Building Up Co-Crystals: Structural Motif Consistencies Across Families of Co-Crystals †


Abstract
:1. Introduction
2. Computational Methodology
3. Results and Discussion
3.1. Co-Former and Compositional Analysis
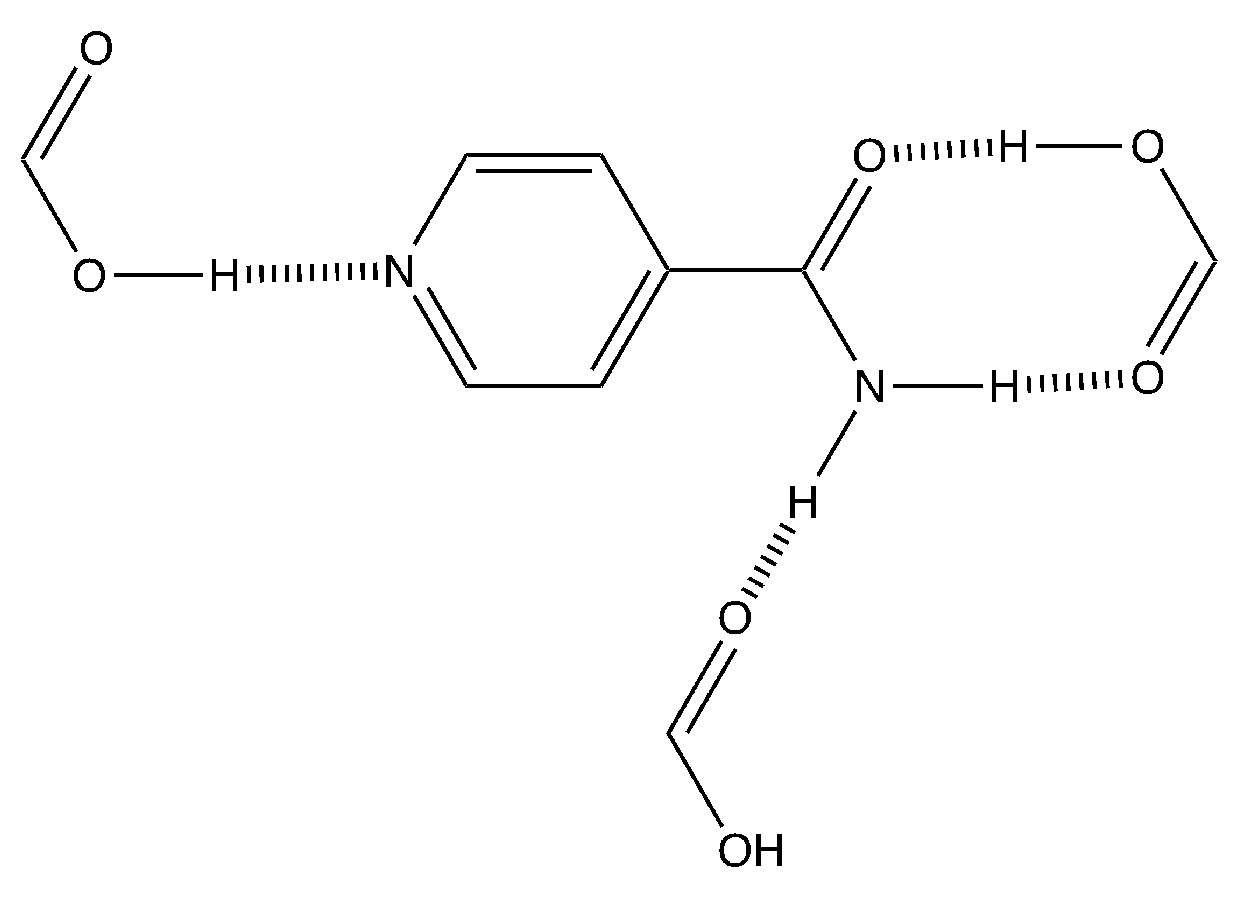
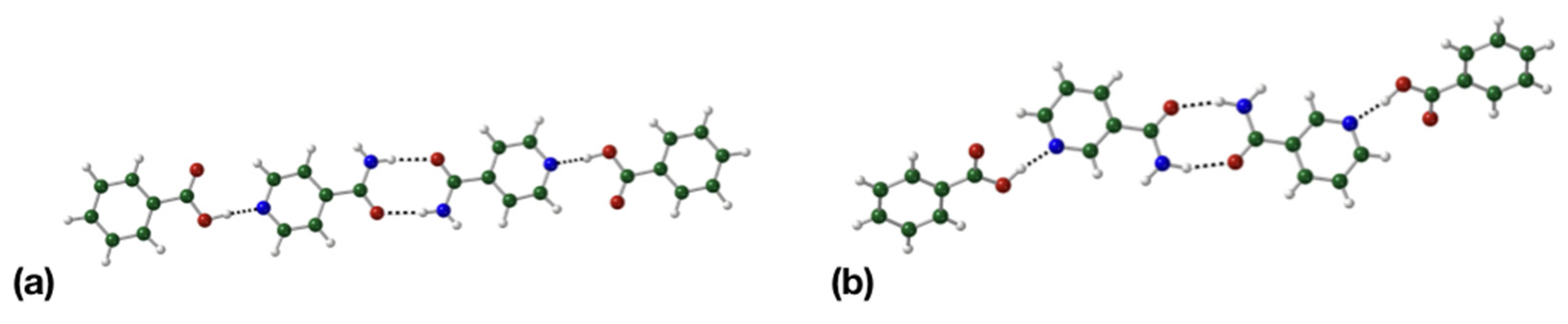
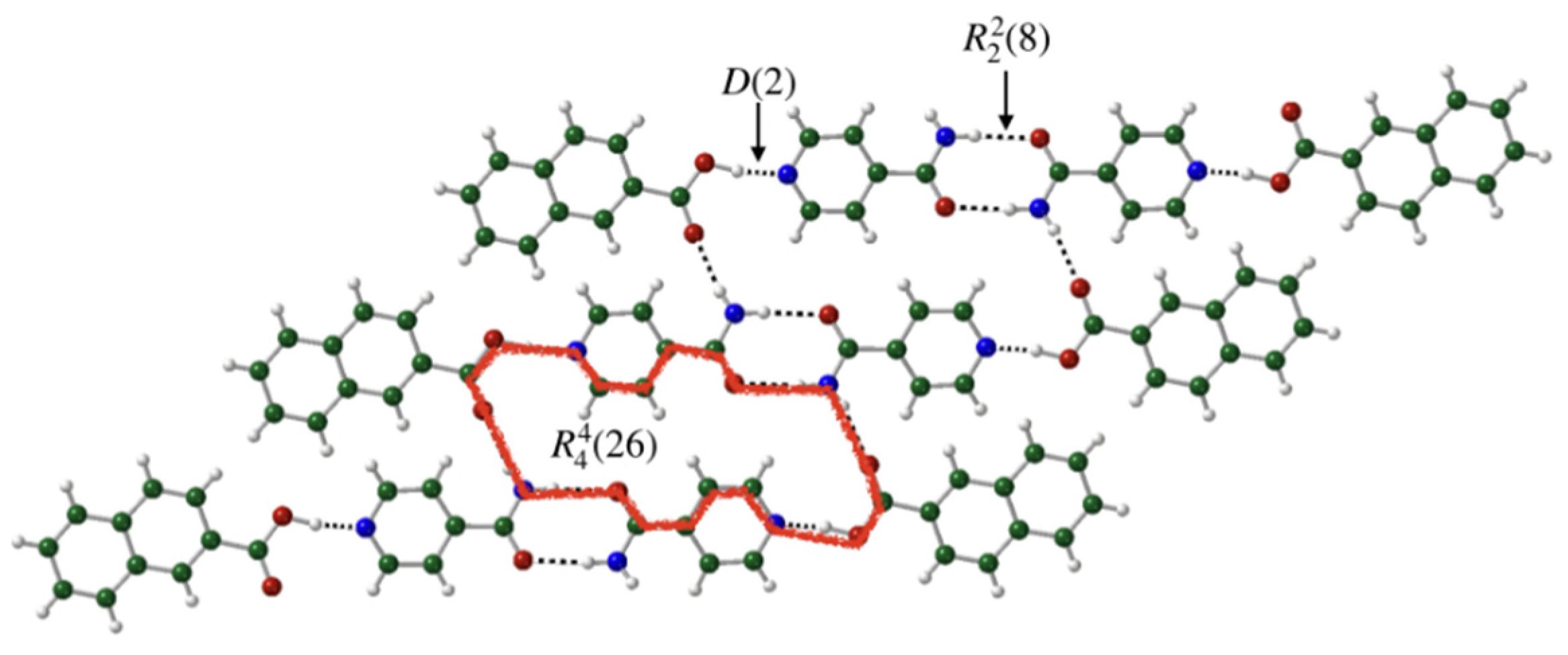
3.2. Crystal Packing Analysis
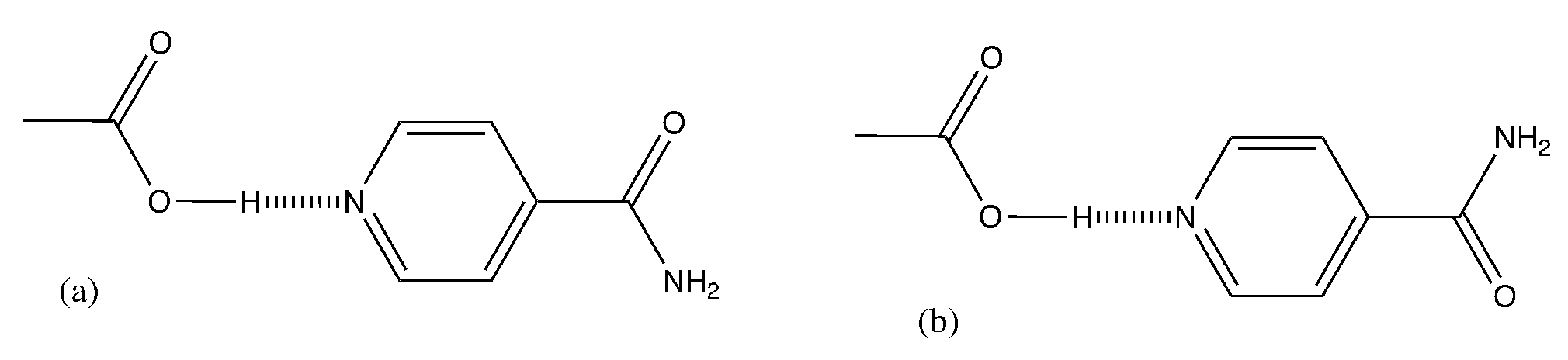
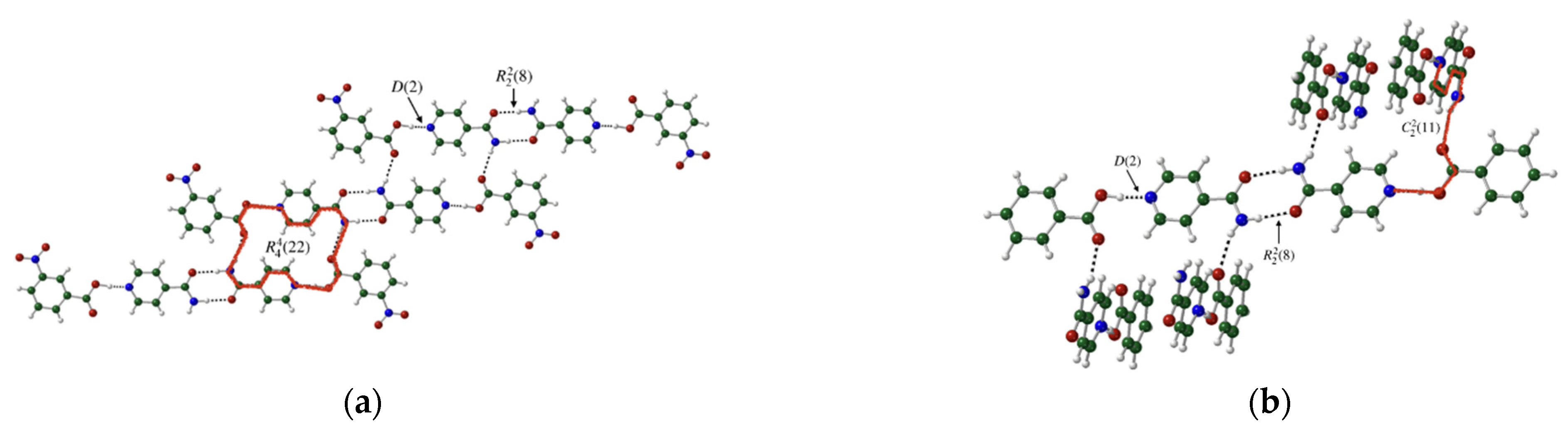
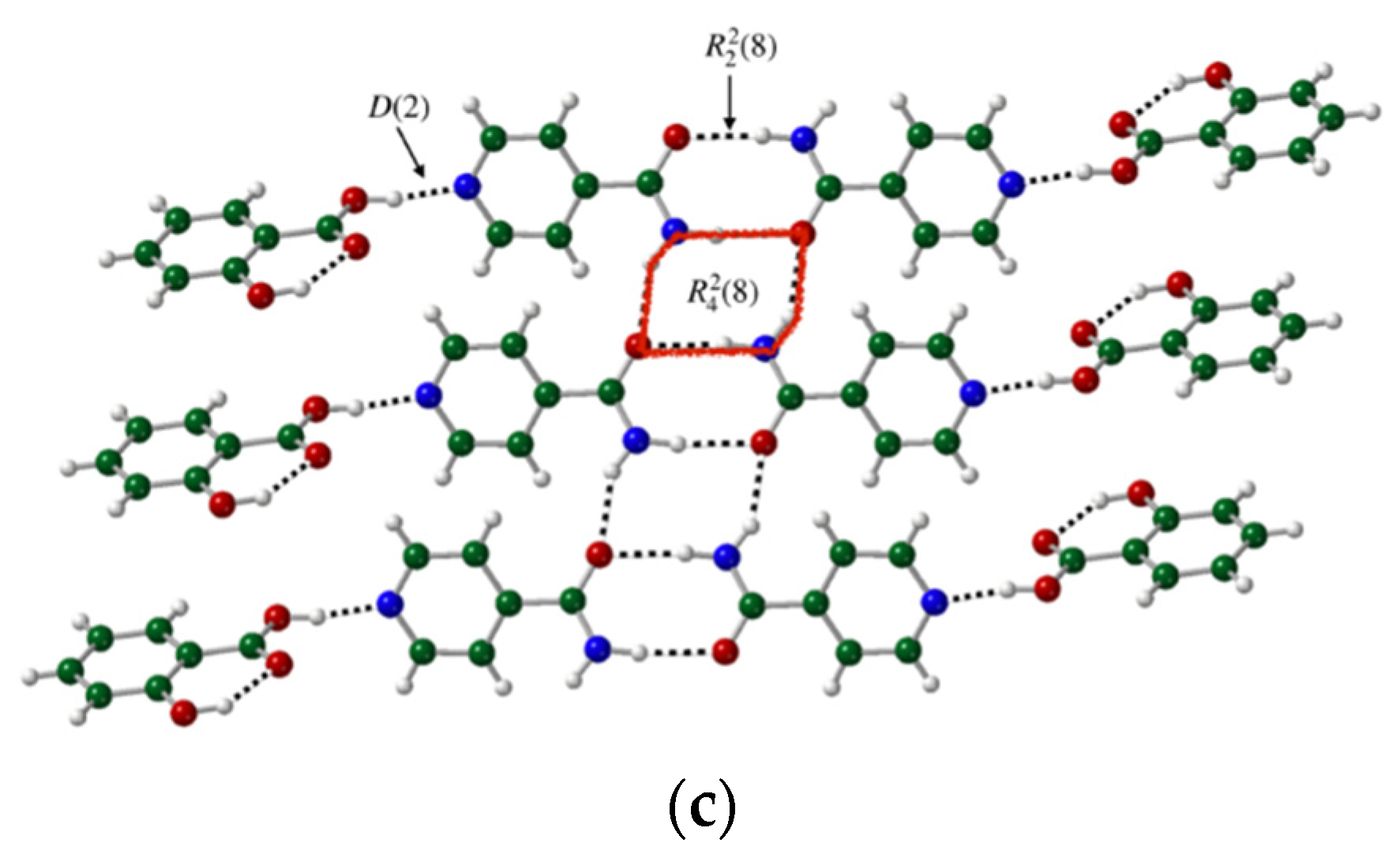
3.3. Binding Interactions
4. Conclusions
Conflicts of Interest
References
- Bolla, G.; Nangia, A. Pharmaceutical cocrystals: Walking the talk. Chem. Commun. 2016, 52, 8342–8360. [Google Scholar] [CrossRef] [PubMed]
- Schultheiss, N.; Henck, J.-O. Role of Co-crystals in the Pharmaceutical Development Continuum. In Pharmaceutical Salts and Co-Crystals; Royal Society of Chemistry: Cambridge, UK, 2011; pp. 110–127. [Google Scholar]
- Trask, A.V.; Motherwell, W.D.S.; Jones, W. Pharmaceutical Cocrystallization: Engineering a Remedy for Caffeine Hydration. Cryst. Growth Des. 2005, 5, 1013–1021. [Google Scholar] [CrossRef]
- Pagire, S.K.; Seaton, C.C.; Paradkar, A. Improving Stability of Effervescent Products by Co-Crystal Formation: A Novel Application of Crystal Engineered Citric Acid. Cryst. Growth Des. 2020, 20, 4839–4844. [Google Scholar] [CrossRef]
- Stahly, G.P. Diversity in Single- and Multiple-Component Crystals. The Search for and Prevalence of Polymorphs and Cocrystals. Cryst. Growth Des. 2007, 7, 1007–1026. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Wicker, J.G.P.; Crowley, L.M.; Robshaw, O.; Little, E.J.; Stokes, S.P.; Cooper, R.I.; Lawrence, S.E. Will they co-crystallize? CrystEngComm 2017, 19, 5336–5340. [Google Scholar] [CrossRef]
- Krishna, G.R.; Ukrainczyk, M.; Zeglinski, J.; Rasmuson, Å.C. Prediction of Solid State Properties of Cocrystals Using Artificial Neural Network Modeling. Cryst. Growth Des. 2018, 18, 133–144. [Google Scholar] [CrossRef]
- Devogelaer, J.J.; Meekes, H.; Tinnemans, P.; Vlieg, E.; Gelder, R. Co-crystal Prediction by Artificial Neural Networks. Angew. Chem. Int. Ed. 2020, 59, 21711–21718. [Google Scholar] [CrossRef]
- Zhang, S.-W.; Brunskill, A.P.J.; Schwartz, E.; Sun, S. Celecoxib–Nicotinamide Cocrystal Revisited: Can Entropy Control Cocrystal Formation? Cryst. Growth Des. 2017, 17, 2836–2843. [Google Scholar] [CrossRef]
- Perlovich, G.L. Formation Thermodynamics of Two-Component Molecular Crystals: Polymorphism, Stoichiometry, and Impact of Enantiomers. Cryst. Growth Des. 2020, 20, 5526–5537. [Google Scholar] [CrossRef]
- Musumeci, D.; Hunter, C.A.; Prohens, R.; Scuderi, S.; McCabe, J.F. Virtual cocrystal screening. Chem. Sci. 2011, 2, 883–890. [Google Scholar] [CrossRef]
- Báthori, N.B.; Lemmerer, A.; Venter, G.A.; Bourne, S.A.; Caira, M.R. Pharmaceutical Co-crystals with Isonicotinamide—Vitamin B3, Clofibric Acid, and Diclofenac—And Two Isonicotinamide Hydrates. Cryst. Growth Des. 2011, 11, 75–87. [Google Scholar] [CrossRef]
- Soares, F.L.F.; Carneiro, R.L. Green Synthesis of Ibuprofen–Nicotinamide Cocrystals and In-Line Evaluation by Raman Spectroscopy. Cryst. Growth Des. 2013, 13, 1510–1517. [Google Scholar] [CrossRef]
- Seaton, C.C.; Parkin, A.; Wilson, C.C.; Blagden, N. Controlling the Formation of Benzoic Acid: Isonicotinamide Molecular Complexes. Cryst. Growth Des. 2009, 9, 47–56. [Google Scholar] [CrossRef]
- Lemmerer, A.; Báthori, N.B.; Bourne, S.A. Chiral carboxylic acids and their effects on melting-point behaviour in co-crystals with isonicotinamide. Acta Crystallogr. Sect. B Struct. Sci. 2008, 64, 780–790. [Google Scholar] [CrossRef]
- Tothadi, S.; Desiraju, G.R. Unusual co-crystal of isonicotinamide: The structural landscape in crystal engineering. Philos. Trans. R. Soc. A 2012, 370, 2900–2915. [Google Scholar] [CrossRef]
- Seaton, C.C.; Parkin, A. Making Benzamide Cocrystals with Benzoic Acids: The Influence of Chemical Structure. Cryst. Growth Des. 2011, 11, 1502–1511. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 389–397. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1059. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2017, 8, e1327-6. [Google Scholar] [CrossRef]
- Zheng, J.; Xu, X.; Truhlar, D.G. Minimally augmented Karlsruhe basis sets. Theor. Chem. Acc. 2010, 128, 295–305. [Google Scholar] [CrossRef]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer17; University of Western Australia: Perth, Australia, 2017; Available online: https://hirshfeldsurface.net.
- Price, K.V. Eliminating Drift Bias from the Differential Evolution Algorithm. In Advances in Differential Evolution; Springer: Berlin/Heidelberg, Germany, 2008; pp. 33–38. [Google Scholar]
- Gavezzotti, A. Efficient computer modeling of organic materials. The atom–atom, Coulomb–London–Pauli (AA-CLP) model for intermolecular electrostatic-polarization, dispersion and repulsion energies. New J. Chem. 2011, 35, 1360–1368. [Google Scholar] [CrossRef]
- Svoboda, V.; MacFhionnghaile, P.; McGinty, J.; Connor, L.E.; Oswald, I.D.H.; Sefcik, J. Continuous Cocrystallization of Benzoic Acid and Isonicotinamide by Mixing-Induced Supersaturation: Exploring Opportunities between Reactive and Antisolvent Crystallization Concepts. Cryst. Growth Des. 2017, 17, 1902–1909. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Beatty, A.M.; Helfrich, B.A. A High-Yielding Supramolecular Reaction. J. Am. Chem. Soc. 2002, 124, 14425–14432. [Google Scholar] [CrossRef] [PubMed]
- Vishweshwar, P.; Nangia, A.; Lynch, V.M. Molecular Complexes of Homologous Alkanedicarboxylic Acids with Isonicotinamide: X-ray Crystal Structures, Hydrogen Bond Synthons, and Melting Point Alternation. Cryst. Growth Des. 2003, 3, 783–790. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Class | (1:1) | (2:1) | (1:2) | (1:1:1) | Other Compositions § | Totals |
|---|---|---|---|---|---|---|
| ArCO2H…INA | 44 | 9 | 5 | 15 | 1 | 74 (34%) |
| RCO2H…INA | 44 | 3 | 22 | 2 | 0 | 71 (33%) |
| ROH…INA | 18 | 1 | 10 | 1 | 4 | 34 (16%) |
| Other Functional Groups | 22 | 2 | 4 | 6 | 2 | 36 (17%) |
| Totals | 128 (60%) | 15 (7%) | 41 (19%) | 24 (11%) | 7 (3%) | 215 |
| Class | (1:1) | (2:1) | (1:2) | (1:1:1) | Other Compositions § | Totals |
|---|---|---|---|---|---|---|
| ArCO2H…NA | 30 | 3 | 3 | 10 | 3 | 49 (31%) |
| RCO2H…NA | 27 | 3 | 9 | 5 | 4 | 48 (30%) |
| ROH…NA | 17 | 2 | 6 | 2 | 6 | 33 (21%) |
| Other Functional Groups | 16 | 1 | 3 | 4 | 4 | 28 (18%) |
| Totals | 90 (58%) | 9 (6%) | 21 (13%) | 21 (13%) | 17 (10%) | 158 |
| Groups—INA | (1:1) | (2:1) | (1:2) | Groups—NA | (1:1) | (2:1) | (1:2) |
|---|---|---|---|---|---|---|---|
| CO2H | 69 | 6 | 22 | CO2H | 36 | 4 | 11 |
| CO2H/OH | 13 | 4 | 4 | CO2H/OH | 14 | 2 | 1 |
| CO2H/NH2 | 6 | 1 | 0 | CO2H/NH2 | 3 | 0 | 0 |
| CO2H/CONHR | 1 | 1 | 0 | CO2H/CONHR | 1 | 0 | 0 |
| CO2H/SO2NH2 | 0 | 1 | 0 | CO2H/SO2NH2 | 2 | 0 | 1 |
| OH | 14 | 1 | 10 | OH | 15 | 1 | 4 |
| OH/CO2H | 3 | 0 | 0 | OH/CO2H | 2 | 0 | 0 |
| OH/CONHR | 2 | 0 | 0 | OH/CONHR | 0 | 0 | 1 |
| CONH2 | 4 | 1 | 0 | CONH2 | 2 | 1 | 2 |
| SO2NHR/CO2H | 1 | 0 | 1 | SO2NHR | 3 | 0 | 0 |
| NH2 | 5 | 0 | 0 | NH2 | 3 | 0 | 0 |
| All other functional groups | 10 | 0 | 4 | All other functional groups | 8 | 0 | 1 |
| System | D(2)R22(8) | D(2)R22(x) | D(2) | D(2)C(x) |
|---|---|---|---|---|
| ArCO2H---INA | 41 | 0 | 2 | 1 |
| RCO2H---INA | 20 | 0 | 21 | 3 |
| ROH---INA | 7 | 5 | 5 | 2 |
| Other Functional Groups | 9 | 0 | 4 | 10 |
| Totals | 77 (59%) | 5 (4%) | 32 (25%) | 16 (12%) |
| ArCO2H---NA | 11 | 0 | 7 | 16 |
| RCO2H---NA | 12 | 0 | 11 | 5 |
| ROH---NA | 5 | 0 | 8 | 7 |
| Other Functional Groups | 7 | 0 | 1 | 8 |
| Totals | 35 (36%) | 0 (0%) | 27 (28%) | 36 (36%) |
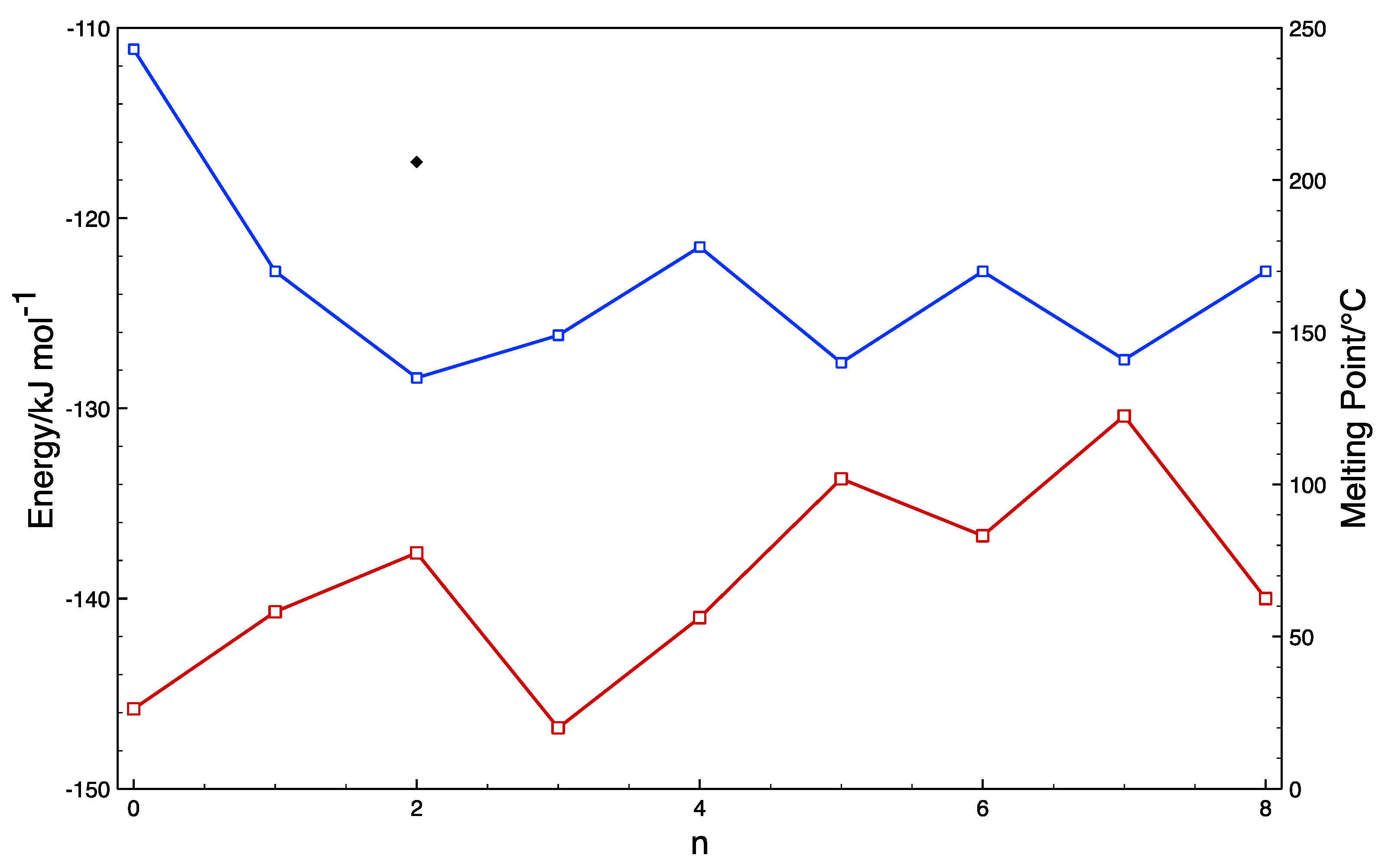
| System | Acid…Amide Motif kJ mol−1 | Acid…Npyr Motif kJ mol−1 | Difference kJ mol−1 |
|---|---|---|---|
| Oxalic acid | −36.145 | −32.556 | −3.589 |
| Malonic acid | −67.531 | −51.742 | −15.789 |
| Succinic acid | −60.365 | −45.143 | −17.814 |
| Glutaric acid | −61.957 | −44.143 | −16.959 |
| Adipic acid | −57.152 | −40.193 | −16.959 |
| Pimelic acid | −57.793 | −50.027 | −7.765 |
| INA | −56.703 | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seaton, C.C. Building Up Co-Crystals: Structural Motif Consistencies Across Families of Co-Crystals. Proceedings 2021, 78, 45. https://doi.org/10.3390/IECP2020-08708
Seaton CC. Building Up Co-Crystals: Structural Motif Consistencies Across Families of Co-Crystals. Proceedings. 2021; 78(1):45. https://doi.org/10.3390/IECP2020-08708
Chicago/Turabian StyleSeaton, Colin C. 2021. "Building Up Co-Crystals: Structural Motif Consistencies Across Families of Co-Crystals" Proceedings 78, no. 1: 45. https://doi.org/10.3390/IECP2020-08708
APA StyleSeaton, C. C. (2021). Building Up Co-Crystals: Structural Motif Consistencies Across Families of Co-Crystals. Proceedings, 78(1), 45. https://doi.org/10.3390/IECP2020-08708