Modelling of Void Collapse with Molecular Dynamics in Pure Sn †


 ,
, {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
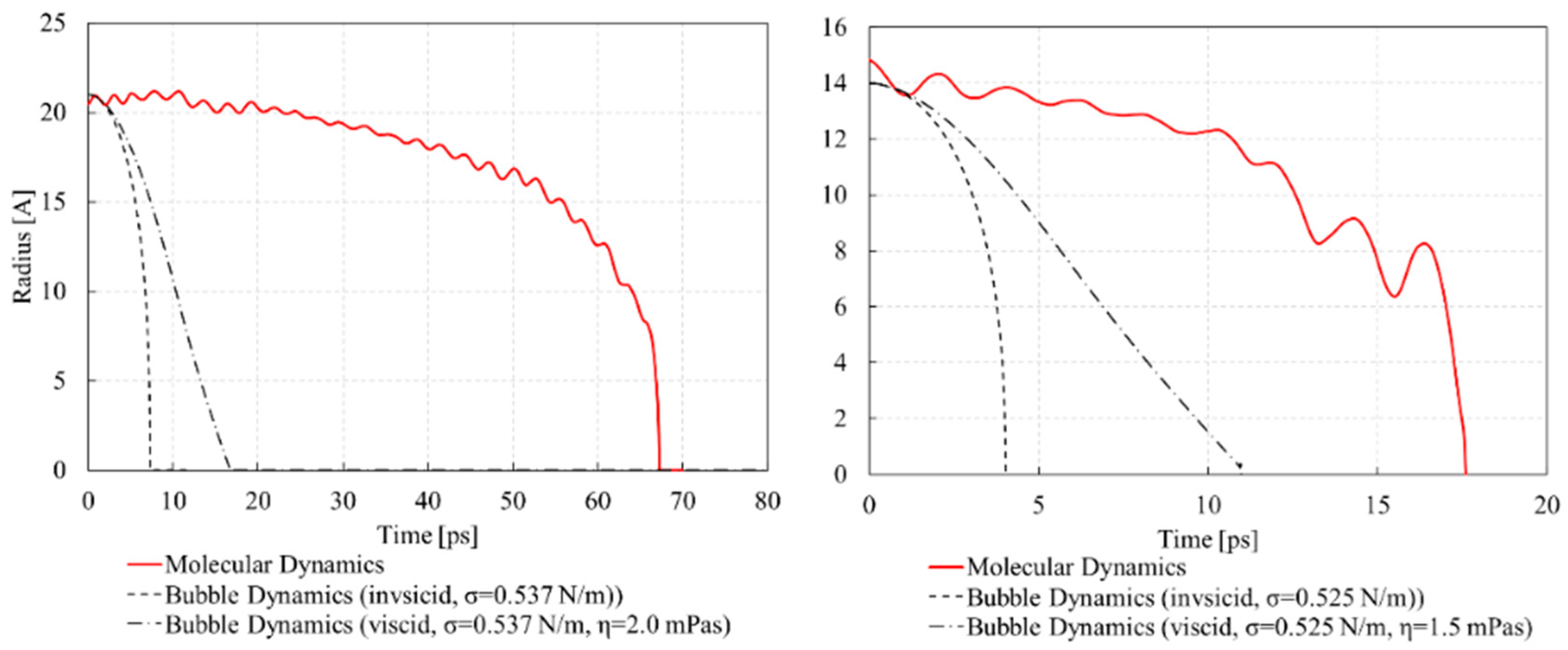
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
References
- van Dijk, N.; van der Zwaag, S. Self-healing materials are coming of age. Adv. Mater. Interfaces 2018, 1800226, 1–13. [Google Scholar]
- Siroky, G.; Kraker, E.; Kieslinger, D.; Kozeschnik, E.; Ecker, W. Micromechanics-based damage model for liquid-assisted healing. Int. J. Damage Mech 2020, 29, 1–22. [Google Scholar] [CrossRef]
- Ravelo, R. Equilibrium and Thermodynamic Properties of Grey, White, and Liquid Tin. Phys Rev Lett 1997, 79, 2482–2485. [Google Scholar] [CrossRef]
- Bourasseau, E.; Filippini, G.; Ghoufi, A.; Malfreyt, P. Calculation of the surface tension of pure tin from atomistic simulations of liquid–vapour systems. Mol. Phys. 2014, 112, 2654–2657. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siroky, G.; Kraker, E.; Kieslinger, D.; Romaner, L.; Kozeschnik, E.; Ecker, W. Modelling of Void Collapse with Molecular Dynamics in Pure Sn. Proceedings 2020, 56, 29. https://doi.org/10.3390/proceedings2020056029
Siroky G, Kraker E, Kieslinger D, Romaner L, Kozeschnik E, Ecker W. Modelling of Void Collapse with Molecular Dynamics in Pure Sn. Proceedings. 2020; 56(1):29. https://doi.org/10.3390/proceedings2020056029
Chicago/Turabian StyleSiroky, Georg, Elke Kraker, Dietmar Kieslinger, Lorenz Romaner, Ernst Kozeschnik, and Werner Ecker. 2020. "Modelling of Void Collapse with Molecular Dynamics in Pure Sn" Proceedings 56, no. 1: 29. https://doi.org/10.3390/proceedings2020056029
APA StyleSiroky, G., Kraker, E., Kieslinger, D., Romaner, L., Kozeschnik, E., & Ecker, W. (2020). Modelling of Void Collapse with Molecular Dynamics in Pure Sn. Proceedings, 56(1), 29. https://doi.org/10.3390/proceedings2020056029