Inhibition of TNF-Alpha Using Plant-Derived Small Molecules for Treatment of Inflammation-Mediated Diseases †

 , , ,
, , , 
Abstract
1. Introduction
2. Methodology
2.1. Ligand Preparation
2.2. Protein Preparation and Receptor Grid Generation
2.3. XP Docking and MM-GBSA Rescoring Workflow
2.4. Induced-Fit Docking
2.5. ADME/T Calculation
2.6. Molecular Dynamics
2.7. DCCM and PCA Analysis
2.8. MM-PBSA Calculation
3. Results
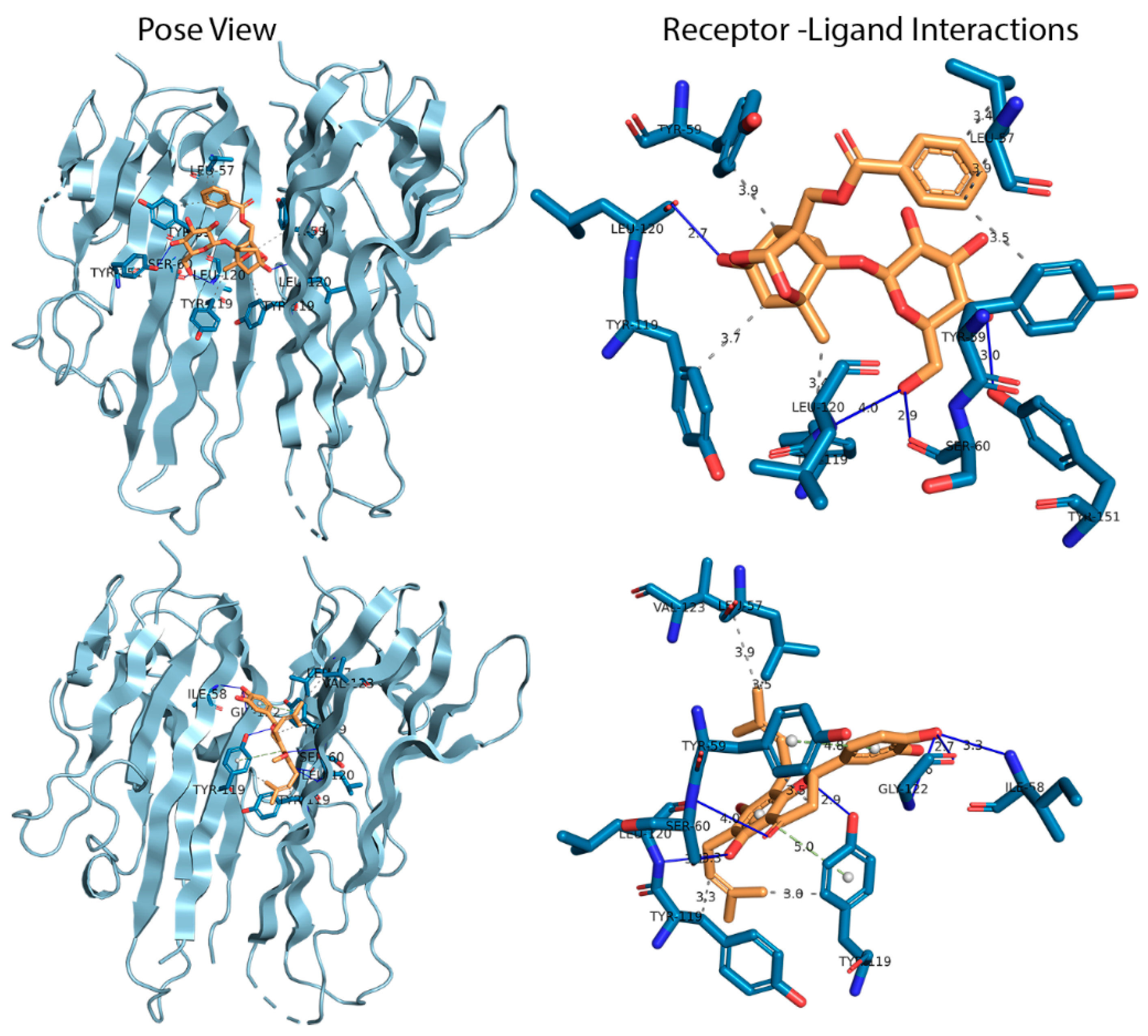
3.1. Docking Analysis
3.2. ADME/T Analysis
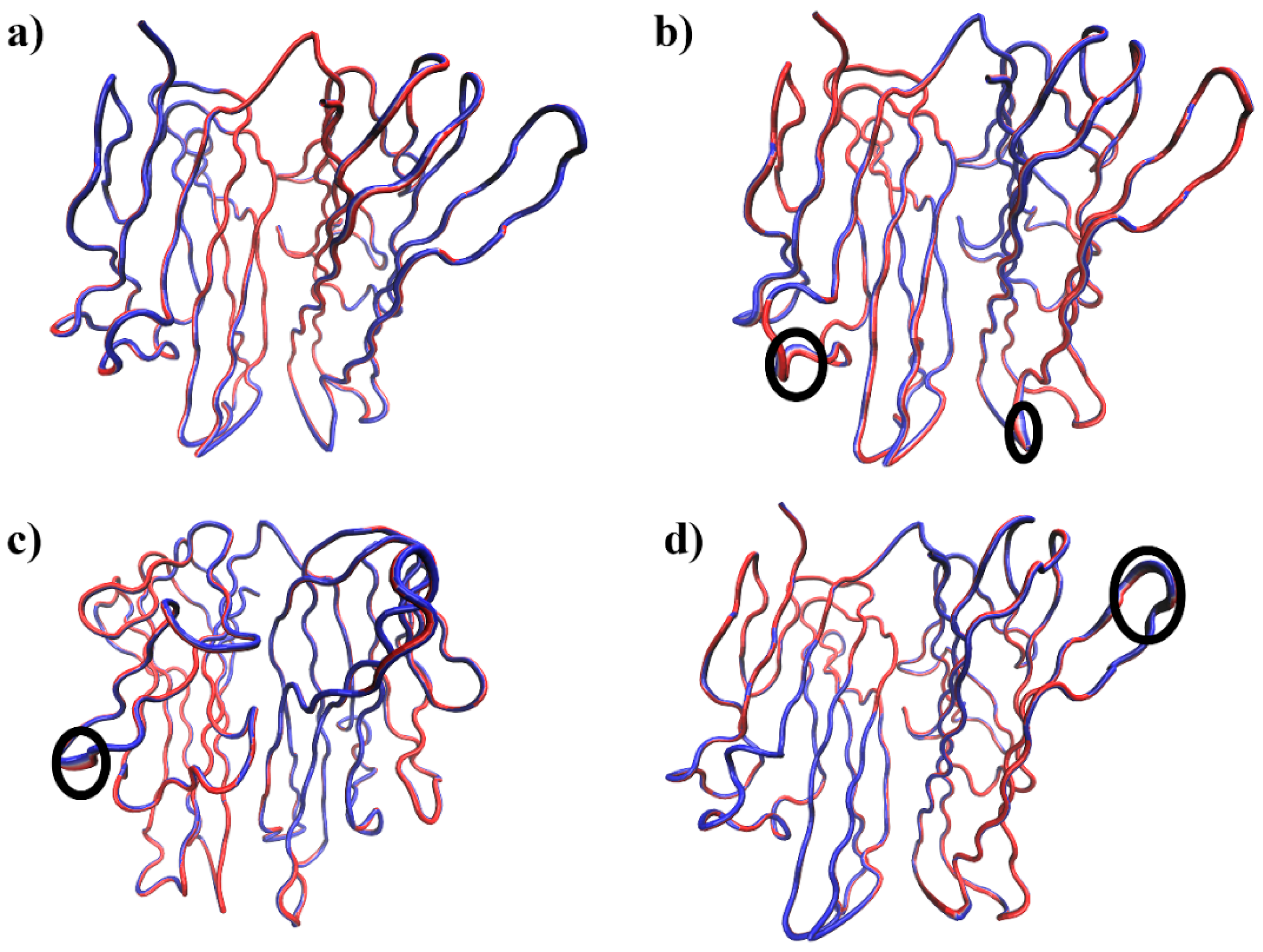
3.2.1. Molecular Dynamics Simulation Analysis
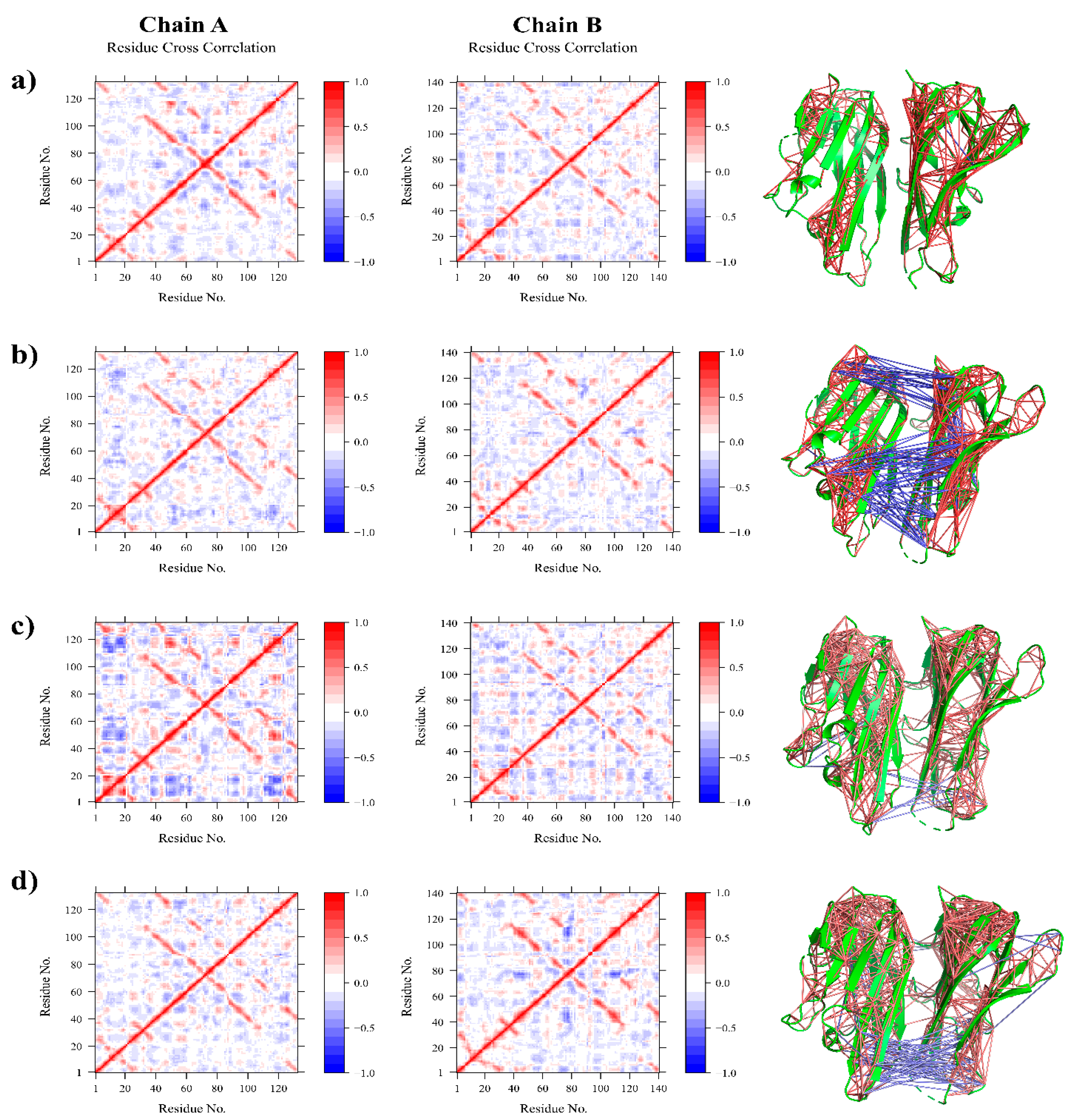
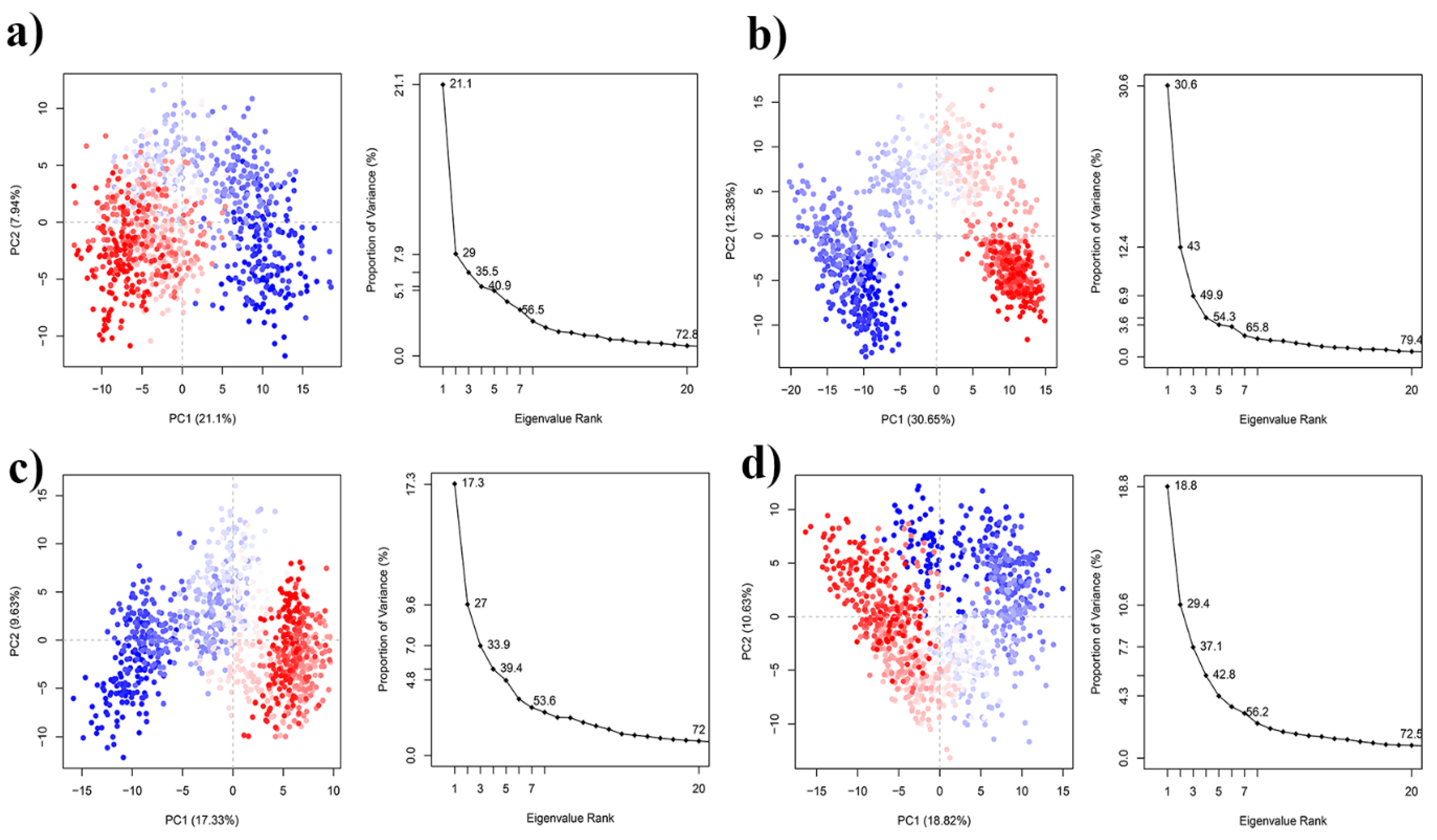
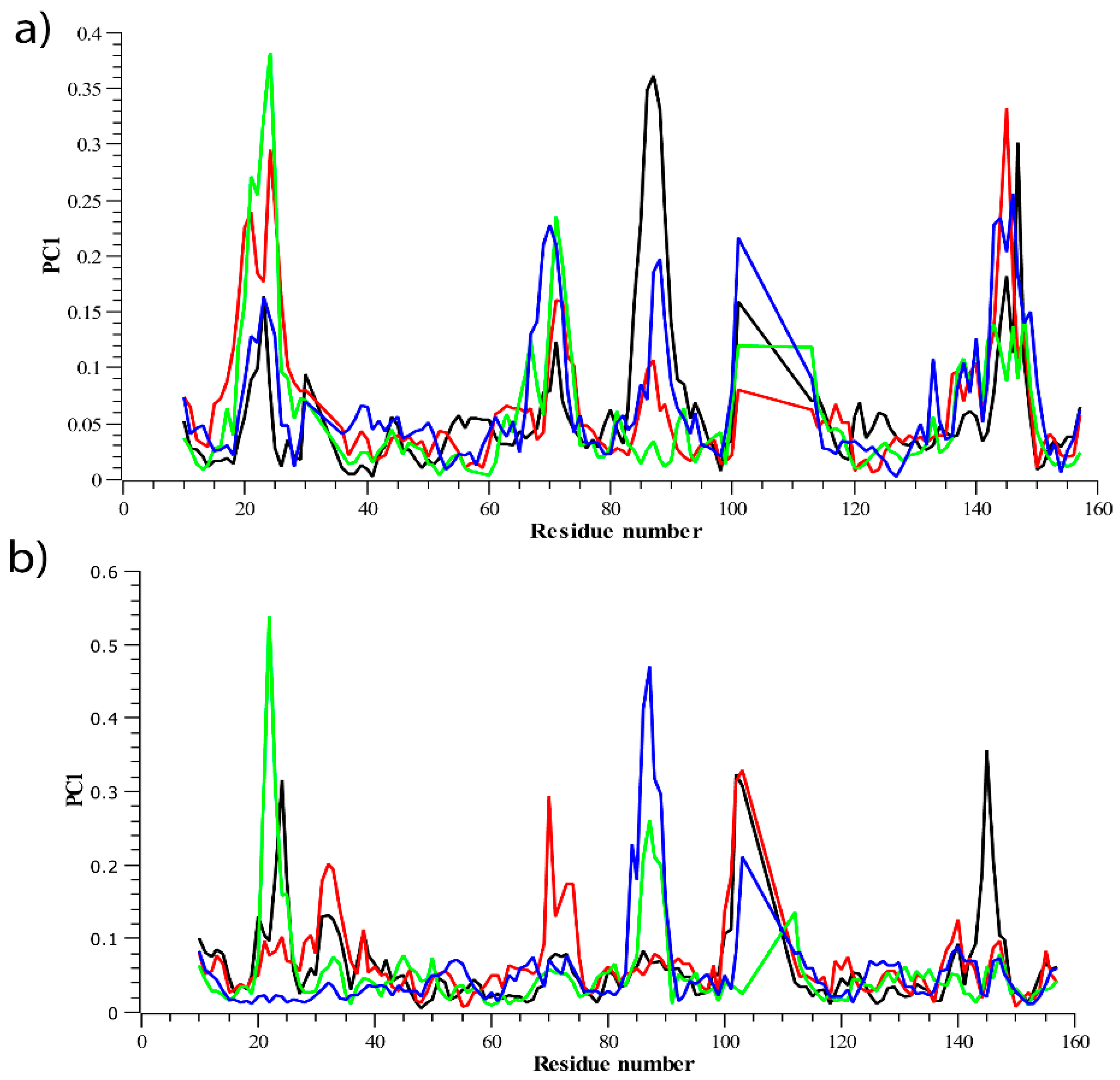
3.2.2. Residue Flexibility and Motion Analysis
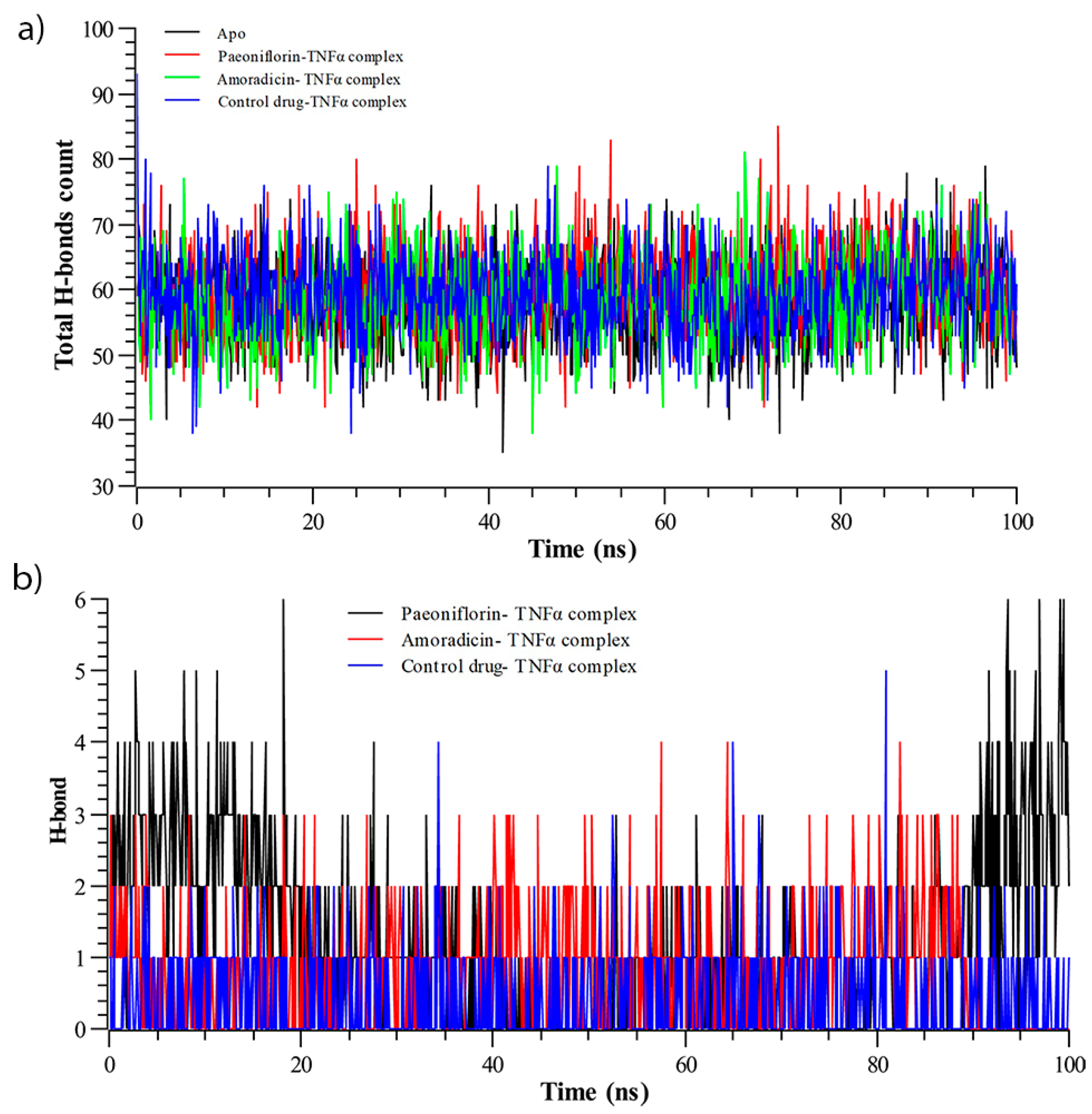
3.2.3. Hydrogen Bond Analysis
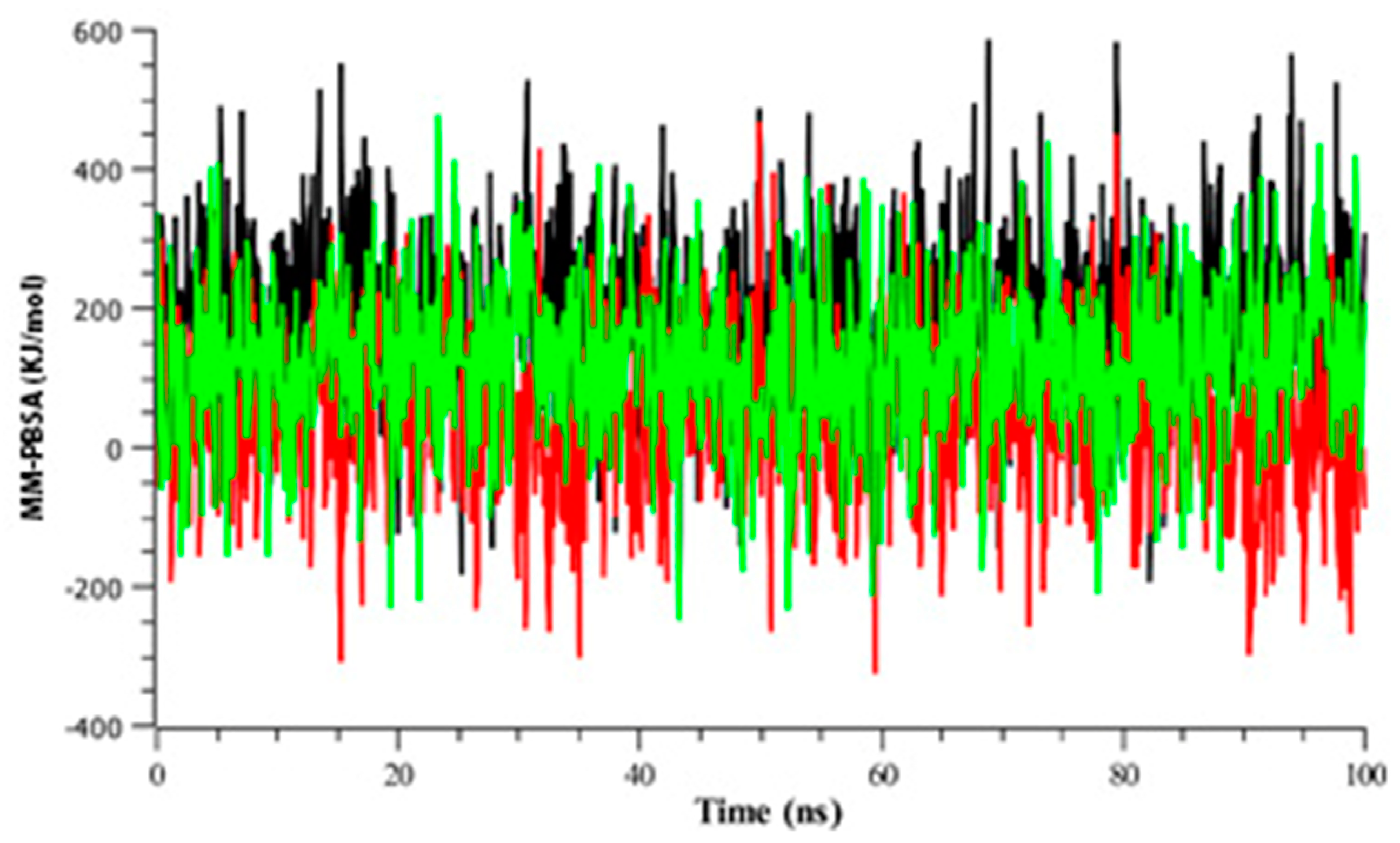
3.2.4. Binding Free Energy Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Feldman, A.M.; Combes, A.; Wagner, D.; Kadakomi, T.; Kubota, T.; Li, Y.Y.; McTiernan, C. The role of tumor necrosis factor in the pathophysiology of heart failure. J. Am. Coll. Cardiol. 2000, 35, 537–544. [Google Scholar] [CrossRef]
- Sethi, G.; Sung, B.; Aggarwal, B.B. TNF: A master switch for inflammation to cancer. Front. Biosci. 2008, 13, 5094–5107. [Google Scholar] [CrossRef] [PubMed]
- Clowes, J.A.; Riggs, B.L.; Khosla, S. The role of the immune system in the pathophysiology of osteoporosis. Immunol. Rev. 2005, 208, 207–227. [Google Scholar] [CrossRef]
- Lin, A.; Dibling, B. The true face of JNK activation in apoptosis. Aging Cell 2002, 1, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [PubMed]
- Kamata, H.; Honda, S.-I.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef]
- Bradley, J.R. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Michalaki, V.; Syrigos, K.; Charles, P.; Waxman, J. Serum levels of IL-6 and TNF-alpha correlate with clinicopathological features and patient survival in patients with prostate cancer. Br. J. Cancer 2004, 90, 2312–2316. [Google Scholar] [CrossRef]
- Ferrajoli, A.; Keating, M.J.; Manshouri, T.; Giles, F.J.; Dey, A.; Estrov, Z.; Koller, C.A.; Kurzrock, R.; Thomas, D.A.; Faderl, S.; et al. The clinical significance of tumor necrosis factor-alpha plasma level in patients having chronic lymphocytic leukemia. Blood. 2002, 100, 1215–1219. [Google Scholar] [CrossRef]
- Berberoglu, U.; Yildirim, E.; Celen, O. Serum levels of tumor necrosis factor alpha correlate with response to neoadjuvant chemotherapy in locally advanced breast cancer. Int. J. Biol. Markers 2004, 19, 130–134. [Google Scholar] [CrossRef]
- Taylor, P.C. Anti-tumor necrosis factor therapies. Curr. Opin. Rheumatol. 2001, 13, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Kroot, E.J.; Van Leeuwen, M.A.; Van Rijswijk, M.H.; Prevoo, M.L.; Van’t Hof, M.A.; Van de Putte, L.B.; Van Riel, P.L. No increased mortality in patients with rheumatoid arthritis: Up to 10 years of follow up from disease onset. Ann. Rheum. Dis. 2000, 59, 954–958. [Google Scholar] [CrossRef] [PubMed]
- Baecklund, E.; Sundstrom, C.; Ekbom, A.; Catrina, A.I.; Biberfeld, P.; Feltelius, N.; Klareskog, L. Lymphoma subtypes in patients with rheumatoid arthritis: Increased proportion of diffuse large B cell lymphoma. Arthritis Rheum. 2003, 48, 1543–1550. [Google Scholar] [CrossRef] [PubMed]
- Ekström, K.; Hjalgrim, H.; Brandt, L.; Baecklund, E.; Klareskog, L.; Ekbom, A.; Askling, J. Risk of malignant lymphomas in patients with rheumatoid arthritis and in their first-degree relatives. Arthritis Rheum. Off. J. Am. Coll. Rheumatol. 2003, 48, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Palladino, M.A.; Bahjat, F.R.; Theodorakis, E.A.; Moldawer, L.L. Anti-TNF-alpha therapies: The next generation. Nat. Rev. Drug Discov. 2003, 2, 736–746. [Google Scholar] [CrossRef]
- Bongartz, T.; Sutton, A.J.; Sweeting, M.J.; Buchan, I.; Matteson, E.L.; Montori, V. Anti-TNF antibody therapy in rheumatoid arthritis and the risk of serious infections and malignancies: Systematic review and meta-analysis of rare harmful effects in randomized controlled trials. JAMA 2006, 295, 2275–2285. [Google Scholar] [CrossRef]
- Guo, Z. The modification of natural products for medical use. Acta Pharm. Sin. B 2017, 7, 119–136. [Google Scholar] [CrossRef]
- Aung, T.N.; Qu, Z.; Kortschak, R.D.; Adelson, D.L. Understanding the effectiveness of natural compound mixtures in cancer through their molecular mode of action. Int. J. Mol. Sci. 2017, 18, 656. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKaprediction and protonation state generation for drug-like molecules. J. Comput.-Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Berman, H.M.; Bourne, P.E.; Westbrook, J.; Zardecki, C. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2015, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinform. 2011, 79, 2794–2812. [Google Scholar] [CrossRef]
- Riza, Y.M.; Parves, M.R.; Tithi, F.A.; Alam, S. Quantum chemical calculation and binding modes of H1R; a combined study of molecular docking and DFT for suggesting therapeutically potent H1R antagonist. Silico Pharmacol. 2019, 7, 1. [Google Scholar] [CrossRef]
- Ignjatović, M.M.; Caldararu, O.; Dong, G.; Muñoz-Gutierrez, C.; Adasme-Carreño, F.; Ryde, U. Binding-affinity predictions of HSP90 in the D3R Grand Challenge 2015 with docking, MM/GBSA, QM/MM, and free-energy simulations. J. Comput.-Aided Mol. Des. 2016, 30, 707–730. [Google Scholar] [CrossRef]
- Vijayakumar, B.; Umamaheswari, A.; Puratchikody, A.; Velmurugan, D. Selection of an improved HDAC8 inhibitor through structure-based drug design. Bioinformation 2011, 7, 134–141. [Google Scholar] [CrossRef][Green Version]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. AdmetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Krieger, E.; Darden, T.; Nabuurs, S.B.; Finkelstein, A.; Vriend, G. Making optimal use of empirical energy functions: Force-field parameterization in crystal space. Proteins Struct. Funct. Bioinform. 2004, 57, 678–683. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., 3; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.J.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Krieger, E.; Nielsen, J.E.; Spronk, C.A.E.M.; Vriend, G. Fast empirical pKa prediction by Ewald summation. J. Mol. Graph. Model. 2006, 25, 481–486. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3D: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Ichiye, T.; Karplus, M. Collective motions in proteins: A covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins Struct. Funct. Bioinform. 1991, 11, 205–217. [Google Scholar] [CrossRef]
- Shlens, J. A tutorial on principal component analysis. arXiv 2014, arXiv:1404.1100. [Google Scholar]
- Salmas, R.E.; Yurtsever, M.; Durdagi, S. Investigation of Inhibition Mechanism of Chemokine Receptor CCR5 by Micro-second Molecular Dynamics Simulations. Sci. Rep. 2015, 5, 13180. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Schrodinger LLC. The PyMOL Molecular Graphics System, Version 2.2.3. 2019. Available online: https://pymol.org/2/ (accessed on 1 July 2019).
- Yan, F.; Liu, X.; Zhang, S.; Su, J.; Zhang, Q.; Chen, J. Electrostatic interaction-mediated conformational changes of adipocyte fatty acid binding protein probed by molecular dynamics simulation. J. Biomol. Struct. Dyn. 2018, 37, 3583–3595. [Google Scholar] [CrossRef] [PubMed]
- Shukla, R.; Singh, T.R. Virtual screening, pharmacokinetics, molecular dynamics and binding free energy analysis for small natural molecules against cyclin-dependent kinase 5 for Alzheimer’s disease. J. Biomol. Struct. Dyn. 2019, 38, 248–262. [Google Scholar] [CrossRef] [PubMed]
- García, A.E. Large-amplitude nonlinear motions in proteins. Phys. Rev. Lett. 1992, 68, 2696–2699. [Google Scholar] [CrossRef]
- Kitao, A.; Go, N. Investigating protein dynamics in collective coordinate space. Curr. Opin. Struct. Biol. 1999, 9, 164–169. [Google Scholar] [CrossRef]
- Kitao, A.; Hirata, F.; Gō, N. The effects of solvent on the conformation and the collective motions of protein: Normal mode analysis and molecular dynamics simulations of melittin in water and in vacuum. Chem. Phys. 1991, 158, 447–472. [Google Scholar] [CrossRef]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Principal component analysis for protein folding dynamics. J. Mol. Biol. 2009, 385, 312–329. [Google Scholar] [CrossRef]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Relation between free energy landscapes of proteins and dynamics. J. Chem. Theory Comput. 2010, 6, 583–595. [Google Scholar] [CrossRef]
- Grottesi, A.; Domene, C.; Hall, B.; Sansom, M.S.P. Conformational Dynamics of M2 Helices in KirBac Channels: Helix Flexibility in Relation to Gating via Molecular Dynamics Simulations. Biochemistry 2005, 44, 14586–14594. [Google Scholar] [CrossRef] [PubMed]
- Spellmon, N.; Sun, X.; Sirinupong, N.; Edwards, B.; Li, C.; Yang, Z. Molecular Dynamics Simulation Reveals Correlated Inter-Lobe Motion in Protein Lysine Methyltransferase SMYD2. PLoS ONE 2016, 10, e0145758. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Gu, Z. Screening of bioactive compounds from moutan cortex and their anti-inflammatory activities in rat synoviocytes. Evid.-Based. Complement. Altern. Med. 2009, 6, 57–63. [Google Scholar] [CrossRef] [PubMed]
- van Ostade, X.; Vandenabeele, P.; Tavernier, J.; Fiers, W. Human tumor necrosis factor mutants with preferential binding to and activity on either the R55 or R75 receptor. Eur. J. Biochem. 1994, 220, 771–779. [Google Scholar] [CrossRef]
- Agullo, G.; Gamet-Payrastre, L.; Manenti, S.; Viala, C.; Rémésy, C.; Chap, H.; Payrastre, B. Relationship between flavonoid structure and inhibition of phosphatidylinositol 3-kinase: A comparison with tyrosine kinase and protein kinase C inhibition. Biochem. Pharmacol. 1997, 53, 1649–1657. [Google Scholar] [CrossRef]
- Cho, J.Y.; Kim, P.S.; Park, J.; Yoo, E.S.; Baik, K.U.; Kim, Y.-K.; Park, M.H. Inhibitor of tumor necrosis factor-α production in lipopolysaccharide-stimulated RAW264.7 cells from Amorpha fruticosa. J. Ethnopharmacol. 2000, 70, 127–133. [Google Scholar] [CrossRef]
- Kuppusamy, U.R.; Khoo, H.E.; Das, N.P. Structure-activity studies of flavonoids as inhibitors of hyaluronidase. Biochem. Pharmacol. 1990, 40, 397–401. [Google Scholar] [CrossRef]
- Beutler, B.; Cerami, A. Tumor necrosis, cachexia, shock, and inflammation: A common mediator. Annu. Rev. Biochem. 1988, 57, 505–518. [Google Scholar] [CrossRef]
- Eck, M.J.; Sprang, S.R. The structure of tumor necrosis factor-alpha at 2.6 A resolution. Implications for receptor binding. J. Biol. Chem. 1989, 264, 17595–17605. [Google Scholar] [CrossRef]
- Jackson, R.W.; Gelinas, R.; Baughman, T.A.; Cox, T.; Howbert, J.J.; Kucera, K.A.; Latham, J.A.; Ramsdell, F.; Singh, D.; Darwish, I.S. Benzobicyclooctanes as novel inhibitors of TNF-alpha signaling. Bioorg. Med. Chem. Lett. 2002, 12, 1093–1097. [Google Scholar] [CrossRef]
- Jackson, R.W.; Tabone, J.C.; Howbert, J.J. Identification of TNF-alpha inhibitors from a split-pool library based on a tyrosine-proline peptidomimetic scaffold. Bioorg. Med. Chem. Lett. 2003, 13, 205–208. [Google Scholar] [CrossRef]
- Deng, X.; Zhang, X.; Tang, B.; Liu, H.; Shen, Q.; Liu, Y.; Lai, L. Design, Synthesis, and Evaluation of Dihydrobenzo[cd]indole-6-sulfonamide as TNF-α Inhibitors. Front. Chem. 2018, 6, 98. Available online: https://www.frontiersin.org/article/10.3389/fchem.2018.00098 (accessed on 7 September 2019). [CrossRef] [PubMed]
- Melagraki, G.; Ntougkos, E.; Papadopoulou, D.; Rinotas, V.; Leonis, G.; Douni, E.; Afantitis, A.; Kollias, G. In Silico Discovery of Plant-Origin Natural Product Inhibitors of Tumor Necrosis Factor (TNF) and Receptor Activator of NF-κB Ligand (RANKL). Front. Pharmacol. 2018, 9, 800. Available online: https://www.frontiersin.org/article/10.3389/fphar.2018.00800 (accessed on 7 September 2019). [CrossRef] [PubMed]
- Malik, A.; Arooj, M.; Butt, T.T.; Zahid, S.; Zahid, F.; Jafar, T.H.; Waquar, S.; Gan, S.H.; Ahmad, S.; Mirza, M.U. In silico and in vivo characterization of cabralealactone, solasodin and salvadorin in a rat model: Potential anti-inflammatory agents. Drug Des. Devel. Ther. 2018, 12, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Maiorov, E.G.; Ozbabacan, S.A.; Keskin, O.; Gursoy, A.; Nussinov, R. Structural Pathways of Cytokines May Illuminate Their Roles in Regulation of Cancer Development and Immunotherapy. Cancers 2014, 6, 663–683. [Google Scholar] [CrossRef] [PubMed]
- He, M.M.; Smith, A.S.; Oslob, J.D.; Flanagan, W.M.; Braisted, A.C.; Whitty, A.; Cancilla, M.T.; Wang, J.; Lugovskoy, A.A.; Yoburn, J.C.; et al. Small-Molecule Inhibition of TNF-α. Science 2005, 310, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Blevitt, J.M.; Hack, M.D.; Herman, K.L.; Jackson, P.F.; Krawczuk, P.J.; Lebsack, A.D.; Liu, A.X.; Mirzadegan, T.; Nelen, M.I.; Patrick, A.N.; et al. Structural Basis of Small-Molecule Aggregate Induced Inhibition of a Protein–Protein Interaction. J. Med. Chem. 2017, 60, 3511–3517. [Google Scholar] [CrossRef]
- Saddala, M.S.; Huang, H. Identification of novel inhibitors for TNFα, TNFR1 and TNFα-TNFR1 complex using pharmacophore-based approaches. J. Transl. Med. 2019, 17, 215. [Google Scholar] [CrossRef]
- Chen, S.; Feng, Z.; Wang, Y.; Ma, S.; Hu, Z.; Yang, P.; Chai, Y.; Xie, X. Discovery of Novel Ligands for TNF-alpha and TNF Receptor-1 through Structure-Based Virtual Screening and Biological Assay. J. Chem. Inf. Model. 2017, 57, 1101–1111. [Google Scholar] [CrossRef]
- Geng, L.; Li, X.; Feng, X.; Zhang, J.; Wang, D.; Chen, J.; Liu, R.; Chen, H.; Sun, L. Association of TNF-α with Impaired Migration Capacity of Mesenchymal Stem Cells in Patients with Systemic Lupus Erythematosus. J. Immunol. Res. 2014, 2014, 169082. [Google Scholar] [CrossRef]
- Cao, Y.; Li, Y.-H.; Lv, D.-Y.; Chen, X.-F.; Chen, L.-D.; Zhu, Z.-Y.; Chai, Y.-F.; Zhang, J.-P. Identification of a ligand for tumor necrosis factor receptor from Chinese herbs by combination of surface plasmon resonance biosensor and UPLC-MS. Anal. Bioanal. Chem. 2016, 408, 5359–5367. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Cavalli, A.; Poluzzi, E.; de Ponti, F.; Recanatini, M. Toward a pharmacophore for drugs inducing the long QT syndrome: Insights from a CoMFA study of HERG K(+) channel blockers. J. Med. Chem. 2002, 45, 3844–3853. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today. Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F. An in silico evaluation of the ADMET profile of the StreptomeDB database. Springerplus 2013, 2, 353. [Google Scholar] [CrossRef] [PubMed]
- Witchel, H.J.; Hancox, J.C. Brief review familial and acquired long QT syndrome and the cardiac rapid delayed rectifier potassium current. Clin. Exp. Pharmacol. Physiol. 2000, 27, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Hengstler, J.G.; Oesch, F. Ames Test. In Encyclopedia of Genetics; Brenner, S., Miller, J.H., Eds.; Academic Press: New York, NY, USA, 2001; pp. 51–54. [Google Scholar] [CrossRef]
- Huang, S.; Strong, J.M.; Zhang, L.; Reynolds, K.S.; Nallani, S.; Temple, R.; Abraham, S.; al Habet, S.; Baweja, R.K.; Burckart, G.J. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J. Clin. Pharmacol. 2008, 48, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Hollenberg, P.F. Characteristics and common properties of inhibitors, inducers, and activators of CYP enzymes. Drug Metab. Rev. 2002, 34, 17–35. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Mahmud, S.; Parves, R.; Riza, Y.M.; Mahmud, K.; Ray, S.; Tithi, F.A.; Zaoti, Z.F.; Absar, N. Exploring the potent inhibitors and binding modes of phospholipase A2 through in silico investigation. J. Biomol. Struct. Dyn. 2020, 38, 4221–4231. [Google Scholar] [CrossRef]
- Mitra, S.; Dash, R. Structural dynamics and quantum mechanical aspects of shikonin derivatives as CREBBP bromodomain inhibitors. J. Mol. Graph. Model. 2018, 83, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Bogatyreva, N.S.; Ivankov, D.N. The relationship between the solvent-accessible surface area of a protein and the number of native contacts in its structure. Mol. Biol. 2008, 42, 932. [Google Scholar] [CrossRef]
- Gilis, D.; Rooman, M. Stability changes upon mutation of solvent-accessible residues in proteins evaluated by database-derived potentials. J. Mol. Biol. 1996, 257, 1112–1126. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Docking Score | Glide Ligand Efficiency | Glide Evdw | Glide Ecoul | Glide Energy | Glide Emodel | MMGBSA ΔG Bind |
|---|---|---|---|---|---|---|---|
| Kaempferol | −10.677 | −0.254 | −40.764 | −14.462 | −55.226 | −73.529 | −61.26 |
| Corilagin | −10.325 | −0.229 | −31.526 | −16.372 | −47.897 | −63.724 | −29.57 |
| Amoradicin | −8.149 | −0.255 | −35.262 | −6.188 | −41.45 | −55.702 | −58.69 |
| Paeoniflorin | −7.696 | −0.226 | −31.778 | −8.588 | −40.366 | −51.405 | −40.42 |
| Quercetin | −7.591 | −0.345 | −29.553 | −7.52 | −37.073 | −42.579 | −41.9 |
| Myricetin | −7.52 | −0.327 | −20.062 | −8.104 | −28.166 | −38.118 | −26.69 |
| Eriodictyol | −7.42 | −0.353 | −16.679 | −15.465 | −32.145 | −36.398 | −36.53 |
| Luteolin | −7.241 | −0.345 | −28.16 | −6.815 | −34.976 | −44.758 | −37.48 |
| Curcumin | −6.515 | −0.241 | −27.64 | −8.604 | −36.244 | −48.232 | −40.98 |
| Compound Name | IFD Score (kcal/mol) | Prime Energy | Glide Score | Glide Ecoul |
|---|---|---|---|---|
| Paeoniflorin | −554.98 | −10699.63 | −11.327 | −9.291 |
| Amoradicin | −546.31 | −10883.87 | −10.790 | −7.678 |
| Parameters | Paeoniflorin | Amoradicin | Control Drug |
|---|---|---|---|
| CNS permeability | −3.914 | −2.769 | −0.686 |
| Blood Brain Barrier permeability | −1.352 | −1.294 | 0.147 |
| Total clearance | 0.645 | 0.32 | 0.79 |
| Renal OCT2 substrate | No | No | No |
| hERG inhibitor | No | No | Yes |
| Hepatotoxicity | No | No | Yes |
| % of Human Oral absorption | 48.350 | 100 | 84.485 |
| % of Human Intestinal absorption | 67.873 | 88.712 | 93.083 |
| QPlogPo/W | 0.256 | 4.998 | 6.700 |
| QPlogS | −2.365 | −6.934 | −6.985 |
| Solvent Accessible Surface Area (SASA) | 654.688 | 761.367 | 684.054 |
| AMES toxicity | No | No | Yes |
| Hydrogen bond donors | 5 | 3 | 0 |
| Hydrogen bond acceptors | 11 | 6 | 5 |
| Lipinski | Yes | Yes | No |
| Bioavailability | 0.55 | 0.55 | 0.55 |
| Molecular Weight | 480.46 g/mol | 438.51 g/mol | 547.621 g/mol |
| CYP2D6 substrate | No | No | Yes |
| CYP3A4 substrate | No | Yes | Yes |
| CYP1A2 inhibitor | No | No | Yes |
| CYP2C9 inhibitor | No | Yes | No |
| CYP2D6 inhibitor | No | No | Yes |
| CYP3A4 inhibitor | No | Yes | Yes |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parves, M.R.; Mahmud, S.; Riza, Y.M.; Sujon, K.M.; Uddin, M.A.R.; Chowdhury, M.I.A.; Islam, M.J.; Tithi, F.A.; Alam, M.; Jui, N.R.; et al. Inhibition of TNF-Alpha Using Plant-Derived Small Molecules for Treatment of Inflammation-Mediated Diseases. Proceedings 2021, 79, 13. https://doi.org/10.3390/IECBM2020-08586
Parves MR, Mahmud S, Riza YM, Sujon KM, Uddin MAR, Chowdhury MIA, Islam MJ, Tithi FA, Alam M, Jui NR, et al. Inhibition of TNF-Alpha Using Plant-Derived Small Molecules for Treatment of Inflammation-Mediated Diseases. Proceedings. 2021; 79(1):13. https://doi.org/10.3390/IECBM2020-08586
Chicago/Turabian StyleParves, Md. Rimon, Shafi Mahmud, Yasir Mohamed Riza, Khaled Mahmud Sujon, Mohammad Abu Raihan Uddin, Md. Iftekhar Alam Chowdhury, Md. Jahirul Islam, Fahmida Alam Tithi, Mosharaf Alam, Nabila Rahman Jui, and et al. 2021. "Inhibition of TNF-Alpha Using Plant-Derived Small Molecules for Treatment of Inflammation-Mediated Diseases" Proceedings 79, no. 1: 13. https://doi.org/10.3390/IECBM2020-08586
APA StyleParves, M. R., Mahmud, S., Riza, Y. M., Sujon, K. M., Uddin, M. A. R., Chowdhury, M. I. A., Islam, M. J., Tithi, F. A., Alam, M., Jui, N. R., Islam, S., & Absar, N. (2021). Inhibition of TNF-Alpha Using Plant-Derived Small Molecules for Treatment of Inflammation-Mediated Diseases. Proceedings, 79(1), 13. https://doi.org/10.3390/IECBM2020-08586