

Navigating the Chemical Space and Chemical Multiverse of a Unified Latin American Natural Product Database: LANaPDB

 , , ,
, , ,  ,
,  , , , ,
, , , ,  , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Bioactive Compounds from Latin American Natural Product Databases
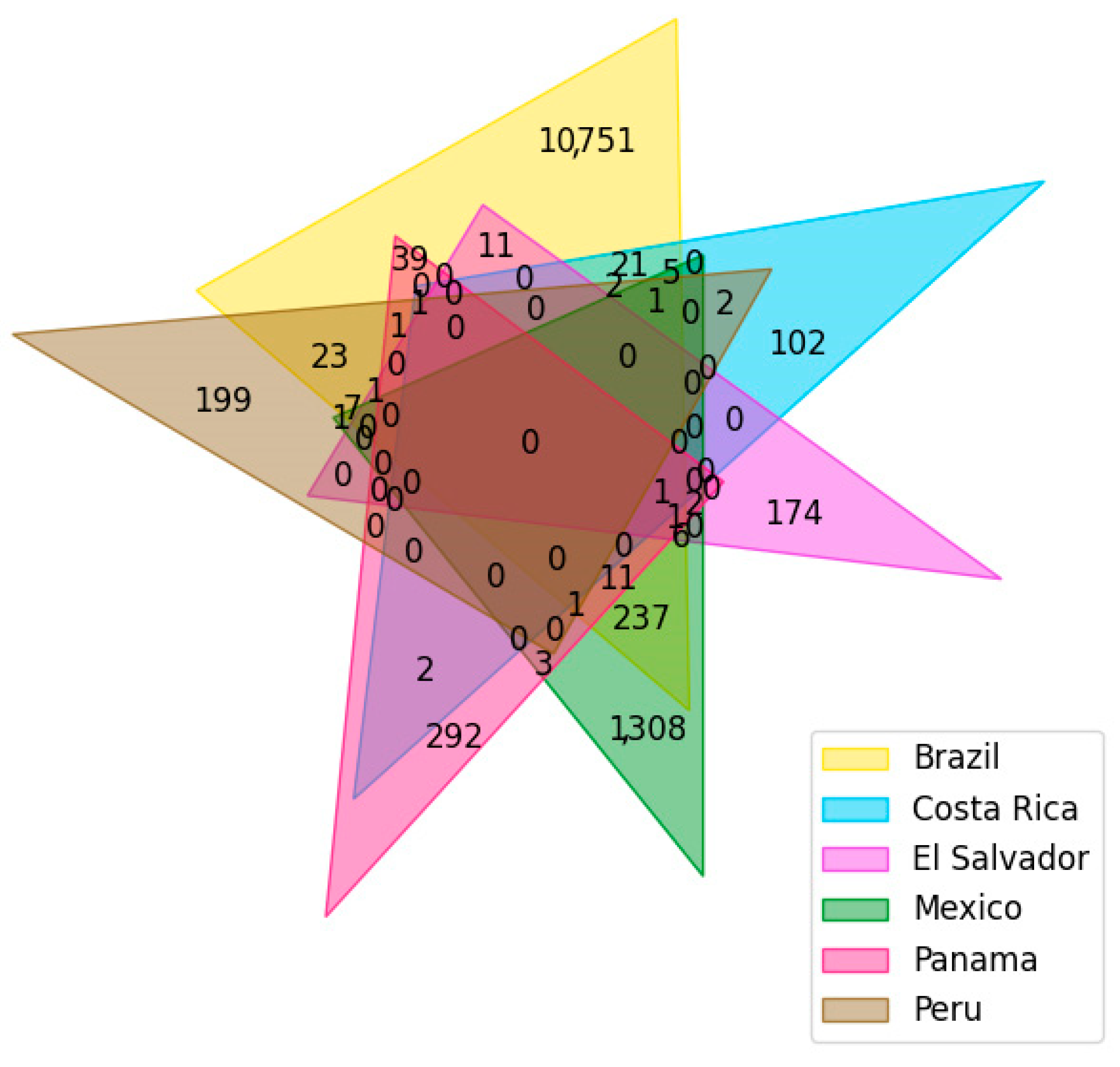
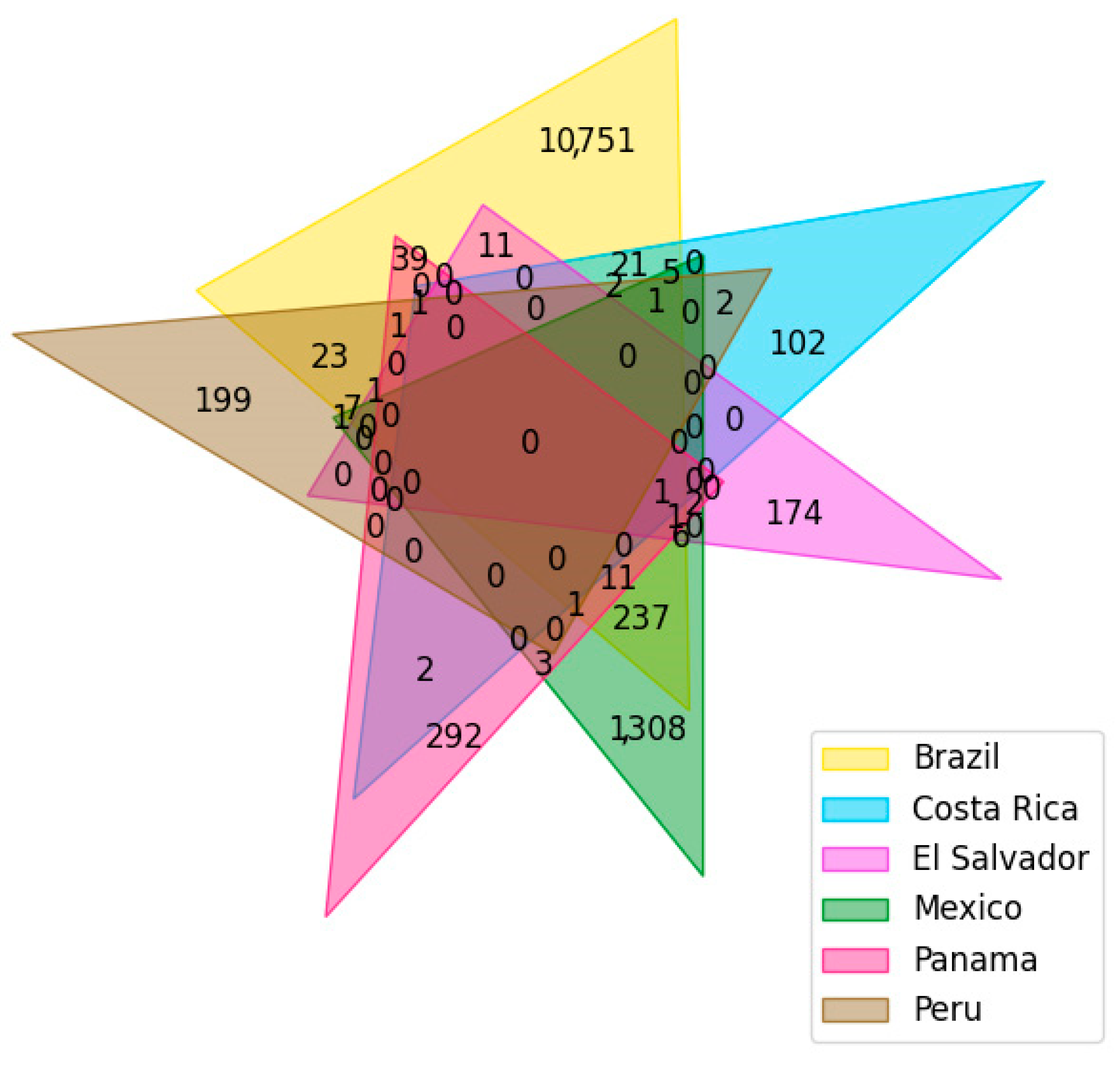
2.2. Dataset Curation
2.3. Structural Classification
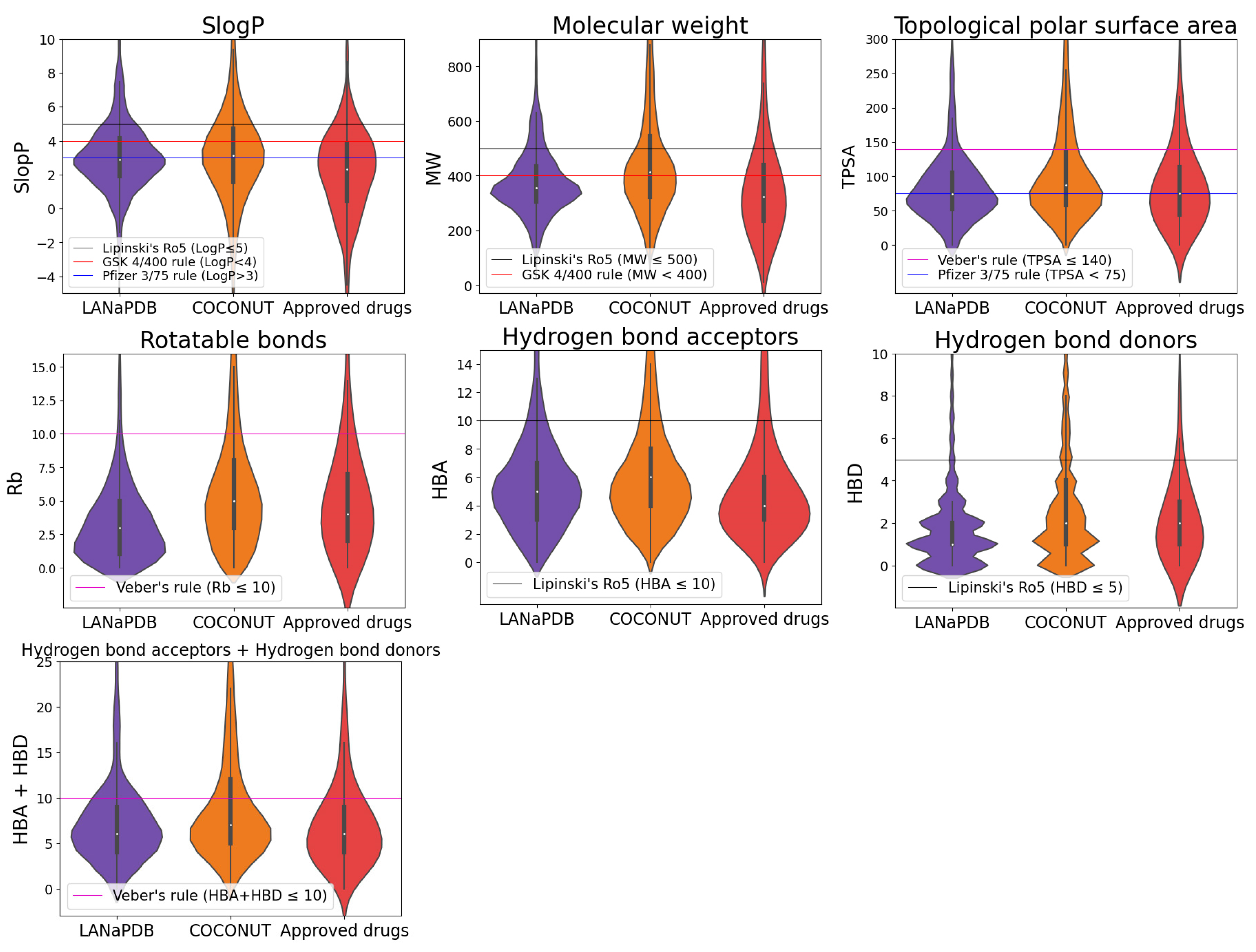
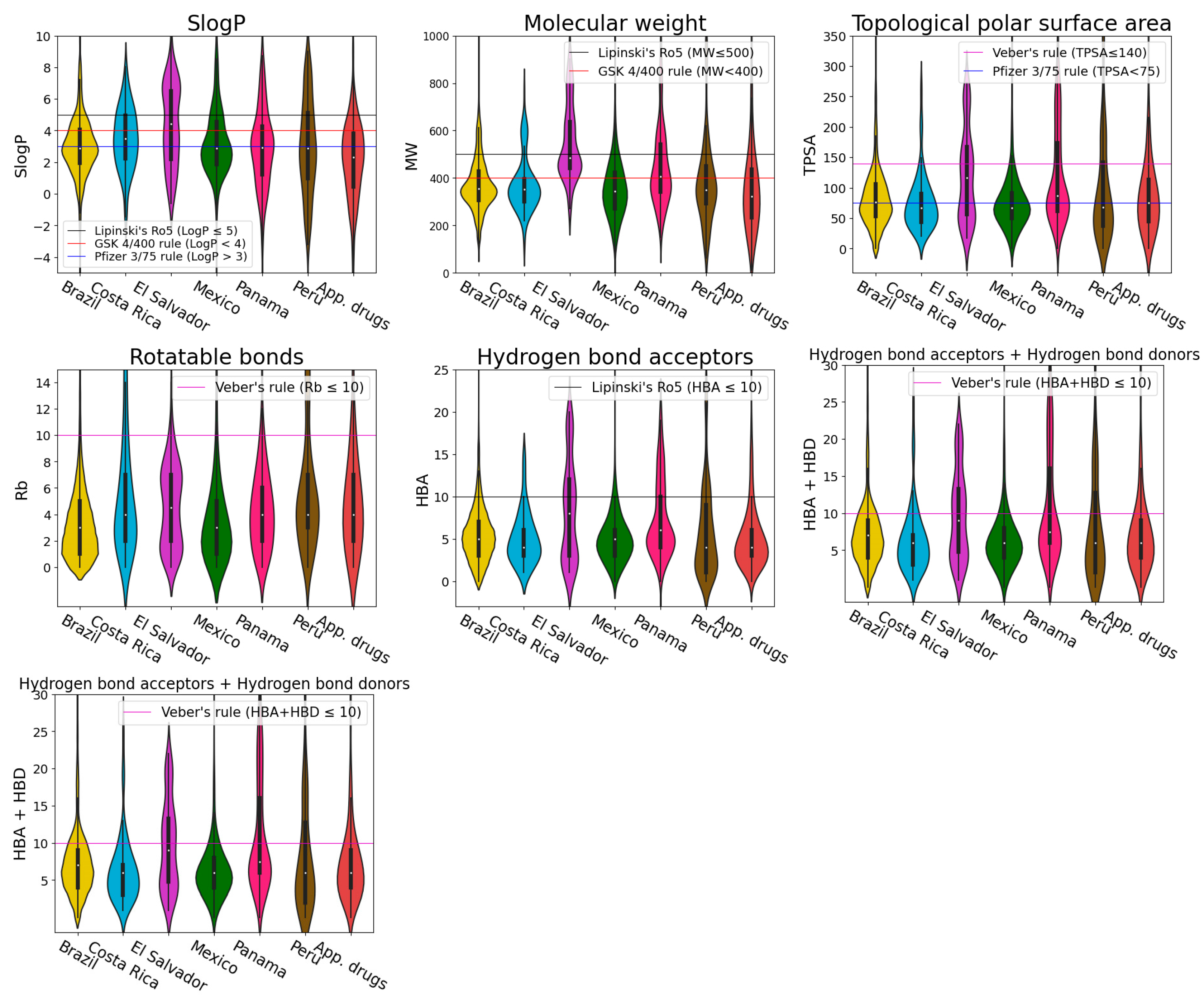
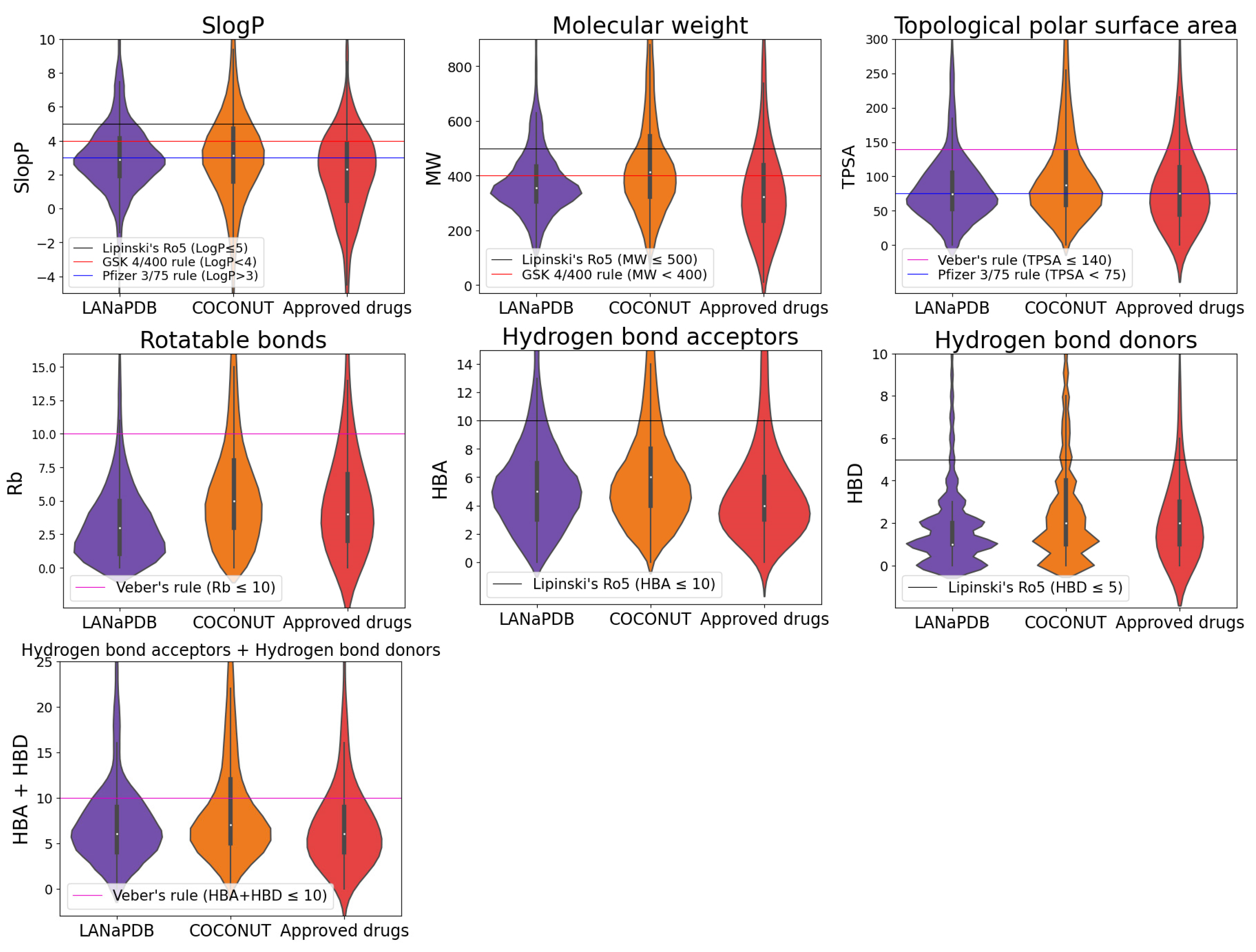
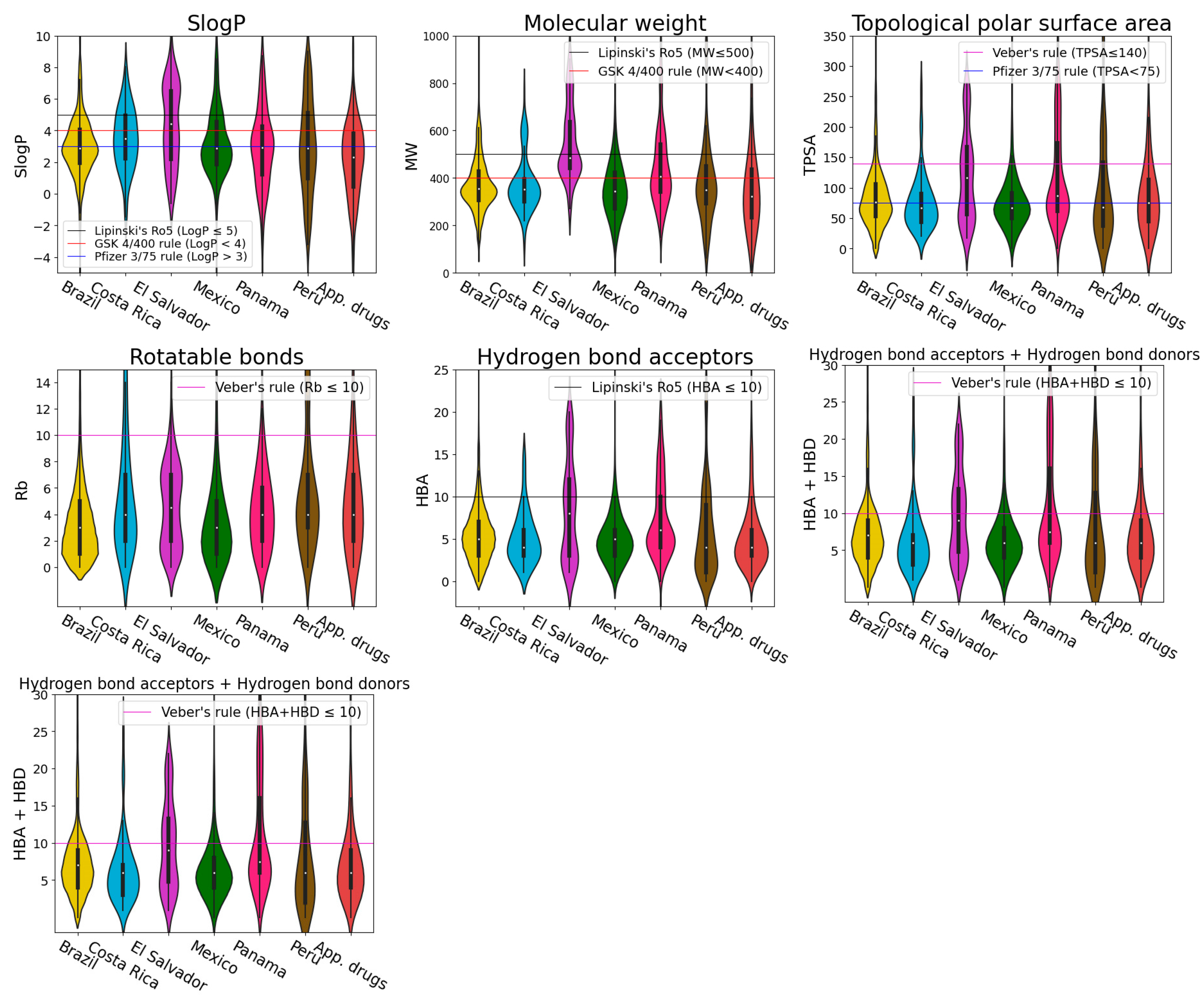
2.4. Physicochemical Properties
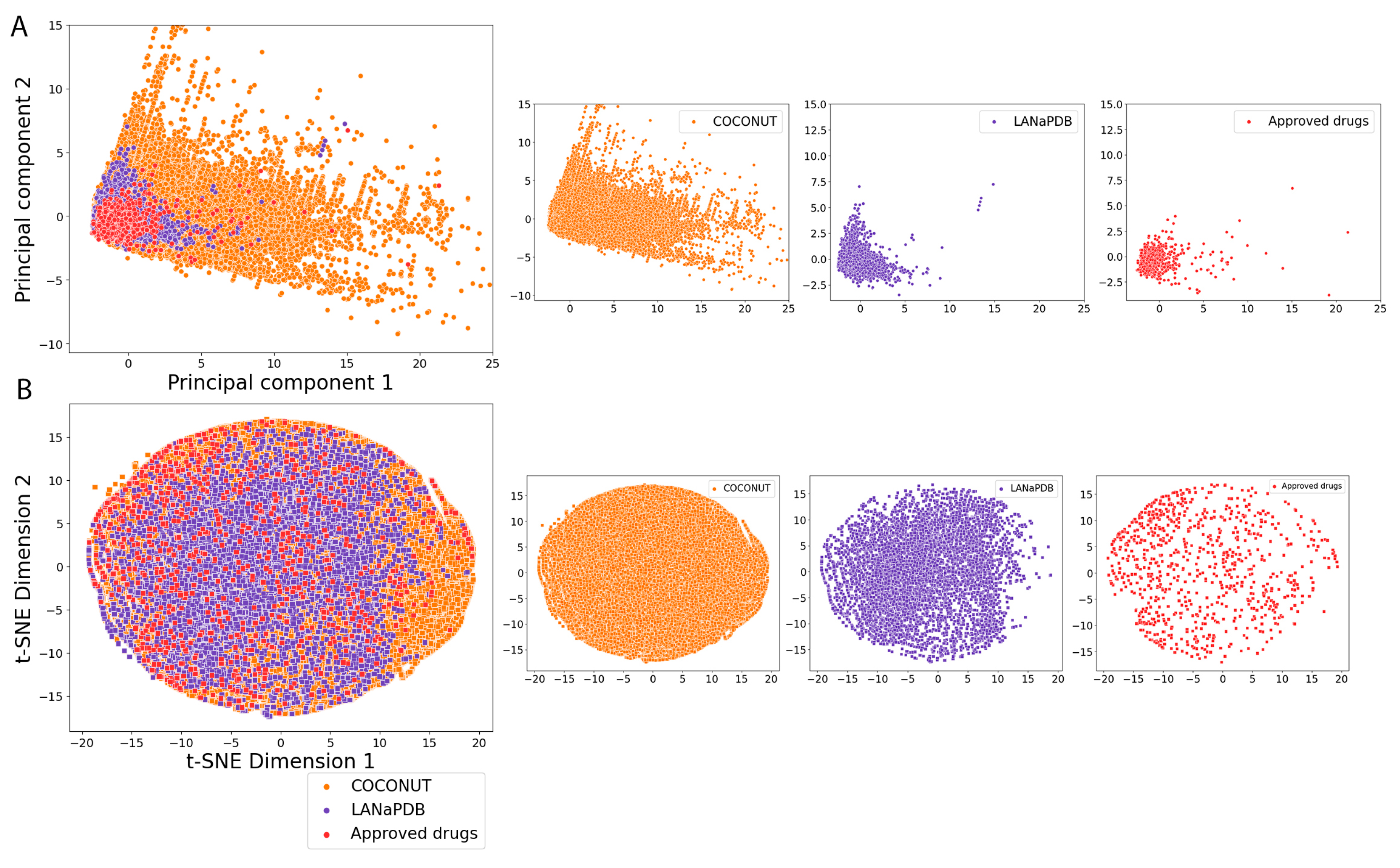
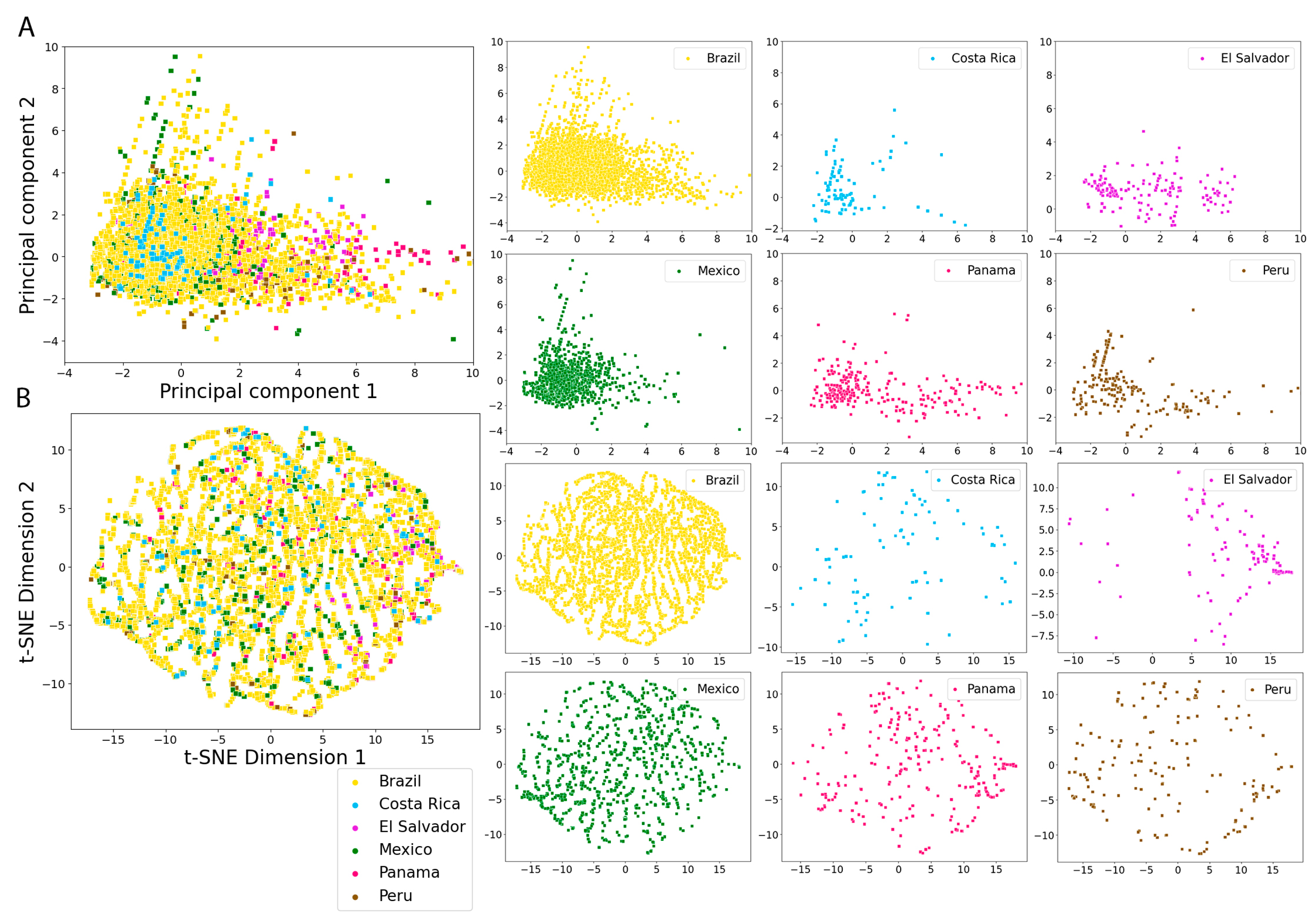
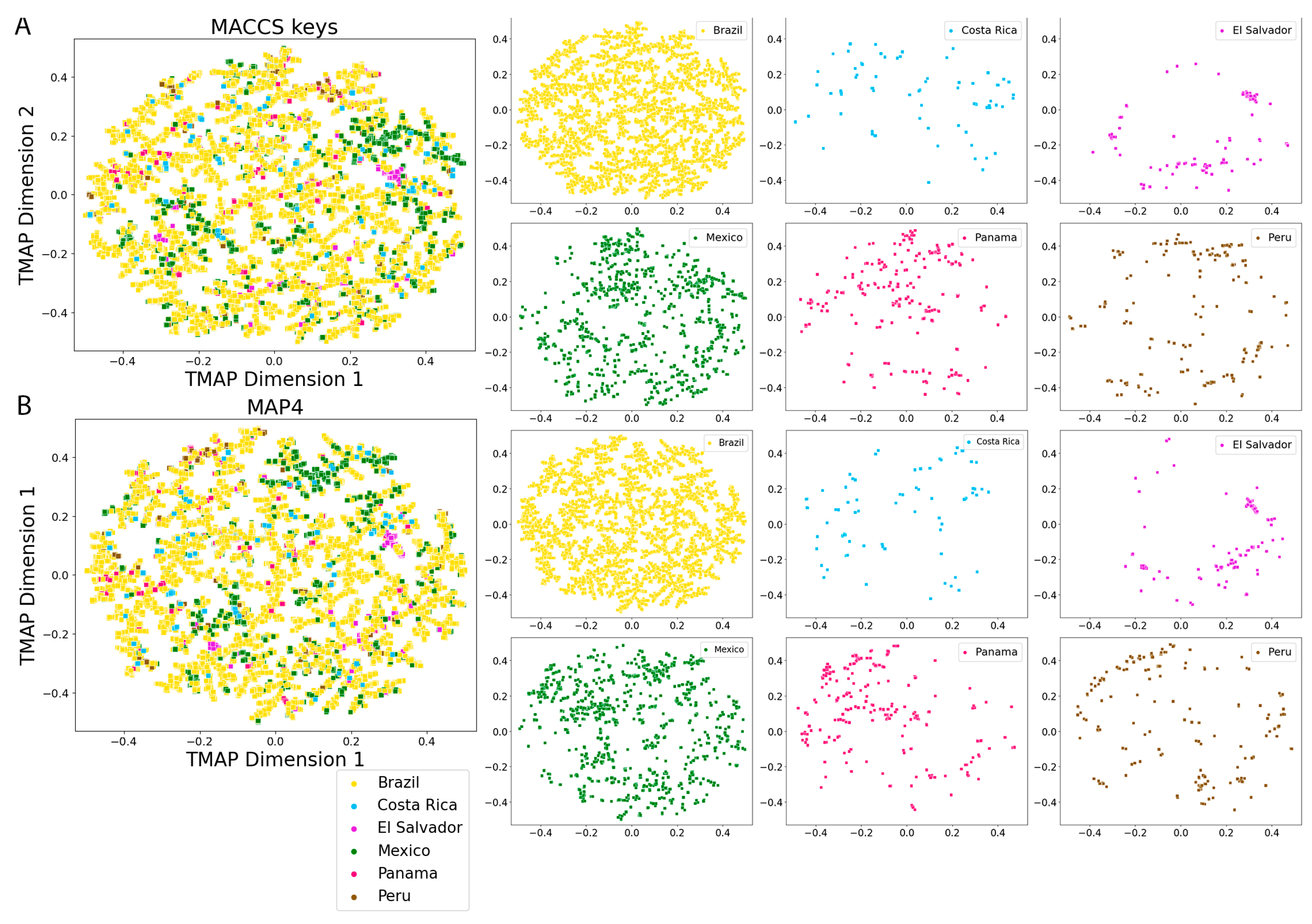
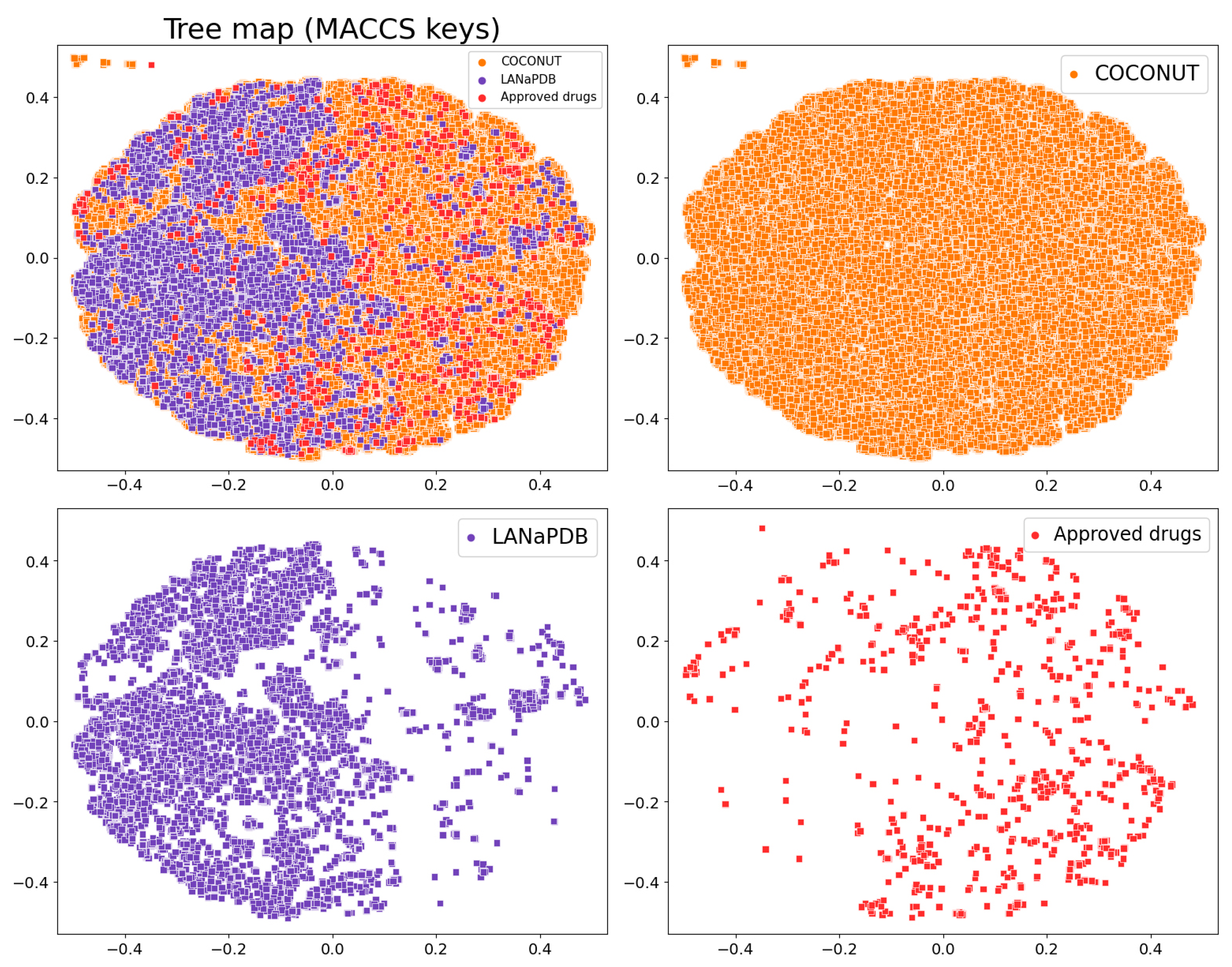
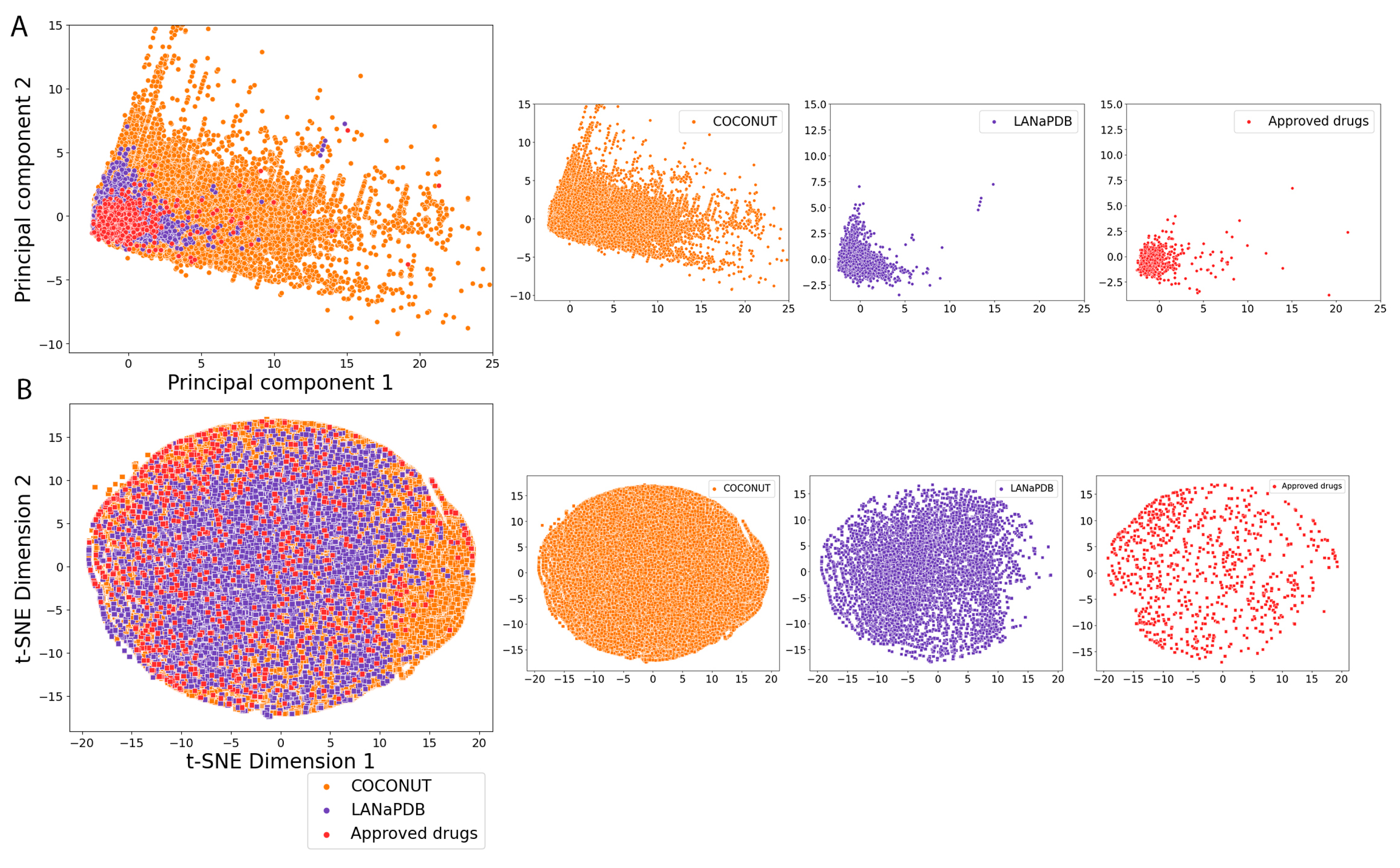
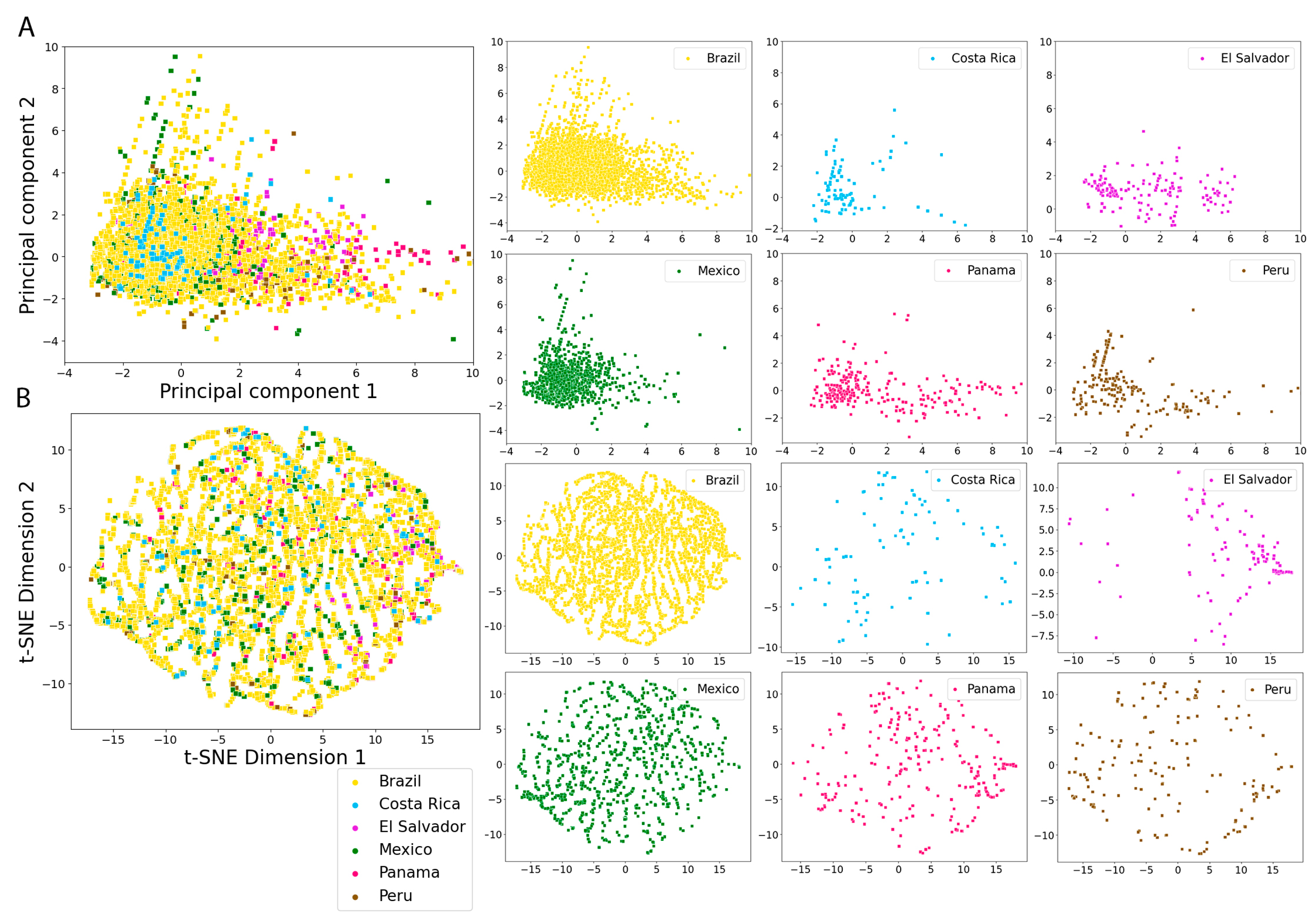
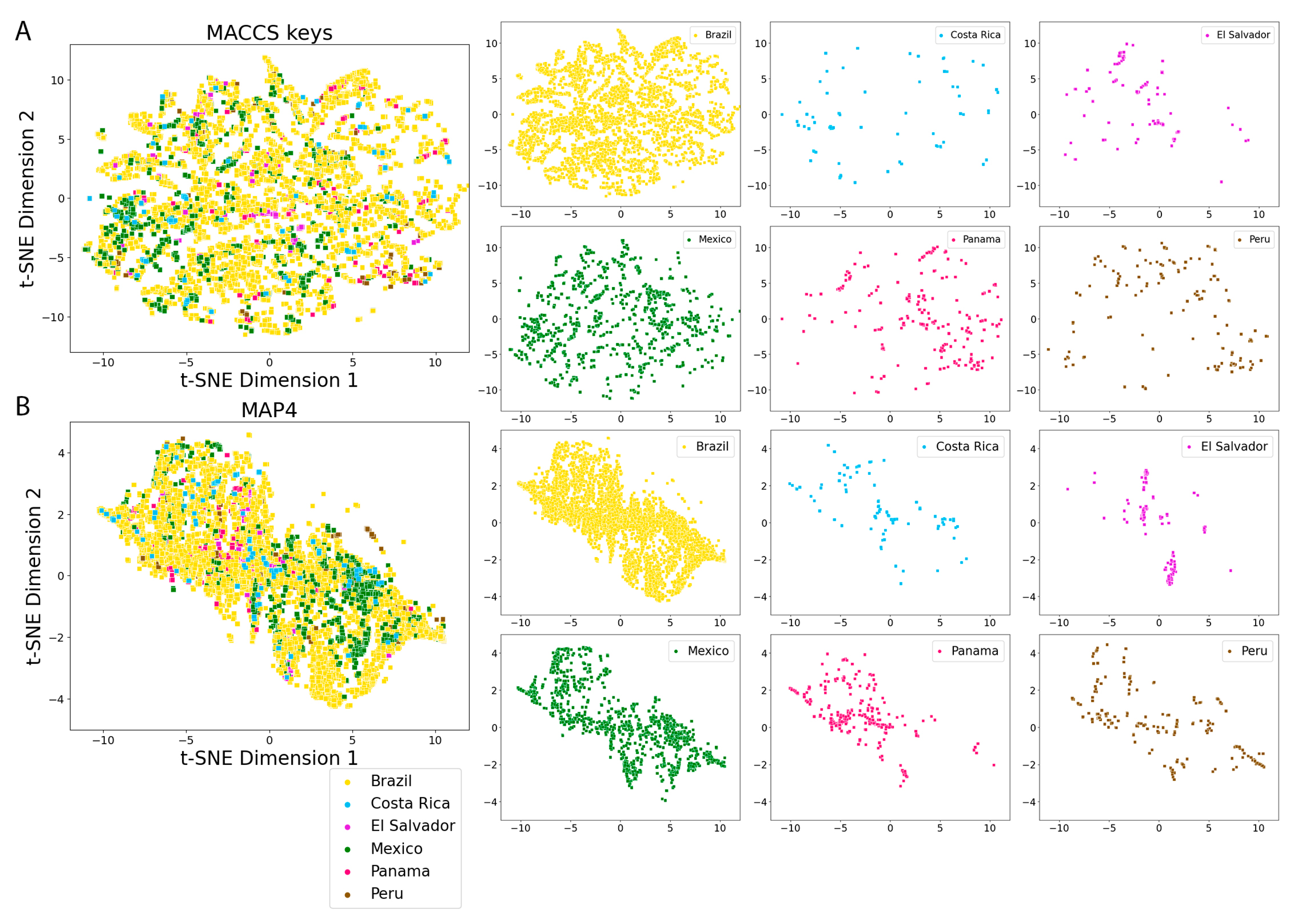
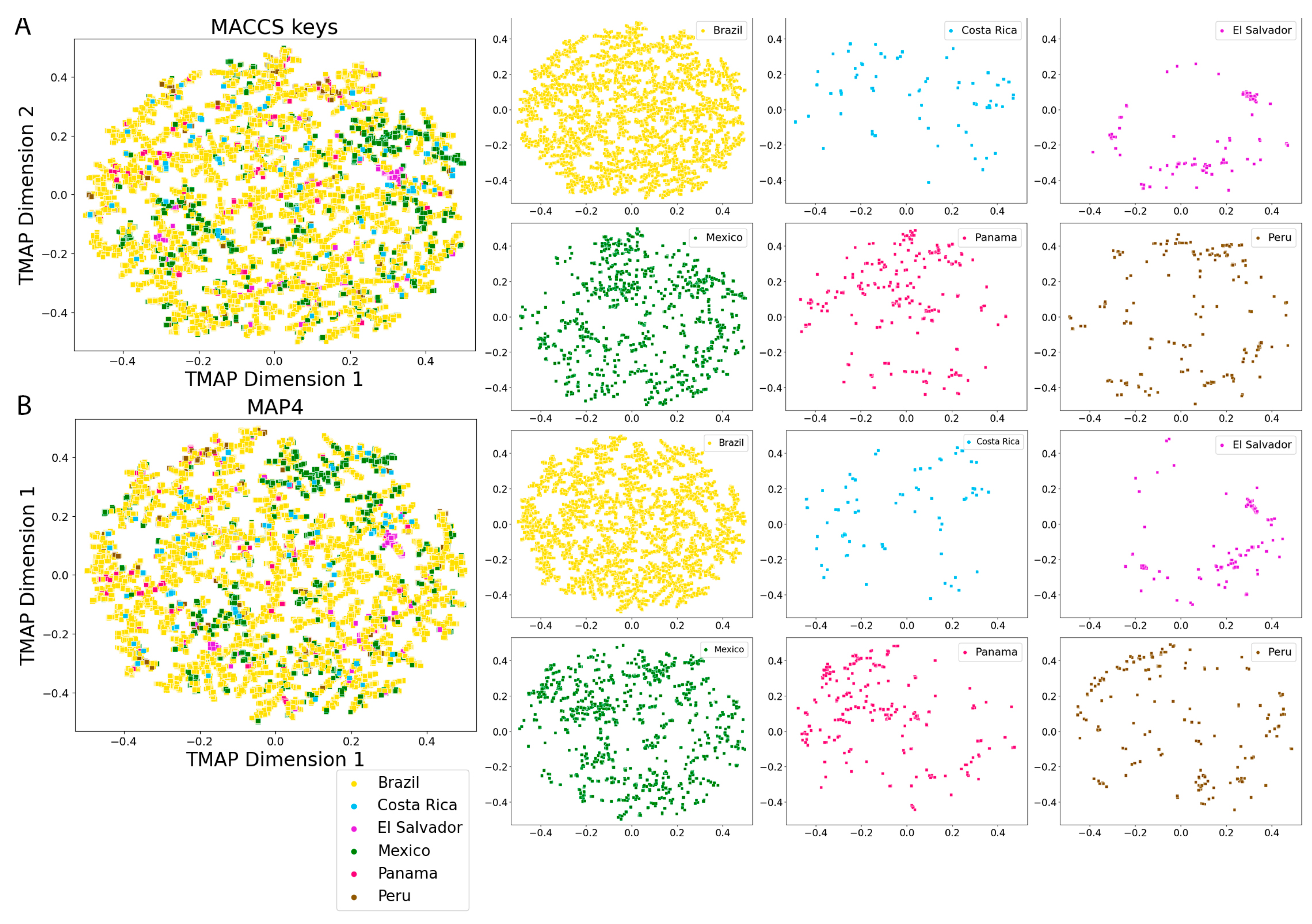
2.5. Molecular Fingerprints
3. Materials and Methods
3.1. Dataset Curation
3.2. Structural Classification
3.3. Physicochemical Properties
3.4. Molecular Fingerprints
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Porras-Alcalá, C.; Moya-Utrera, F.; García-Castro, M.; Sánchez-Ruiz, A.; López-Romero, J.M.; Pino-González, M.S.; Díaz-Morilla, A.; Kitamura, S.; Wolan, D.W.; Prados, J.; et al. The development of the bengamides as new antibiotics against drug-resistant bacteria. Mar. Drugs 2022, 20, 373. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.-L.; Hu, B.-Y.; Qi, Z.-H.; Wang, X.-N.; Xie, T.-Z.; Wang, Z.-J.; Ma, D.-Y.; Zeng, Q.; Luo, X.-D. Chemistry and bioactivities of natural steroidal alkaloids. Nat. Prod. Bioprospect. 2022, 12, 23. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-W. Chemical ecology-driven discovery of bioactive marine natural products as potential drug leads. Chin. J. Nat. Med. 2020, 18, 837–838. [Google Scholar] [CrossRef]
- Banerjee, P.; Mandhare, A.; Bagalkote, V. Marine natural products as source of new drugs: An updated patent review (July 2018–July 2021). Expert Opin. Ther. Pat. 2022, 32, 317–363. [Google Scholar] [CrossRef]
- Singh, A.; Singh, D.K.; Kharwar, R.N.; White, J.F.; Gond, S.K. Fungal endophytes as efficient sources of plant-derived bioactive compounds and their prospective applications in natural product drug discovery: Insights, avenues, and challenges. Microorganisms 2021, 9, 197. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.; Bae, H. Endophytic fungi: Key insights, emerging prospects, and challenges in natural product drug discovery. Microorganisms 2022, 10, 360. [Google Scholar] [CrossRef]
- Foxfire, A.; Buhrow, A.R.; Orugunty, R.S.; Smith, L. Drug discovery through the isolation of natural products from Burkholderia. Expert Opin. Drug Discov. 2021, 16, 807–822. [Google Scholar] [CrossRef]
- Porras, G.; Chassagne, F.; Lyles, J.T.; Marquez, L.; Dettweiler, M.; Salam, A.M.; Samarakoon, T.; Shabih, S.; Farrokhi, D.R.; Quave, C.L. Ethnobotany and the role of plant natural products in antibiotic drug discovery. Chem. Rev. 2021, 121, 3495–3560. [Google Scholar] [CrossRef]
- Zhang, L.; Song, J.; Kong, L.; Yuan, T.; Li, W.; Zhang, W.; Hou, B.; Lu, Y.; Du, G. The strategies and techniques of drug discovery from natural products. Pharmacol. Ther. 2020, 216, 107686. [Google Scholar] [CrossRef]
- Bordon, K.; de, C.F.; Cologna, C.T.; Fornari-Baldo, E.C.; Pinheiro-Júnior, E.L.; Cerni, F.A.; Amorim, F.G.; Anjolette, F.A.P.; Cordeiro, F.A.; Wiezel, G.A.; et al. From animal poisons and venoms to medicines: Achievements, challenges and perspectives in drug discovery. Front. Pharmacol. 2020, 11, 1132. [Google Scholar] [CrossRef] [PubMed]
- Hussain, H.; Mamadalieva, N.Z.; Hussain, A.; Hassan, U.; Rabnawaz, A.; Ahmed, I.; Green, I.R. Fruit peels: Food waste as a valuable source of bioactive natural products for drug discovery. Curr. Issues Mol. Biol. 2022, 44, 1960–1994. [Google Scholar] [CrossRef] [PubMed]
- Shams, U.L.; Hassan, S.; Jin, H.-Z.; Abu-Izneid, T.; Rauf, A.; Ishaq, M.; Suleria, H.A.R. Stress-driven discovery in the natural products: A gateway towards new drugs. Biomed. Pharmacother. 2019, 109, 459–467. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Zhang, Y. Teaching an old dog new tricks: Drug discovery by repositioning natural products and their derivatives. Drug Discov. Today 2022, 27, 1936–1944. [Google Scholar] [CrossRef]
- Evans, B.E.; Rittle, K.E.; Bock, M.G.; DiPardo, R.M.; Freidinger, R.M.; Whitter, W.L.; Lundell, G.F.; Veber, D.F.; Anderson, P.S.; Chang, R.S. Methods for drug discovery: Development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 1988, 31, 2235–2246. [Google Scholar] [CrossRef]
- Davison, E.K.; Brimble, M.A. Natural product derived privileged scaffolds in drug discovery. Curr. Opin. Chem. Biol. 2019, 52, 1–8. [Google Scholar] [CrossRef]
- Karageorgis, G.; Foley, D.J.; Laraia, L.; Brakmann, S.; Waldmann, H. Pseudo natural products-chemical evolution of natural product structure. Angew. Chem. Int. Ed. 2021, 60, 15705–15723. [Google Scholar] [CrossRef]
- Karageorgis, G.; Foley, D.J.; Laraia, L.; Waldmann, H. Principle and design of pseudo-natural products. Nat. Chem. 2020, 12, 227–235. [Google Scholar] [CrossRef]
- Cremosnik, G.S.; Liu, J.; Waldmann, H. Guided by evolution: From biology oriented synthesis to pseudo natural products. Nat. Prod. Rep. 2020, 37, 1497–1510. [Google Scholar] [CrossRef]
- Guo, Z. The modification of natural products for medical use. Acta Pharm. Sin. B 2017, 7, 119–136. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.M.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Roy, K. How far can virtual screening take us in drug discovery? Expert Opin. Drug Discov. 2013, 8, 245–261. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef]
- Vijayan, R.S.K.; Kihlberg, J.; Cross, J.B.; Poongavanam, V. Enhancing preclinical drug discovery with artificial intelligence. Drug Discov. Today 2022, 27, 967–984. [Google Scholar] [CrossRef]
- Grover, I.; Singh, I.; Bakshi, I. Quantitative structure-property relationships in pharmaceutical research-Part 1. Pharm. Sci. Technol. Today 2000, 3, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Di Filippo, J.I. Artificial intelligence in the early stages of drug discovery. Arch. Biochem. Biophys. 2021, 698, 108730. [Google Scholar] [CrossRef]
- Shen, C.; Hu, Y.; Wang, Z.; Zhang, X.; Zhong, H.; Wang, G.; Yao, X.; Xu, L.; Cao, D.; Hou, T. Can machine learning consistently improve the scoring power of classical scoring functions? Insights into the role of machine learning in scoring functions. Brief. Bioinform. 2021, 22, 497–514. [Google Scholar] [CrossRef]
- Ain, Q.U.; Aleksandrova, A.; Roessler, F.D.; Ballester, P.J. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2015, 5, 405–424. [Google Scholar] [CrossRef]
- Saldívar-González, F.I.; Aldas-Bulos, V.D.; Medina-Franco, J.L.; Plisson, F. Natural product drug discovery in the artificial intelligence era. Chem. Sci. 2022, 13, 1526–1546. [Google Scholar] [CrossRef]
- Jeon, J.; Kang, S.; Kim, H.U. Predicting biochemical and physiological effects of natural products from molecular structures using machine learning. Nat. Prod. Rep. 2021, 38, 1954–1966. [Google Scholar] [CrossRef] [PubMed]
- de Sousa Luis, J.A.; Barros, R.P.C.; de Sousa, N.F.; Muratov, E.; Scotti, L.; Scotti, M.T. Virtual screening of natural products database. Mini Rev. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Gangadevi, S.; Badavath, V.N.; Thakur, A.; Yin, N.; De Jonghe, S.; Acevedo, O.; Jochmans, D.; Leyssen, P.; Wang, K.; Neyts, J.; et al. Kobophenol A inhibits binding of host ace2 receptor with spike RBD domain of SARS-CoV-2, a lead compound for blocking COVID-19. J. Phys. Chem. Lett. 2021, 12, 1793–1802. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Hsu, H.-J.; Wu, T.-Y.; Liou, J.-W. Computer-aided discovery, design, and investigation of COVID-19 therapeutics. Tzu Chi Med. J. 2022, 34, 276–286. [Google Scholar] [CrossRef]
- Siva Kumar, B.; Anuragh, S.; Kammala, A.K.; Ilango, K. Computer aided drug design approach to screen phytoconstituents of adhatoda vasica as potential inhibitors of SARS-CoV-2 main protease enzyme. Life 2022, 12, 315. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Dai, R.; Su, R. Computer-aided drug design for the pain-like protease (PLpro) inhibitors against SARS-CoV-2. Biomed. Pharmacother. 2023, 159, 114247. [Google Scholar] [CrossRef]
- Cerón-Carrasco, J.P. When virtual screening yields inactive drugs: Dealing with false theoretical friends. ChemMedChem 2022, 17, e202200278. [Google Scholar] [CrossRef]
- Dantas, R.F.; Evangelista, T.C.S.; Neves, B.J.; Senger, M.R.; Andrade, C.H.; Ferreira, S.B.; Silva-Junior, F.P. Dealing with frequent hitters in drug discovery: A multidisciplinary view on the issue of filtering compounds on biological screenings. Expert Opin. Drug Discov. 2019, 14, 1269–1282. [Google Scholar] [CrossRef]
- Sorokina, M.; Steinbeck, C. Review on natural products databases: Where to find data in 2020. J. Cheminf. 2020, 12, 20. [Google Scholar] [CrossRef]
- Sorokina, M.; Merseburger, P.; Rajan, K.; Yirik, M.A.; Steinbeck, C. COCONUT online: Collection of Open Natural Products database. J. Cheminf. 2021, 13, 2. [Google Scholar] [CrossRef]
- Gu, J.; Gui, Y.; Chen, L.; Yuan, G.; Lu, H.-Z.; Xu, X. Use of natural products as chemical library for drug discovery and network pharmacology. PLoS ONE 2013, 8, e62839. [Google Scholar] [CrossRef] [PubMed]
- ISDB. A database of In-Silico predicted MS/MS spectrum of Natural Products. Available online: http://oolonek.github.io/ISDB/ (accessed on 12 June 2023).
- Gallo, K.; Kemmler, E.; Goede, A.; Becker, F.; Dunkel, M.; Preissner, R.; Banerjee, P. SuperNatural 3.0-a database of natural products and natural product-based derivatives. Nucleic Acids Res. 2023, 51, D654–D659. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC 15--ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Ye, L.; Kang, H.; Zhang, D.; Tao, L.; Tang, K.; Liu, X.; Zhu, R.; Liu, Q.; Chen, Y.Z.; et al. HIT: Linking herbal active ingredients to targets. Nucleic Acids Res. 2011, 39, D1055–D1059. [Google Scholar] [CrossRef]
- Kang, H.; Tang, K.; Liu, Q.; Sun, Y.; Huang, Q.; Zhu, R.; Gao, J.; Zhang, D.; Huang, C.; Cao, Z. HIM-herbal ingredients in-vivo metabolism database. J. Cheminf. 2013, 5, 28. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Ma, C.; Zhao, X.; Hu, Z.; Du, T.; Xu, X.; Wang, Z.; Lin, J. YaTCM: Yet another traditional chinese medicine database for drug discovery. Comput. Struct. Biotechnol. J. 2018, 16, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Ru, J.; Li, P.; Wang, J.; Zhou, W.; Li, B.; Huang, C.; Li, P.; Guo, Z.; Tao, W.; Yang, Y.; et al. TCMSP: A database of systems pharmacology for drug discovery from herbal medicines. J. Cheminf. 2014, 6, 13. [Google Scholar] [CrossRef]
- Kim, S.-K.; Nam, S.; Jang, H.; Kim, A.; Lee, J.-J. TM-MC: A database of medicinal materials and chemical compounds in Northeast Asian traditional medicine. BMC Complement. Altern. Med. 2015, 15, 218. [Google Scholar] [CrossRef]
- Xu, H.-Y.; Zhang, Y.-Q.; Liu, Z.-M.; Chen, T.; Lv, C.-Y.; Tang, S.-H.; Zhang, X.-B.; Zhang, W.; Li, Z.-Y.; Zhou, R.-R.; et al. ETCM: An encyclopaedia of traditional Chinese medicine. Nucleic Acids Res. 2019, 47, D976–D982. [Google Scholar] [CrossRef]
- Fang, X.; Shao, L.; Zhang, H.; Wang, S. CHMIS-C: A comprehensive herbal medicine information system for cancer. J. Med. Chem. 2005, 48, 1481–1488. [Google Scholar] [CrossRef]
- Qiao, X.; Hou, T.; Zhang, W.; Guo, S.; Xu, X. A 3D structure database of components from Chinese traditional medicinal herbs. J. Chem. Inf. Comput. Sci. 2002, 42, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zheng, Y.; Wu, W.; Xie, T.; Yao, H.; Pang, X.; Sun, F.; Ouyang, L.; Wang, J. CEMTDD: The database for elucidating the relationships among herbs, compounds, targets and related diseases for Chinese ethnic minority traditional drugs. Oncotarget 2015, 6, 17675–17684. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.-C. TCM Database@Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef]
- Mohanraj, K.; Karthikeyan, B.S.; Vivek-Ananth, R.P.; Chand, R.P.B.; Aparna, S.R.; Mangalapandi, P.; Samal, A. IMPPAT: A curated database of Indian medicinal plants, phytochemistry and therapeutics. Sci. Rep. 2018, 8, 4329. [Google Scholar] [CrossRef] [PubMed]
- Potshangbam, A.M.; Polavarapu, R.; Rathore, R.S.; Naresh, D.; Prabhu, N.P.; Potshangbam, N.; Kumar, P.; Vindal, V. MedPServer: A database for identification of therapeutic targets and novel leads pertaining to natural products. Chem. Biol. Drug Des. 2019, 93, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Bultum, L.E.; Woyessa, A.M.; Lee, D. ETM-DB: Integrated Ethiopian traditional herbal medicine and phytochemicals database. BMC Complement. Altern. Med. 2019, 19, 212. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Onguéné, P.A.; Scharfe, M.; Owono Owono, L.C.; Megnassan, E.; Mbaze, L.M.; Sippl, W.; Efange, S.M.N. ConMedNP: A natural product library from Central African medicinal plants for drug discovery. RSC Adv. 2014, 4, 409–419. [Google Scholar] [CrossRef]
- Ibezim, A.; Debnath, B.; Ntie-Kang, F.; Mbah, C.J.; Nwodo, N.J. Binding of anti-Trypanosoma natural products from African flora against selected drug targets: A docking study. Med. Chem. Res. 2017, 26, 562–579. [Google Scholar] [CrossRef]
- Onguéné, P.A.; Ntie-Kang, F.; Mbah, J.A.; Lifongo, L.L.; Ndom, J.C.; Sippl, W.; Mbaze, L.M. The potential of anti-malarial compounds derived from African medicinal plants, part III: An in silico evaluation of drug metabolism and pharmacokinetics profiling. Org. Med. Chem. Lett. 2014, 4, 6. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Nwodo, J.N.; Ibezim, A.; Simoben, C.V.; Karaman, B.; Ngwa, V.F.; Sippl, W.; Adikwu, M.U.; Mbaze, L.M. Molecular modeling of potential anticancer agents from African medicinal plants. J. Chem. Inf. Model. 2014, 54, 2433–2450. [Google Scholar] [CrossRef]
- Ntie-Kang, F.; Amoa Onguéné, P.; Fotso, G.W.; Andrae-Marobela, K.; Bezabih, M.; Ndom, J.C.; Ngadjui, B.T.; Ogundaini, A.O.; Abegaz, B.M.; Meva’a, L.M. Virtualizing the p-ANAPL library: A step towards drug discovery from African medicinal plants. PLoS ONE 2014, 9, e90655. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Zofou, D.; Babiaka, S.B.; Meudom, R.; Scharfe, M.; Lifongo, L.L.; Mbah, J.A.; Mbaze, L.M.; Sippl, W.; Efange, S.M.N. AfroDb: A select highly potent and diverse natural product library from African medicinal plants. PLoS ONE 2013, 8, e78085. [Google Scholar] [CrossRef]
- Ionov, N.; Druzhilovskiy, D.; Filimonov, D.; Poroikov, V. Phyto4Health: Database of phytocomponents from russian pharmacopoeia plants. J. Chem. Inf. Model. 2023, 63, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Raven, P.H.; Gereau, R.E.; Phillipson, P.B.; Chatelain, C.; Jenkins, C.N.; Ulloa Ulloa, C. The distribution of biodiversity richness in the tropics. Sci. Adv. 2020, 6, eabc6228. [Google Scholar] [CrossRef] [PubMed]
- Mittermeier, R.A.; Turner, W.R.; Larsen, F.W.; Brooks, T.M.; Gascon, C. Global biodiversity conservation: The critical role of Hotspots. In Biodiversity Hotspots; Zachos, F.E., Habel, J.C., Eds.; Springer: Berlin, Heidelberg, 2011; pp. 3–22. ISBN 978-3-642-20991-8. [Google Scholar]
- NaturAr. Available online: https://naturar.quimica.unlp.edu.ar/es/ (accessed on 9 December 2022).
- Valli, M.; dos Santos, R.N.; Figueira, L.D.; Nakajima, C.H.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. Development of a natural products database from the biodiversity of Brazil. J. Nat. Prod. 2013, 76, 439–444. [Google Scholar] [CrossRef]
- Pilon, A.C.; Valli, M.; Dametto, A.C.; Pinto, M.E.F.; Freire, R.T.; Castro-Gamboa, I.; Andricopulo, A.D.; Bolzani, V.S. NuBBEDB: An updated database to uncover chemical and biological information from Brazilian biodiversity. Sci. Rep. 2017, 7, 7215. [Google Scholar] [CrossRef]
- Scotti, M.T.; Herrera-Acevedo, C.; Oliveira, T.B.; Costa, R.P.O.; Santos, S.Y.K.; de, O.; Rodrigues, R.P.; Scotti, L.; Da-Costa, F.B. SistematX, an online web-based cheminformatics tool for data management of secondary metabolites. Molecules 2018, 23, 103. [Google Scholar] [CrossRef]
- Costa, R.P.O.; Lucena, L.F.; Silva, L.M.A.; Zocolo, G.J.; Herrera-Acevedo, C.; Scotti, L.; Da-Costa, F.B.; Ionov, N.; Poroikov, V.; Muratov, E.N.; et al. The Sistematx web portal of natural products: An update. J. Chem. Inf. Model. 2021, 61, 2516–2522. [Google Scholar] [CrossRef]
- UEFS Natural Products. Available online: http://zinc12.docking.org/catalogs/uefsnp (accessed on 2 December 2022).
- Olmedo, D.A.; González-Medina, M.; Gupta, M.P.; Medina-Franco, J.L. Cheminformatic characterization of natural products from Panama. Mol. Divers. 2017, 21, 779–789. [Google Scholar] [CrossRef]
- Olmedo, A.D.; Medina-Franco, L.J. Chemoinformatic approach: The case of natural products of Panama. In Cheminformatics and Its Applications; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Barazorda-Ccahuana, H.L.; Ranilla, L.G.; Candia-Puma, M.A.; Cárcamo-Rodriguez, E.G.; Centeno-Lopez, A.E.; Davila-Del-Carpio, G.; Medina-Franco, J.L.; Chávez-Fumagalli, M.A. PeruNPDB: The Peruvian natural products database for in silico drug screening. Sci. Rep. 2023, 13, 7577. [Google Scholar] [CrossRef]
- UNIIQUIM. Available online: https://uniiquim.iquimica.unam.mx/ (accessed on 6 December 2022).
- Pilón-Jiménez, B.A.; Saldívar-González, F.I.; Díaz-Eufracio, B.I.; Medina-Franco, J.L. BIOFACQUIM: A mexican compound database of natural products. Biomolecules 2019, 9, 31. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Cruz, N.; Pilón-Jiménez, B.A.; Medina-Franco, J.L. Functional group and diversity analysis of BIOFACQUIM: A Mexican natural product database. F1000Research 2019, 8, 2071. [Google Scholar] [CrossRef]
- Gómez-García, A.; Medina-Franco, J.L. Progress and impact of latin american natural product databases. Biomolecules 2022, 12, 1202. [Google Scholar] [CrossRef] [PubMed]
- do Carmo Santos, N.; da Paixão, V.G.; da Rocha Pita, S.S. New Trypanosoma cruzi Trypanothione Reductase Inhibitors Identification using the Virtual Screening in Database of Nucleus Bioassay, Biosynthesis and Ecophysiology (NuBBE). Anti-Infect. Agents 2019, 17, 138–149. [Google Scholar] [CrossRef]
- Acevedo, C.H.; Scotti, L.; Scotti, M.T. In Silico studies designed to select sesquiterpene lactones with potential antichagasic activity from an in-house asteraceae database. ChemMedChem 2018, 13, 634–645. [Google Scholar] [CrossRef]
- Antunes, S.S.; Won-Held Rabelo, V.; Romeiro, N.C. Natural products from Brazilian biodiversity identified as potential inhibitors of PknA and PknB of M. tuberculosis using molecular modeling tools. Comput. Biol. Med. 2021, 136, 104694. [Google Scholar] [CrossRef]
- Herrera-Acevedo, C.; Dos Santos Maia, M.; Cavalcanti, É.B.V.S.; Coy-Barrera, E.; Scotti, L.; Scotti, M.T. Selection of antileishmanial sesquiterpene lactones from SistematX database using a combined ligand-/structure-based virtual screening approach. Mol. Divers. 2021, 25, 2411–2427. [Google Scholar] [CrossRef]
- Barazorda-Ccahuana, H.L.; Goyzueta-Mamani, L.D.; Candia Puma, M.A.; Simões de Freitas, C.; de Sousa Vieria Tavares, G.; Pagliara Lage, D.; Ferraz Coelho, E.A.; Chávez-Fumagalli, M.A. Computer-aided drug design approaches applied to screen natural product’s structural analogs targeting arginase in Leishmania spp. F1000Research 2023, 12, 93. [Google Scholar] [CrossRef]
- Menezes, R.P.B.; de Viana, J.; de, O.; Muratov, E.; Scotti, L.; Scotti, M.T. Computer-assisted discovery of alkaloids with schistosomicidal activity. Curr. Issues Mol. Biol. 2022, 44, 383–408. [Google Scholar] [CrossRef]
- Rodrigues, G.C.S.; Dos Santos Maia, M.; de Menezes, R.P.B.; Cavalcanti, A.B.S.; de Sousa, N.F.; de Moura, É.P.; Monteiro, A.F.M.; Scotti, L.; Scotti, M.T. Ligand and structure-based virtual screening of lamiaceae diterpenes with potential activity against a novel coronavirus (2019-nCoV). Curr. Top. Med. Chem. 2020, 20, 2126–2145. [Google Scholar] [CrossRef]
- Przybyłek, M. Application 2D descriptors and artificial neural networks for beta-glucosidase inhibitors screening. Molecules 2020, 25, 5942. [Google Scholar] [CrossRef]
- Martinez-Mayorga, K.; Marmolejo-Valencia, A.F.; Cortes-Guzman, F.; García-Ramos, J.C.; Sánchez-Flores, E.I.; Barroso-Flores, J.; Medina-Franco, J.L.; Esquivel-Rodriguez, B. Toxicity assessment of structurally relevant natural products from Mexican plants with antinociceptive activity. J. Mex. Chem. Soc. 2017, 61, 186–196. [Google Scholar] [CrossRef]
- Barrera-Vázquez, O.S.; Gómez-Verjan, J.C.; Magos-Guerrero, G.A. Chemoinformatic screening for the selection of potential senolytic compounds from natural products. Biomolecules 2021, 11, 467. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Acevedo, C.; Perdomo-Madrigal, C.; Herrera-Acevedo, K.; Coy-Barrera, E.; Scotti, L.; Scotti, M.T. Machine learning models to select potential inhibitors of acetylcholinesterase activity from SistematX: A natural products database. Mol. Divers. 2021, 25, 1553–1568. [Google Scholar] [CrossRef] [PubMed]
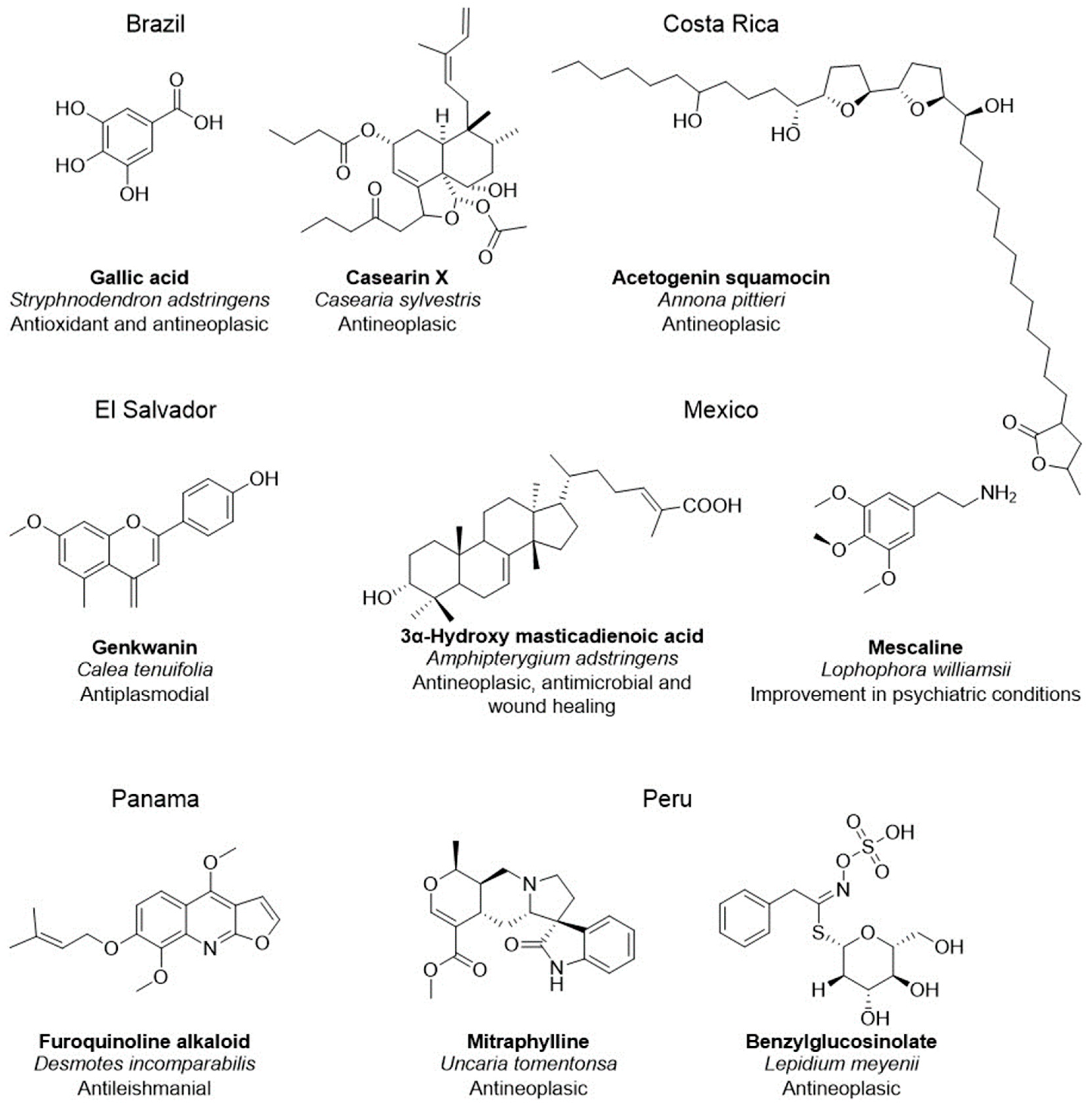
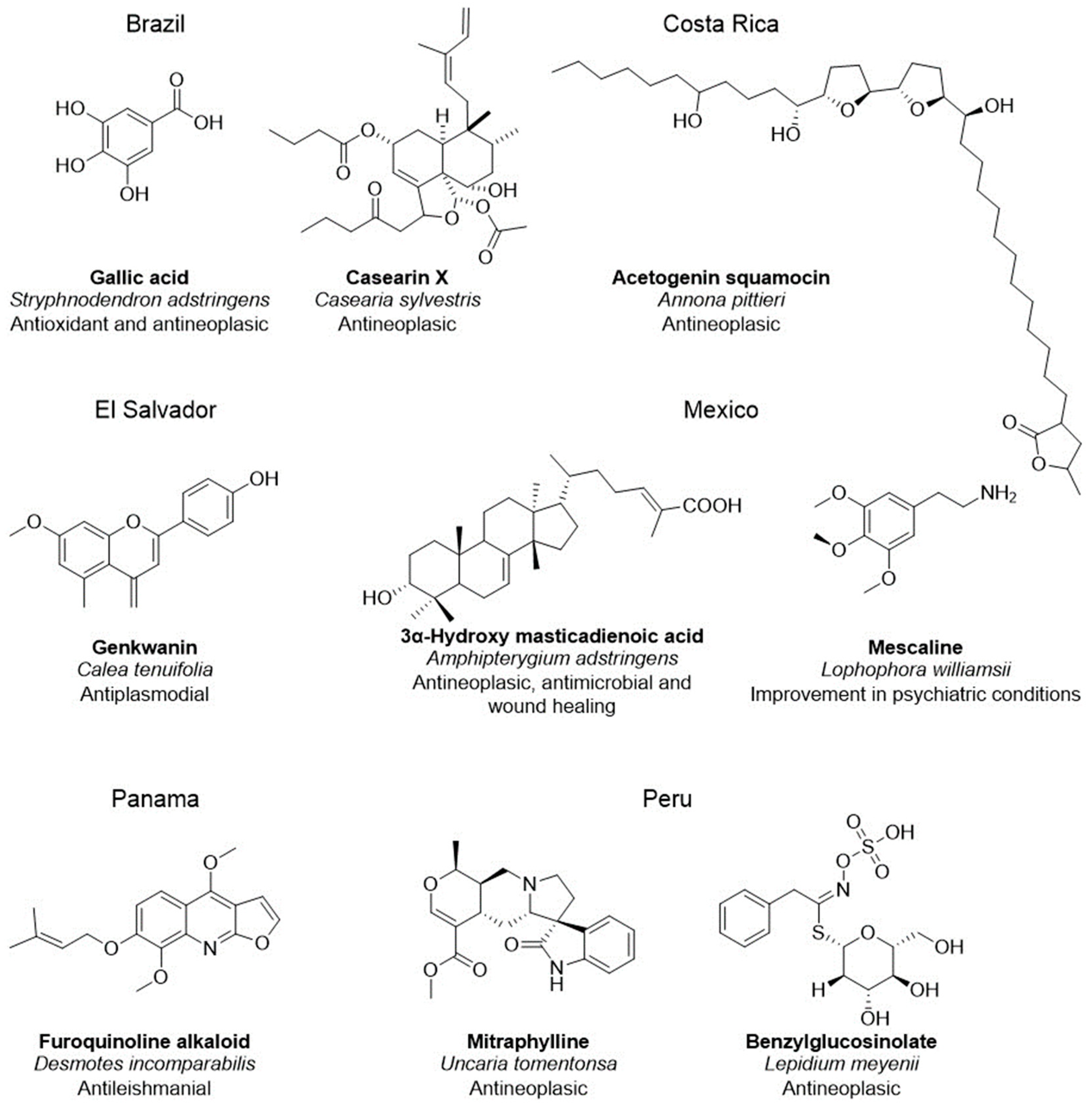
- de Souza Ribeiro, M.M.; dos Santos, L.C.; de Novais, N.S.; Viganó, J.; Veggi, P.C. An evaluative review on Stryphnodendron adstringens extract composition: Current and future perspectives on extraction and application. Ind. Crops Prod. 2022, 187, 115325. [Google Scholar] [CrossRef]
- Li, R.; Morris-Natschke, S.L.; Lee, K.-H. Clerodane diterpenes: Sources, structures, and biological activities. Nat. Prod. Rep. 2016, 33, 1166–1226. [Google Scholar] [CrossRef]
- Parra, J.; Ford, C.D.; Murillo, R. Phytochemical study of endemic Costa rican annonaceae species annona pittieri and Cymbopetalum costaricense. J. Chil. Chem. Soc. 2021, 66, 5047–5050. [Google Scholar] [CrossRef]
- Köhler, I.; Jenett-Siems, K.; Siems, K.; Hernández, M.A.; Ibarra, R.A.; Berendsohn, W.G.; Bienzle, U.; Eich, E. In vitro antiplasmodial investigation of medicinal plants from El Salvador. Z. Naturforsch. C. J. Biosci. 2002, 57, 277–281. [Google Scholar] [CrossRef]
- Sotelo-Barrera, M.; Cília-García, M.; Luna-Cavazos, M.; Díaz-Núñez, J.L.; Romero-Manzanares, A.; Soto-Hernández, R.M.; Castillo-Juárez, I. Amphipterygium adstringens (Schltdl.) Schiede ex Standl (Anacardiaceae): An Endemic Plant with Relevant Pharmacological Properties. Plants 2022, 11, 1766. [Google Scholar] [CrossRef]
- Agin-Liebes, G.; Haas, T.F.; Lancelotta, R.; Uthaug, M.V.; Ramaekers, J.G.; Davis, A.K. Naturalistic use of mescaline is associated with self-reported psychiatric improvements and enduring positive life changes. ACS Pharmacol. Transl. Sci. 2021, 4, 543–552. [Google Scholar] [CrossRef]
- Cubilla-Rios, L.; Chérigo, L.; Ríos, C.; Togna, G.D.; Gerwick, W.H. Phytochemical analysis and antileishmanial activity of Desmotes incomparabilis, an endemic plant from Panama. Planta Med. 2008, 74, PA98. [Google Scholar] [CrossRef]
- García Giménez, D.; García Prado, E.; Sáenz Rodríguez, T.; Fernández Arche, A.; De la Puerta, R. Cytotoxic effect of the pentacyclic oxindole alkaloid mitraphylline isolated from Uncaria tomentosa bark on human Ewing’s sarcoma and breast cancer cell lines. Planta Med. 2010, 76, 133–136. [Google Scholar] [CrossRef]
- Gonzales, G.F.; Valerio, L.G. Medicinal plants from Peru: A review of plants as potential agents against cancer. Anticancer. Agents Med. Chem. 2006, 6, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; Chávez-Hernández, A.L.; López-López, E.; Saldívar-González, F.I. Chemical multiverse: An expanded view of chemical space. Mol. Inf. 2022, 41, e2200116. [Google Scholar] [CrossRef] [PubMed]
- Isah, M.B.; Tajuddeen, N.; Umar, M.I.; Alhafiz, Z.A.; Mohammed, A.; Ibrahim, M.A. Terpenoids as emerging therapeutic agents: Cellular targets and mechanisms of action against protozoan parasites. In Studies in Natural Products Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; ISBN 9780444641793. [Google Scholar]
- Wildman, S.A.; Crippen, G.M. Prediction of physicochemical parameters by atomic contributions. J. Chem. Inf. Comput. Sci. 1999, 39, 868–873. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; Decrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef] [PubMed]
- Ntie-Kang, F.; Nyongbela, K.D.; Ayimele, G.A.; Shekfeh, S. “Drug-likeness” properties of natural compounds. Phys. Sci. Rev. 2019, 4, 20180169. [Google Scholar] [CrossRef]
- Probst, D.; Reymond, J.-L. Visualization of very large high-dimensional data sets as minimum spanning trees. J. Cheminf. 2020, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Capecchi, A.; Probst, D.; Reymond, J.-L. One molecular fingerprint to rule them all: Drugs, biomolecules, and the metabolome. J. Cheminform. 2020, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Durant, J.L.; Leland, B.A.; Henry, D.R.; Nourse, J.G. Reoptimization of MDL keys for use in drug discovery. J. Chem. Inf. Comput. Sci. 2002, 42, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Waskom, M. Seaborn: Statistical data visualization. J. Open Source Softw. 2021, 6, 3021. [Google Scholar] [CrossRef]
- Open-Source Chemoinformatics and Machine Learning. RDKit: Open-Source Cheminformatics Software. Available online: https://www.rdkit.org (accessed on 8 February 2023).
- MolVS: Molecule Validation and Standardization. Available online: https://molvs.readthedocs.io/en/latest/index.html (accessed on 9 February 2023).
- Kim, H.W.; Wang, M.; Leber, C.A.; Nothias, L.-F.; Reher, R.; Kang, K.B.; van der Hooft, J.J.J.; Dorrestein, P.C.; Gerwick, W.H.; Cottrell, G.W. NPClassifier: A deep neural network-based structural classification tool for natural products. J. Nat. Prod. 2021, 84, 2795–2807. [Google Scholar] [CrossRef]
- Plotly Technologies Inc. Collaborative Data Science Publisher: Plotly Technologies Inc.; Plotly Technologies Inc.: Montréal, QC, USA, Canada, 2015. [Google Scholar]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Thiel, K.; Wiswedel, B. KNIME-the Konstanz information miner. SIGKDD Explor. Newsl. 2009, 11, 26. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Müller, A.; Nothman, J.; Louppe, G.; et al. Scikit-learn: Machine Learning in Python. arXiv 2012. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Bajusz, D.; Rácz, A.; Héberger, K. Chemical data formats, fingerprints, and other molecular descriptions for database analysis and searching. In Comprehensive Medicinal Chemistry III.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 329–378. ISBN 9780128032015. [Google Scholar]
- Willighagen, E.L.; Mayfield, J.W.; Alvarsson, J.; Berg, A.; Carlsson, L.; Jeliazkova, N.; Kuhn, S.; Pluskal, T.; Rojas-Chertó, M.; Spjuth, O.; et al. The Chemistry Development Kit (CDK) v2.0: Atom typing, depiction, molecular formulas, and substructure searching. J. Cheminform. 2017, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Salas, J.; Peña-Varas, C.; Valenzuela Martínez, I.; Olmedo, D.A.; Zamora, W.J.; Chávez-Fumagalli, M.A.; Azevedo, D.Q.; Castilho, R.O.; Maltarollo, V.G.; Ramírez, D.; et al. Trends and challenges in chemoinformatics research in Latin America. Artif. Intell. Life Sci. 2023, 3, 100077. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
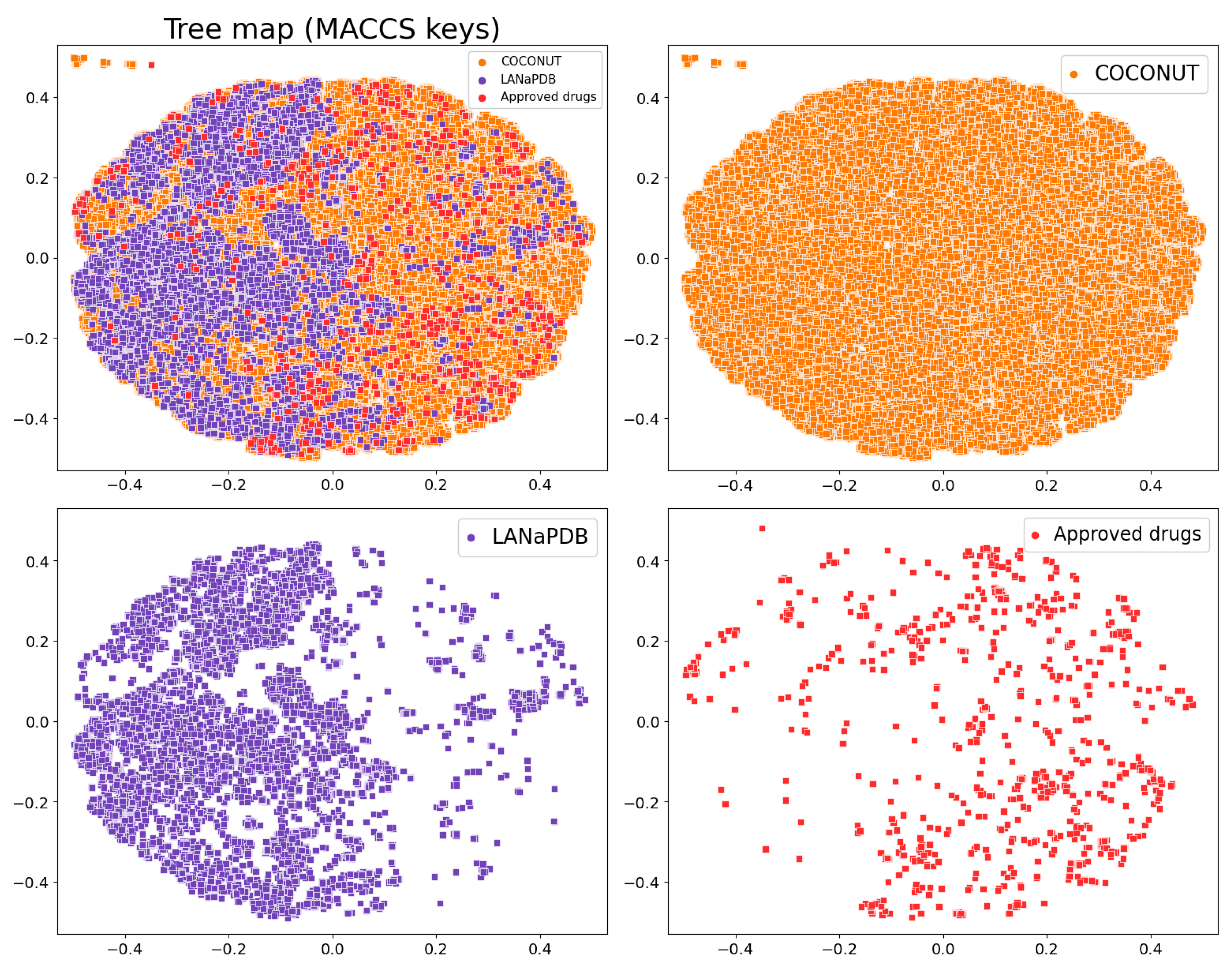{kind=link}
| Database Name | Disease or Symptom | Number of Identified Compounds | Reference |
|---|---|---|---|
| NuBBEDB | Chagas disease | 10 | [80] |
| Tuberculosis | 13 | [82] | |
| SistematX | Chagas disease | 13 | [81] |
| Leishmaniasis | 13 | [83] | |
| Schistosomiasis | 5 | [85] | |
| Coronavirus disease 2019 | 19 | [86] | |
| Alzheimer’s disease | 2 | [90] | |
| UNIIQUIM | Pain | 6 | [88] |
| BIOFACQUIM | Obesity | 8 | [87] |
| Diabetes | |||
| Hyperlipoproteinemia Cancer | |||
| HIV/AIDS * | |||
| Hepatitis B and C. | |||
| Age-related diseases | 3 | [89] |
| Database Name (Country) | Number of Compounds a | Source | General Description | References |
|---|---|---|---|---|
| NuBBEDB (Brazil) | 2223 | Plants Microorganisms Terrestrial and marine animals | Natural products of Brazilian biodiversity. Developed by the São Paulo State University and the University of São Paulo. | [68,69] |
| SistematX (Brazil) | 9514 | Plants | Database composed of secondary metabolites and developed at the Federal University of Paraiba. | [70,71] |
| UEFS (Brazil) | 503 | Plants | Natural products that have been separately published, but there is no common publication nor public database for it. Developed at the State University of Feira de Santana. | [72] |
| NAPRORE-CR (Costa Rica) | 359 | Plants Microorganisms | Developed in the CBio3 and LaToxCIA Laboratories of the University of Costa Rica. | * |
| LAIPNUDELSAV (El Salvador) | 214 | Plants | Developed by the Research Laboratory in Natural Products of the University of El Salvador. | * |
| UNIIQUIM (Mexico) | 1112 | Plants | Natural products isolated and characterized at the Institute of Chemistry of the National Autonomous University of Mexico. | [76] |
| BIOFACQUIM (Mexico) | 553 | Plants Fungus Propolis Marine animals | Natural products isolated and characterized in Mexico at the School of Chemistry of the National Autonomous University of Mexico and other Mexican institutions. | [77,78] |
| CIFPMA (Panama) | 363 | Plants | Natural products that have been tested in over twenty-five in vitro and in vivo bioassays for different therapeutic targets. Developed at the University of Panama. | [73,74] |
| PeruNPDB (Peru) | 280 | Animals Plants | Created and curated at the Catholic University of Santa Maria. | [75] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-García, A.; Jiménez, D.A.A.; Zamora, W.J.; Barazorda-Ccahuana, H.L.; Chávez-Fumagalli, M.Á.; Valli, M.; Andricopulo, A.D.; Bolzani, V.d.S.; Olmedo, D.A.; Solís, P.N.; et al. Navigating the Chemical Space and Chemical Multiverse of a Unified Latin American Natural Product Database: LANaPDB. Pharmaceuticals 2023, 16, 1388. https://doi.org/10.3390/ph16101388
Gómez-García A, Jiménez DAA, Zamora WJ, Barazorda-Ccahuana HL, Chávez-Fumagalli MÁ, Valli M, Andricopulo AD, Bolzani VdS, Olmedo DA, Solís PN, et al. Navigating the Chemical Space and Chemical Multiverse of a Unified Latin American Natural Product Database: LANaPDB. Pharmaceuticals. 2023; 16(10):1388. https://doi.org/10.3390/ph16101388
Chicago/Turabian StyleGómez-García, Alejandro, Daniel A. Acuña Jiménez, William J. Zamora, Haruna L. Barazorda-Ccahuana, Miguel Á. Chávez-Fumagalli, Marilia Valli, Adriano D. Andricopulo, Vanderlan da S. Bolzani, Dionisio A. Olmedo, Pablo N. Solís, and et al. 2023. "Navigating the Chemical Space and Chemical Multiverse of a Unified Latin American Natural Product Database: LANaPDB" Pharmaceuticals 16, no. 10: 1388. https://doi.org/10.3390/ph16101388