on previous studies performed in our laboratory, the first activated carbon to be tested was N-S. We started by trying to adjust the reactant proportions and load of catalyst at room temperature (
Table 6, entries 1–5). The results showed no complete reaction at 24 h, even with a 10:1 AA:BA ratio and a 10 mol% load of catalyst. In order to improve the protocol, we decided to heat the reaction while reducing the amount of AA (
Table 6, entry 6).
Having established the reaction conditions as those of entry 6 in
Table 6, our next step was to check the activity of the other two catalysts prepared and the three commercial carbons. All the modified ACs were acidic, whereas the N was neutral activated carbon, the X was a mildly basic carbon gel, and the M was a basic activated carbon (
Table 6, entries 6–11).
The acetylation reaction was suitable for either acidic or basic catalysis. The modified X-S was the best catalyst, probably due to its acidity. However, N-S was nearly as acidic as X-S and yet its catalytic performance was poorer. This may be explained by the fact that N-S had a larger microporous volume and a smaller mesoporous volume than X-S (see
Table 1). Hence, the larger mesoporous network of X-S facilitated access of the reactants to the active sites. M-S was less acidic, which explains its less satisfactory performance. Within the commercial carbons, M was the most basic, which most likely justifies its catalytic activity; on the other hand, carbons X and N lacked sufficient basicity to act as well as M.
In view of the results, the activated carbon X-S was selected for further testing. Our next goal was to try and reduce the amount of both acetic anhydride and catalyst. We found that when 1 equiv. of BA was reacted with 2.5 equiv. of AA in the presence of 4 mol% of catalyst X-S at 60 °C, the reaction was completed in 75 min. This was only a bit longer than the reaction in which a 1:5 BA:AA ratio in the presence of 10 mol% of catalyst was used. As the saving in both acetic anhydride and catalyst was substantial and the length of the reaction was only slightly longer, we decided that these would be the standard conditions for this protocol for further testing. Therefore, the protocol entailed the use of the best catalyst, X-S, in 4 mol% load at 60 °C for the acetylation of 1 equiv. of the hydroxylated substrate with 2.5 equiv. of acetic anhydride per hydroxyl group. Subsequently, with these reaction conditions, we extended the study to other substrates to establish the scope of this procedure. The primary, secondary, allylic, benzylic, and glycol alcohols tested were acetylated in 3 h or less (
Table 7, entries 1–6). It is worth mentioning that the reaction of benzyl alcohol in the same conditions but without catalyst had not completed after 24 h, thereby confirming the good catalytic performance of the system. β-Naphthol was acetylated in 8 h, whereas phenol took 24 h for complete acetylation. We finally decided to check whether this catalytic system could be applied to the per-
O-acetylation of monosaccharides. For this, a ketohexose (D-fructose), an aldohexose (D-glucose), and an aldopentose (D-xylose) were reacted (
Table 3, entries 9–11, respectively). It was found that D-fructose was peracetylated in 1 h; D-fructose and D-glucose took 7 h and 8 h to yield the pentaacetates, respectively.






{kind=link}










