Microbiome Composition and Borrelia Detection in Ixodes scapularis Ticks at the Northwestern Edge of Their Range


Abstract

1. Introduction
2. Materials and Methods
2.1. Tick Specimens
2.2. Control Samples
2.3. Borrelia qPCR
2.4. 16S rRNA Sequencing
2.5. Sequence Data Processing
2.6. Diversity Measures and Associations with Tck Vriables
3. Results
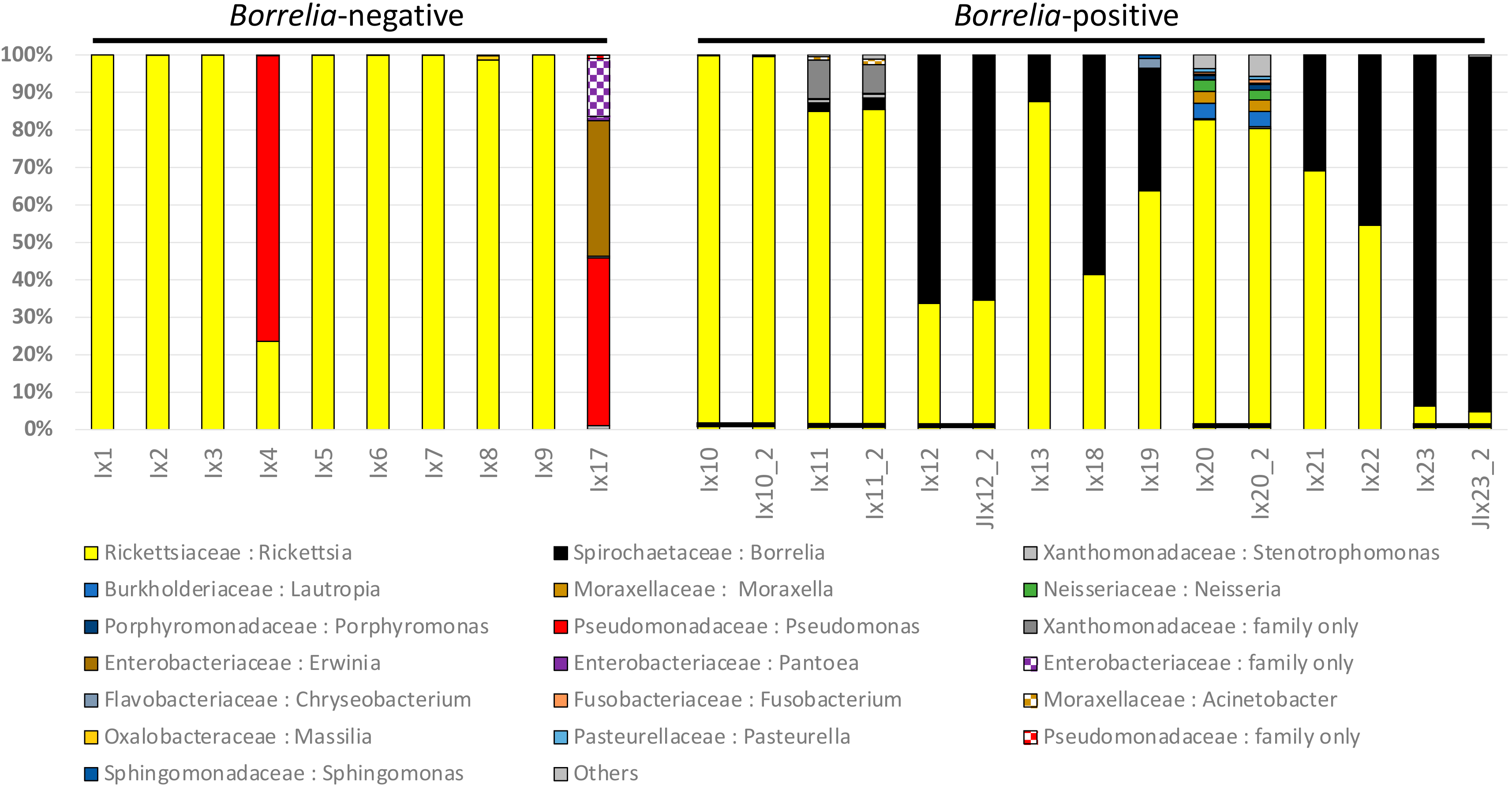
3.1. Ixodes Scapularis Microbiome Characteristics
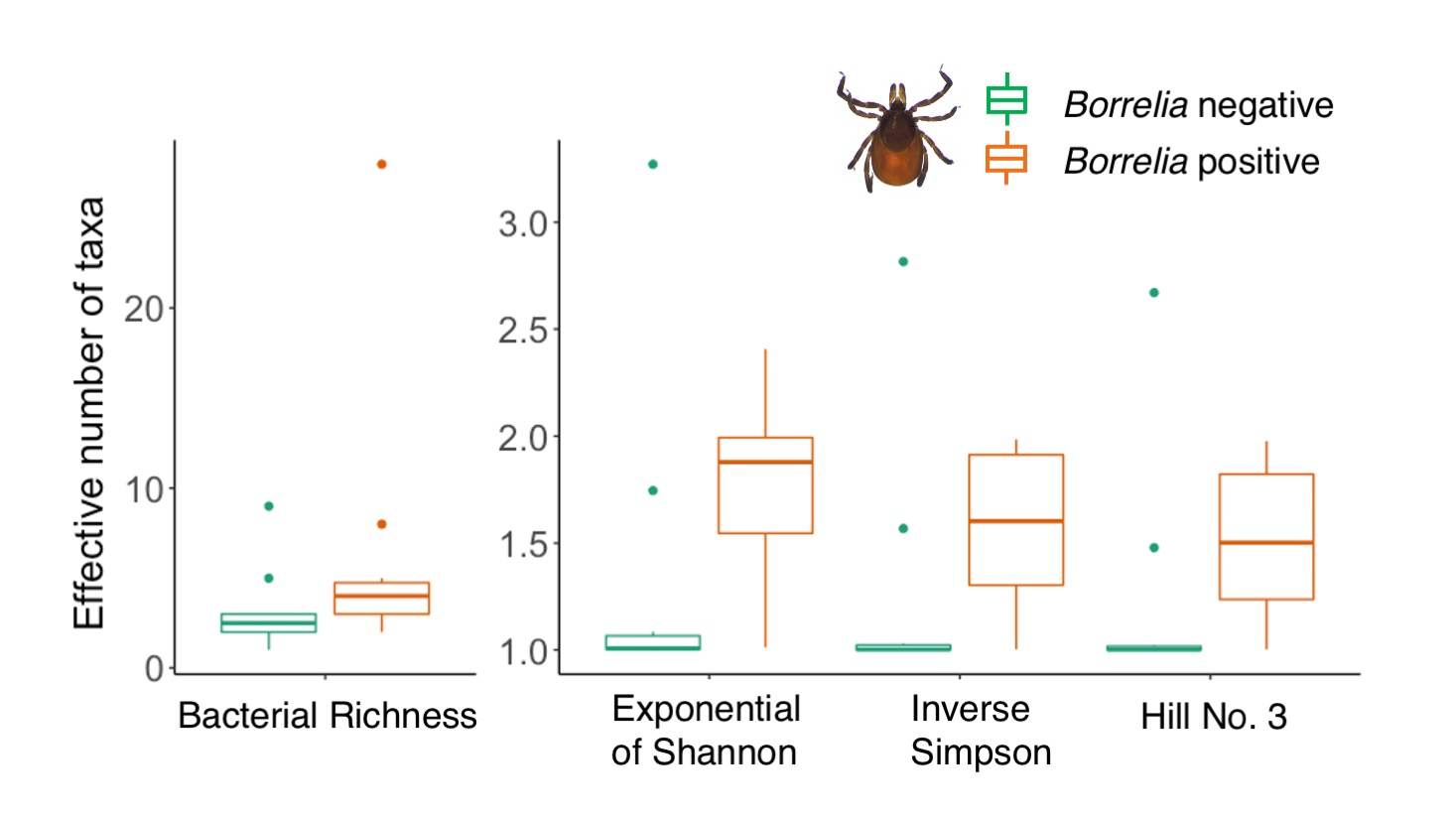
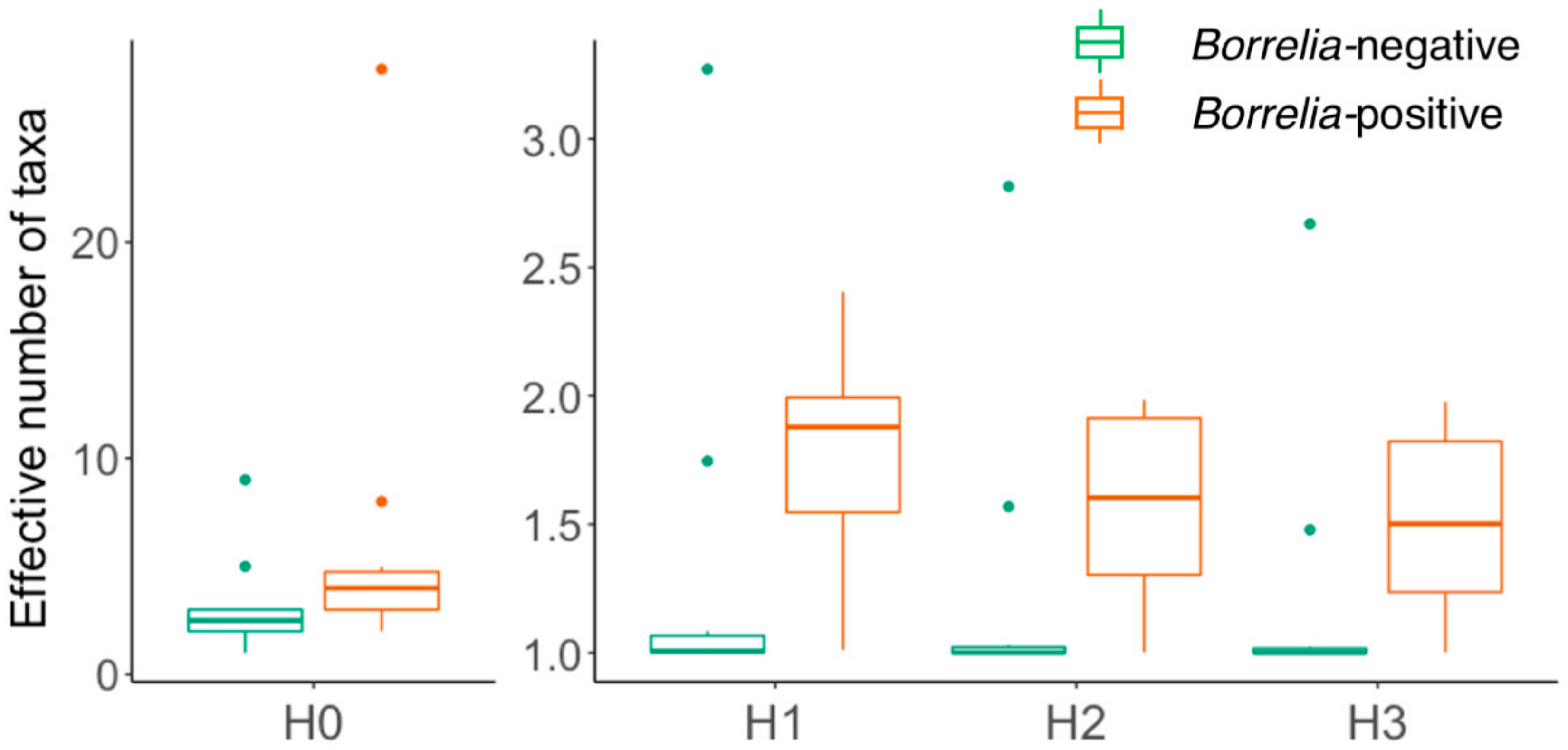
3.2. Comparison of Microbiomes With or Without Borrelia
3.3. Borrelia Detection: 16S Versus qPCR
4. Discussion
4.1. Components of the Microbiome
4.2. Borrelia Infection Status and Tick Microbiome Diversity
4.3. Effectiveness of 16S rRNA Surveys
4.4. Variation in Tick Microbiomes as a Tool for Monitoring Disease Risk
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gondard, M.; Cabezas-Cruz, A.; Charles, R.A.; Vayssier-Taussat, M.; Albina, E.; Moutailler, S. Ticks and Tick-Borne Pathogens of the Caribbean: Current Understanding and Future Directions for More Comprehensive Surveillance. Front. Cell. Infect. Microbiol. 2017, 7, 490. [Google Scholar] [CrossRef]
- Bonnet, S.I.; Binetruy, F.; Hernández-Jarguín, A.; Duron, O. The Tick Microbiome: Why Non-pathogenic Microorganisms Matter in Tick Biology and Pathogen Transmission. Front. Cell. Infect. Microbiol. 2017, 7, 236. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Llanos-Soto, S.G.; Gangloff-Kaufmann, J.L.; Lampman, J.M.; Frye, M.J.; Benedict, M.C.; Tallmadge, R.; Mitchell, P.K.; Anderson, R.R.; Cronk, B.D.; et al. Active surveillance of pathogens from ticks collected in New York State suburban parks and schoolyards. Zoonoses Public Health 2020, 67, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Greay, T.L.; Gofton, A.W.; Paparini, A.; Ryan, U.M.; Oskam, C.L.; Irwin, P.J. Recent insights into the tick microbiome gained through next-generation sequencing. Parasites Vectors 2018, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Dib, L.; Bitam, I.; Tahri, M.; Bensouilah, M.; De Meeûs, T. Competitive Exclusion between Piroplasmosis and Anaplasmosis Agents within Cattle. PLoS Pathog. 2008, 4, e7. [Google Scholar] [CrossRef]
- Galletti, M.F.B.M.; Ueti, M.W.; Knowles, D.P.; Brayton, K.A.; Palmer, G.H. Independence of Anaplasma marginale Strains with High and Low Transmission Efficiencies in the Tick Vector following Simultaneous Acquisition by Feeding on a Superinfected Mammalian Reservoir Host. Infect. Immun. 2009, 77, 1459–1464. [Google Scholar] [CrossRef]
- Genné, D.; Sarr, A.; Rais, O.; Voordouw, M.J. Competition Between Strains of Borrelia afzelii in Immature Ixodes ricinus Ticks Is Not Affected by Season. Front. Cell. Infect. Microbiol. 2019, 9, 431. [Google Scholar] [CrossRef]
- Martins, L.A.; Galletti, M.F.B.D.M.; Ribeiro, J.M.; Fujita, A.; Costa, F.B.; Labruna, M.B.; Daffre, S.; Fogaça, A.C. The Distinct Transcriptional Response of the Midgut of Amblyomma sculptum and Amblyomma aureolatum Ticks to Rickettsia rickettsii Correlates to Their Differences in Susceptibility to Infection. Front. Cell. Infect. Microbiol. 2017, 7, 129. [Google Scholar] [CrossRef]
- Gomes-Solecki, M.; Arnaboldi, P.M.; Backenson, P.B.; Benach, J.L.; Cooper, C.L.; Dattwyler, R.J.; Diuk-Wasser, M.; Fikrig, E.; Hovius, J.W.; Laegreid, W.; et al. Protective Immunity and New Vaccines for Lyme Disease. Clin. Infect. Dis. 2020, 70, 1768–1773. [Google Scholar] [CrossRef]
- Nováková, M.; Šmajs, D. Rickettsial endosymbionts of ticks. In On Ticks and Tick-Borne Pathogens; Abubakar, M., Perera, P.K., Eds.; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Neelakanta, G.; Sultana, H.; Fish, D.; Anderson, J.F.; Fikrig, E. Anaplasma phagocytophilum induces Ixodes scapularis ticks to express an antifreeze glycoprotein gene that enhances their survival in the cold. J. Clin. Investig. 2010, 120, 3179–3190. [Google Scholar] [CrossRef]
- Herrmann, C.; Gern, L. Survival of Ixodes ricinus (Acari: Ixodidae) Under Challenging Conditions of Temperature and Humidity Is Influenced by Borrelia burgdorferi sensu lato Infection. J. Med. Entomol. 2010, 47, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Machado-Ferreira, E.; Vizzoni, V.F.; Piesman, J.; Gazeta, G.S.; Soares, C.A.G. Bacteria associated with Amblyomma cajennense tick eggs. Genet. Mol. Biol. 2015, 38, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, R.; De León, A.A.P.; E Dowd, S.; Guerrero, F.D.; Bendele, K.G.; Scoles, G.A. Assessment of bacterial diversity in the cattle tick Rhipicephalus (Boophilus) microplus through tag-encoded pyrosequencing. BMC Microbiol. 2011, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Carpi, G.; Cagnacci, F.; Wittekindt, N.E.; Zhao, F.; Qi, J.; Tomsho, L.P.; Drautz, D.I.; Rizzoli, A.; Schuster, S.C. Metagenomic Profile of the Bacterial Communities Associated with Ixodes ricinus Ticks. PLoS ONE 2011, 6, e25604. [Google Scholar] [CrossRef]
- Burtis, J.C.; Yavitt, J.B.; Fahey, T.J.; Ostfeld, R.S. Ticks as Soil-Dwelling Arthropods: An Intersection Between Disease and Soil Ecology. J. Med. Entomol. 2019, 56, 1555–1564. [Google Scholar] [CrossRef]
- Pollet, T.; Sprong, H.; Lejal, E.; Krawczyk, A.I.; Moutailler, S.; Cosson, J.-F.; Vayssier-Taussat, M.; Estrada-Peña, A. The scale affects our view on the identification and distribution of microbial communities in ticks. Parasites Vectors 2020, 13, 1–13. [Google Scholar] [CrossRef]
- Vayssier-Taussat, M.; Kazimirova, M.; Hubalek, Z.; Hornok, S.; Farkas, R.; Cosson, J.-F.; Bonnet, S.; Vourch, G.; Gasqui, P.; Mihalca, A.D.; et al. Emerging horizons for tick-borne pathogens: From the ‘one pathogen–one disease’ vision to the pathobiome paradigm. Futur. Microbiol. 2015, 10, 2033–2043. [Google Scholar] [CrossRef]
- Cabezas-Cruz, A.; Vayssier-Taussat, M.; Greub, G. Tick-borne pathogen detection: What’s new? Microbes Infect. 2018, 20, 441–444. [Google Scholar] [CrossRef]
- Modarelli, J.J.; Ferro, P.J.; De León, A.A.P.; Esteve-Gasent, M.D. TickPath Layerplex: Adaptation of a real-time PCR methodology for the simultaneous detection and molecular surveillance of tick-borne pathogens. Sci. Rep. 2019, 9, 6950. [Google Scholar] [CrossRef]
- Sanchez-Vicente, S.; Tagliafierro, T.; Coleman, J.L.; Benach, J.L.; Tokarz, R.; Azad, A.; Fikrig, E.; Munderloh, U.; Telford, S.R. Polymicrobial Nature of Tick-Borne Diseases. mBio 2019, 10, e02055-19. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. 16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef] [PubMed]
- Jian, C.; Luukkonen, P.; Yki-Järvinen, H.; Salonen, A.; Korpela, K. Quantitative PCR provides a simple and accessible method for quantitative microbiota profiling. PLoS ONE 2020, 15, e0227285. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, C.; Dibernardo, A.; Koffi, J.; Wood, H.; A Leighton, P.; Lindsay, L.R. Increased risk of tick-borne diseases with climate and environmental changes. Can. Commun. Dis. Rep. 2019, 45, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Leighton, P.A.; Koffi, J.K.; Pelcat, Y.; Lindsay, L.R.; Ogden, N.H. Predicting the speed of tick invasion: An empirical model of range expansion for the Lyme disease vectorIxodes scapularisin Canada. J. Appl. Ecol. 2012, 49, 457–464. [Google Scholar] [CrossRef]
- Lloyd, V.K.; Hawkins, R.G. Under-Detection of Lyme Disease in Canada. Healthcare 2018, 6, 125. [Google Scholar] [CrossRef]
- Ogden, N.H.; Bouchard, C.; Badcock, J.; Drebot, M.A.; Elias, S.P.; Hatchette, T.F.; Koffi, J.K.; Leighton, P.A.; Lindsay, L.R.; Lubelczyk, C.B.; et al. What is the real number of Lyme disease cases in Canada? BMC Public Health 2019, 19, 1–12. [Google Scholar] [CrossRef]
- Brinkerhoff, R.J.; Clark, C.; Ocasio, K.; Gauthier, D.T.; Hynes, W.L. Factors affecting the microbiome of Ixodes scapularis and Amblyomma americanum. PLoS ONE 2020, 15, e0232398. [Google Scholar] [CrossRef]
- Tokarz, R.; Tagliafierro, T.; Sameroff, S.; Cucura, D.M.; Oleynik, A.; Che, X.; Jain, K.; Lipkin, W.I. Microbiome analysis of Ixodes scapularis ticks from New York and Connecticut. Ticks Tick Borne Dis. 2019, 10, 894–900. [Google Scholar] [CrossRef]
- Fitzgerald, D.T. The Species Composition and Distribution of Ixodidae from Companion Animals in Alberta, Canada. Master’s Thesis, University of Alberta, Edmonton, AB, Canada, 2012. [Google Scholar] [CrossRef]
- Government of Alberta. Surveillance of Ticks on Companion Animals in Alberta. 2018. Available online: https://open.alberta.ca/dataset/b1545e9a-367e-40c8-b153-24f0d1b0b042/resource/f8269b75-14b6-454a-92e5-89d9d0654d60/download/af-tick-summary-2018.pdf (accessed on 11 October 2020).
- Consensus Conference on Lyme Disease. Can. J. Infect. Dis. 1991, 2, 49–54. [CrossRef]
- Government of Canada. Where in Canada are You at Risk? Available online: https://www.canada.ca/en/public-health/services/diseases/lyme-disease/risk-lyme-disease.html#a3 (accessed on 5 November 2020).
- Ogden, N.; Koffi, J.; Pelcat, Y.; Lindsay, L. Environmental risk from Lyme disease in central and eastern Canada: A summary of recent surveillance information. Can. Commun. Dis. Rep. 2014, 40, 74–82. [Google Scholar] [CrossRef]
- Hatchette, T.; Davis, I.; Johnston, B.L. Lyme disease: Clinical diagnosis and treatment. Can. Commun. Dis. Rep. 2014, 40, 194–208. [Google Scholar] [CrossRef]
- Gasmi, S.; Bouchard, C.; Ogden, N.H.; Adam-Poupart, A.; Pelcat, Y.; Rees, E.E.; Milord, F.; Leighton, P.A.; Lindsay, R.L.; Koffi, J.K.; et al. Evidence for increasing densities and geographic ranges of tick species of public health significance other than Ixodes scapularis in Québec, Canada. PLoS ONE 2018, 13, e0201924. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, M.; Kryuchkov, R.; Statculescu, A.; Thickstun, C.; Dibernardo, A.; Lindsay, L.; Talbot, B. Ixodes scapularis tick distribution and infection rates in Ottawa, Ontario, 2017. Can. Commun. Dis. Rep. 2018, 44, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Government of Alberta. Blacklegged Ticks Submitted to the Tick Surveillance Program. 2015. Available online: https://open.alberta.ca/publications/blacklegged-ticks-submitted-to-the-tick-surveillance-program#detailed (accessed on 5 July 2020).
- Government of Alberta. Interactive Health Data Application. 2020. Available online: http://www.ahw.gov.ab.ca/IHDA_Retrieval/redirectToURL.do?cat=324&subCat=1042 (accessed on 5 July 2020).
- Government of Alberta. Alberta Health Tick Surveillance 2013 Summary. 2014. Available online: http://www.health.alberta.ca/documents/Tick-Surveillance-Summary-2013.pdf (accessed on 11 October 2020).
- Keirans, J.E.; Clifford, C.M. The Genus Ixodes in the United States: A Scanning Electron Microscope Study and Key to the Adults1. J. Med Entomol. 1978, 15, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Dibernardo, A. Extraction and Amplification of Tick DNA for the Detection of Tick-Borne Pathogens; Standard Operating Procedure. FS-PR2010-1; National Microbiology Lab.: Winnipeg, MB, Canada, 2010. [Google Scholar]
- Sperling, J.; Silva-Brandão, K.; Brandão, M.; Lloyd, V.; Dang, S.; Davis, C.; Sperling, F.; Magor, K. Comparison of bacterial 16S rRNA variable regions for microbiome surveys of ticks. Ticks Tick Borne Dis. 2017, 8, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Ion Torrent. 16S Metagenomics Kit User Guide (Pub. no MAN0010799 Rev. C). 2015. Available online: https://assets.thermofisher.com/TFS-Assets/LSG/manuals/MAN0010799_Ion_16S_Metagenomics_UG.pdf (accessed on 10 November 2020).
- Chakravorty, S.; Helb, D.; Burday, M.; Connell, N.; Alland, D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J. Microbiol. Methods 2007, 69, 330–339. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. vegan: Community Ecology Package, version:2.5-6. 2019. Available online: https://CRAN.R-project.org/package=vegan (accessed on 10 November 2020).
- Kurtti, T.J.; Felsheim, R.F.; Burkhardt, N.Y.; Oliver, J.D.; Heu, C.C.; Munderloh, U.G. Rickettsia buchneri sp. nov., a rickettsial endosymbiont of the blacklegged tick Ixodes scapularis. Int. J. Syst. Evol. Microbiol. 2015, 65, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Lejal, E.; Estrada-Peña, A.; Marsot, M.; Cosson, J.-F.; Rué, O.; Mariadassou, M.; Midoux, C.; Vayssier-Taussat, M.; Pollet, T. Taxon Appearance From Extraction and Amplification Steps Demonstrates the Value of Multiple Controls in Tick Microbiota Analysis. Front. Microbiol. 2020, 11, 1093. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 1–12. [Google Scholar] [CrossRef]
- Palmer, J.M.; Jusino, M.A.; Banik, M.T.; Lindner, D.L. Non-biological synthetic spike-in controls and the AMPtk software pipeline improve mycobiome data. PeerJ 2018, 6, e4925. [Google Scholar] [CrossRef]
- Van Treuren, W.; Ponnusamy, L.; Brinkerhoff, R.J.; Gonzalez, A.; Parobek, C.M.; Juliano, J.J.; Andreadis, T.G.; Falco, R.C.; Ziegler, L.B.; Hathaway, N.; et al. Variation in the Microbiota of Ixodes Ticks with Regard to Geography, Species, and Sex. Appl. Environ. Microbiol. 2015, 81, 6200–6209. [Google Scholar] [CrossRef] [PubMed]
- Ogden, N.H.; Margos, G.; Aanensen, D.M.; Drebot, M.A.; Feil, E.J.; Hanincová, K.; Schwartz, I.; Tyler, S.; Lindsay, L.R. Investigation of Genotypes of Borrelia burgdorferi in Ixodes scapularis Ticks Collected during Surveillance in Canada. Appl. Environ. Microbiol. 2011, 77, 3244–3254. [Google Scholar] [CrossRef] [PubMed]
- Fryxell, R.T.T.; Steelman, C.D.; Szalanski, A.L.; Billingsley, P.M.; Williamson, P.C. Molecular Detection of Rickettsia Species Within Ticks (Acari: Ixodidae) Collected from Arkansas United States. J. Med Entomol. 2015, 52, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Mignard, S.; Flandrois, J. 16S rRNA sequencing in routine bacterial identification: A 30-month experiment. J. Microbiol. Methods 2006, 67, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, S.; Jung, S. Instruction of microbiome taxonomic profiling based on 16S rRNA sequencing. J. Microbiol. 2020, 58, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.-E.; Dumler, J.S.; Greub, G.; Zhang, J.; Wu, Y.; Raoult, D. Gene Sequence-Based Criteria for Identification of New Rickettsia Isolates and Description of Rickettsia heilongjiangensis sp. nov. J. Clin. Microbiol. 2003, 41, 5456–5465. [Google Scholar] [CrossRef] [PubMed]
- Clow, K.M.; Weese, J.S.; Rousseau, J.; Jardine, C.M. Microbiota of field-collected Ixodes scapularis and Dermacentor variabilis from eastern and southern Ontario, Canada. Ticks Tick Borne Dis. 2018, 9, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Thapa, S.; Zhang, Y.; Allen, M.S. Bacterial microbiomes of Ixodes scapularis ticks collected from Massachusetts and Texas, USA. BMC Microbiol. 2019, 19, 1–12. [Google Scholar] [CrossRef]
- Ruiz-Roldán, L.; Rojo-Bezares, B.; De Toro, M.; López, M.; Toledano, P.; Lozano, C.; Chichón, G.; Alvarez-Erviti, L.; Torres, C.; Sáenz, Y. Antimicrobial resistance and virulence of Pseudomonas spp. among healthy animals: Concern about exolysin ExlA detection. Sci. Rep. 2020, 10, 1–11. [Google Scholar] [CrossRef]
- Haas, D.; Keel, C. Regulation of antibiotic production in root-colonizing Pseudomonass pp. and relevance for biological control of plant disease. Annu. Rev. Phytopathol. 2003, 41, 117–153. [Google Scholar] [CrossRef]
- Verhille, S.; Baida, N.; Dabboussi, F.; Hamze, M.; Izard, D.; Leclerc, H. Pseudomonas gessardii sp. nov. and Pseudomonas migulae sp. nov., two new species isolated from natural mineral waters. Int. J. Syst. Evol. Microbiol. 1999, 49, 1559–1572. [Google Scholar] [CrossRef] [PubMed]
- Palleroni, N. Pseudomonas. In Bergey’s Manual of Systematics of Archaea and Bacteria; John Wiley & Sons, Inc.: New York, NY, USA, 2015; pp. 58–69. [Google Scholar] [CrossRef]
- Stoddard, S.F.; Smith, B.J.; Hein, R.; Roller, B.R.; Schmidt, T.M. rrnDB: Improved tools for interpreting rRNA gene abundance in bacteria and archaea and a new foundation for future development. Nucleic Acids Res. 2015, 43, D593–D598. [Google Scholar] [CrossRef] [PubMed]
- Louca, S.; Doebeli, M.; Parfrey, L.W. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 2018, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Palacio-Bielsa, A.; Roselló, M.; Llop, P.; López, M.M. Erwinia spp. from pome fruit trees: Similarities and differences among pathogenic and non-pathogenic species. Trees 2011, 26, 13–29. [Google Scholar] [CrossRef]
- Harada, H.; Oyaizu, H.; Kosako, Y.; Ishikawa, H. Erwinia aphidicola, a new species isolated from pea aphid, Acyrthosiphon pisum. J. Gen. Appl. Microbiol. 1997, 43, 349–354. [Google Scholar] [CrossRef]
- Skrodenytėarbaciauskienė, V.; Radžiutė, S.; Stunžėnas, V.; Būda, V. Erwinia typographi sp. nov., isolated from bark beetle (Ips typographus) gut. Int. J. Syst. Evol. Microbiol. 2012, 62, 942–948. [Google Scholar] [CrossRef]
- De Vries, E.J.; Jacobs, G.; Breeuwer, J.A. Growth and Transmission of Gut Bacteria in the Western Flower Thrips, Frankliniella occidentalis. J. Invertebr. Pathol. 2001, 77, 129–137. [Google Scholar] [CrossRef]
- Ross, B.D.; Hayes, B.; Radey, M.C.; Lee, X.; Josek, T.; Bjork, J.K.H.; Neitzel, D.; Paskewitz, S.; Chou, S.; Mougous, J.D. Ixodes scapularis does not harbor a stable midgut microbiome. ISME J. 2018, 12, 2596–2607. [Google Scholar] [CrossRef]
- Lindquist, E.; Galloway, T.; Artsob, H.; Lindsay, R.; Drebot, M.; Wood, H.; Robbins, R. A handbook to the ticks (Ixodida: Ixodidae, Argasidae) of Canada. Biol. Surv. Can. 2016. [Google Scholar] [CrossRef]
- Narasimhan, S.; Rajeevan, N.; Liu, L.; Zhao, Y.O.; Heisig, J.; Pan, J.; Eppler-Epstein, R.; DePonte, K.; Fish, D.; Fikrig, E. Gut Microbiota of the Tick Vector Ixodes scapularis Modulate Colonization of the Lyme Disease Spirochete. Cell Host Microbe 2014, 15, 58–71. [Google Scholar] [CrossRef]
- Kurokawa, C.; Lynn, G.E.; Pedra, J.H.F.; Pal, U.; Narasimhan, S.; Fikrig, E. Interactions between Borrelia burgdorferi and ticks. Nat. Rev. Genet. 2020, 18, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, G.; McClure, J.; Hekman, J.; Marsh, P.W.; Bailey, J.A.; Daniels, R.F.; Genereux, D.P.; Karlsson, E.K. Combining Citizen Science and Genomics to Investigate Tick, Pathogen, and Commensal Microbiome at Single-Tick Resolution. Front. Genet. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kwan, J.Y.; Griggs, R.; Chicana, B.; Miller, C.; Swei, A. Vertical vs. horizontal transmission of the microbiome in a key disease vector, Ixodes pacificus. Mol. Ecol. 2017, 26, 6578–6589. [Google Scholar] [CrossRef] [PubMed]
- Reese, A.T.; Dunn, R.R. Drivers of Microbiome Biodiversity: A Review of General Rules, Feces, and Ignorance. mBio 2018, 9. [Google Scholar] [CrossRef]
- Ullmann, A.J.; Lane, R.S.; Kurtenbach, K.; Miller, M.; Schriefer, M.E.; Zeidner, N.; Piesman, J. Bacteriolytic Activity of Selected Vertebrate Sera for Borrelia burgdorferi Sensu Stricto and Borrelia bissettii. J. Parasitol. 2003, 89, 1256–1257. [Google Scholar] [CrossRef]
- Swei, A.; Kwan, J.Y. Tick microbiome and pathogen acquisition altered by host blood meal. ISME J. 2016, 11, 813–816. [Google Scholar] [CrossRef]
- Lane, R.S.; Quistad, G.B. Borreliacidal Factor in the Blood of the Western Fence Lizard (Sceloporus occidentalis). J. Parasitol. 1998, 84, 29–34. [Google Scholar] [CrossRef]
- Kuo, M.M.; Lane, R.S.; Giclas, P.C. A comparative study of mammalian and reptilian alternative pathway of complement-mediated killing of the Lyme disease spirochete (Borrelia burgdorferi). J. Parasitol. 2000, 86, 1223–1228. [Google Scholar] [CrossRef]
- Dunn, J.M.; Krause, P.J.; Davis, S.; Vannier, E.G.; Fitzpatrick, M.C.; Rollend, L.; Belperron, A.A.; States, S.L.; Stacey, A.; Bockenstedt, L.K.; et al. Borrelia burgdorferi Promotes the Establishment of Babesia microti in the Northeastern United States. PLoS ONE 2014, 9, e115494. [Google Scholar] [CrossRef]
- Pokutnaya, D.; Molaei, G.; Weinberger, D.M.; Vossbrinck, C.R.; Diaz, A.J. Prevalence of Infection and Co-Infection and Presence of Rickettsial Endosymbionts in Ixodes scapularis (Acari: Ixodidae) in Connecticut, USA. J. Parasitol. 2020, 106, 30. [Google Scholar] [CrossRef]
- Nieto, N.C.; Foley, J. Meta-Analysis of Coinfection and Coexposure with Borrelia burgdorferi and Anaplasma phagocytophilumin Humans, Domestic Animals, Wildlife, andIxodes ricinus-Complex Ticks. Vector-Borne Zoonotic Dis. 2009, 9, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Abraham, N.M.; Liu, L.; Jutras, B.L.; Yadav, A.K.; Narasimhan, S.; Gopalakrishnan, V.; Ansari, J.M.; Jefferson, K.K.; Cava, F.; Jacobs-Wagner, C.; et al. Pathogen-mediated manipulation of arthropod microbiota to promote infection. Proc. Natl. Acad. Sci. USA 2017, 114, E781–E790. [Google Scholar] [CrossRef] [PubMed]
- Nelder, M.P.; Russell, C.; Lindsay, L.R.; Dibernardo, A.; Brandon, N.C.; Pritchard, J.; Johnson, S.; Cronin, K.; Patel, S.N. Recent Emergence of Anaplasma phagocytophilum in Ontario, Canada: Early Serological and Entomological Indicators. Am. J. Trop. Med. Hyg. 2019, 101, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Parkins, M.D.; Church, D.L.; Jiang, X.Y.; Gregson, D.B. Human granulocytic anaplasmosis: First reported case in Canada. Can. J. Infect. Dis. Med. Microbiol. 2009, 20, e100–e102. [Google Scholar] [CrossRef]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2015, 18, 1403–1414. [Google Scholar] [CrossRef] [PubMed]
- Kralik, P.; Ricchi, M. A Basic Guide to Real Time PCR in Microbial Diagnostics: Definitions, Parameters, and Everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- Auer, L.; Mariadassou, M.; O’Donohue, M.; Klopp, C.; Hernandez-Raquet, G. Analysis of large 16S rRNA Illumina data sets: Impact of singleton read filtering on microbial community description. Mol. Ecol. Resour. 2017, 17, e122–e132. [Google Scholar] [CrossRef]
- Marine, R.L.; Magaña, L.C.; Castro, C.J.; Zhao, K.; Montmayeur, A.M.; Schmidt, A.; Diez-Valcarce, M.; Ng, T.F.F.; Vinjé, J.; Burns, C.C.; et al. Comparison of Illumina MiSeq and the Ion Torrent PGM and S5 platforms for whole-genome sequencing of picornaviruses and caliciviruses. J. Virol. Methods 2020, 280, 113865. [Google Scholar] [CrossRef]
- Song, L.; Huang, W.; Kang, J.; Huang, Y.; Ren, H.; Ding, K. Comparison of error correction algorithms for Ion Torrent PGM data: Application to hepatitis B virus. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Ebbert, M.T.W.; Initiative, F.T.A.D.N.; Wadsworth, M.E.; Staley, L.A.; Hoyt, K.L.; Pickett, B.; Miller, J.; Duce, J.; Kauwe, J.S.K.; Ridge, P.G. Evaluating the necessity of PCR duplicate removal from next-generation sequencing data and a comparison of approaches. BMC Bioinform. 2016, 17, 491–500. [Google Scholar] [CrossRef]
- Pollock, J.; Glendinning, L.; Wisedchanwet, T.; Watson, M. The Madness of Microbiome: Attempting to Find Consensus “Best Practice” for 16S Microbiome Studies. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [PubMed]
- Hornung, B.V.H.; Zwittink, R.D.; Kuijper, E.J. Issues and current standards of controls in microbiome research. FEMS Microbiol. Ecol. 2019, 95, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Laursen, M.F.; Dalgaard, M.D.; Bahl, M.I. Genomic GC-Content Affects the Accuracy of 16S rRNA Gene Sequencing Based Microbial Profiling due to PCR Bias. Front. Microbiol. 2017, 8, 1934. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Clark, K.; Foley, J.E.; Bierman, B.C.; Durden, L.A. Far-Reaching Dispersal of Borrelia burgdorferi Sensu Lato-Infected Blacklegged Ticks by Migratory Songbirds in Canada. Health 2018, 6, 89. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Tick ID # | Date Collected | Outside Alberta | Tick Species | Host Animal | Bb qPCR | 16S Reads-1st prep. | 16S Reads-2nd prep. |
|---|---|---|---|---|---|---|---|
| I × 1 | 2016-05-06 | Yes | I. scapularis | Dog | Neg. | 91,317 | - |
| I × 2 | 2016-05-02 | No | I. scapularis | Cat | Neg. | 71,209 | - |
| I × 3 | 2016-05-13 | Unkn. | I. scapularis | Cat | Neg. | 134,766 | - |
| I × 4 | 2016-05-17 | No | I. scapularis | Dog | Neg. | 109,864 | - |
| I × 5 | 2016-05-18 | No | I. scapularis | Cat | Neg. | 145,698 | - |
| I × 6 | 2016-05-19 | No | I. scapularis | Dog | Neg. | 81,204 | - |
| I × 7 | 2016-05-20 | No | I. scapularis | Dog | Neg. | 62,927 | - |
| I × 8 | 2016-05-25 | No | I. scapularis | Dog | Neg. | 104,139 | - |
| I × 9 | 2016-05-26 | Unkn. | I. scapularis | Dog | Neg. | 110,758 | - |
| I × 10 | 2016-01-17 | Yes | I. scapularis | Dog | Pos. | 118,427 | 289,781 |
| I × 11 | 2016-04-11 | No | I. scapularis | Dog | Pos. | 130,881 | 339,121 |
| I × 12 | 2016-04-18 | No | I. scapularis | Dog | Pos. | 100,333 | 321,740 |
| I × 13 | 2016-04-27 | No | I. scapularis | Dog | Pos. | 104,545 | - |
| I × 17 | 2016-05-17 | No | I. scapularis | Dog | Neg. | 174,362 | - |
| I × 18 | 2016-05-08 | No | I. scapularis | Cat | Pos. | 120,645 | - |
| I × 19 | 2016-05-13 | No | I. scapularis | Dog | Pos. | 123,266 | - |
| I × 20 | 2016-05-31 | No | Ixodes spp. | Dog | Pos. | 126,995 | 347,373 |
| I × 21 | 2016-04-28 | No | I. scapularis | Dog | Pos. | 98,205 | - |
| I × 22 | 2016-05-20 | No | I. scapularis | Dog | Pos. | 127,989 | - |
| I × 23 | 2016-05-04 | Unkn. | Ixodes spp. | Dog | Pos. | 115,995 | 295,902 |
| MOC | n/a | n/a | n/a | n/a | n/a | 95,023 | - |
| SEC | n/a | n/a | n/a | n/a | n/a | 71,920 | - |
| NTC | n/a | n/a | n/a | n/a | n/a | 44,610 | - |
| Ticks | 112,676.3 | 318,783.4 | |||||
| Ticks SE | 5,741.7 | 11,411.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sperling, J.L.H.; Fitzgerald, D.; Sperling, F.A.H.; Magor, K.E. Microbiome Composition and Borrelia Detection in Ixodes scapularis Ticks at the Northwestern Edge of Their Range. Trop. Med. Infect. Dis. 2020, 5, 173. https://doi.org/10.3390/tropicalmed5040173
Sperling JLH, Fitzgerald D, Sperling FAH, Magor KE. Microbiome Composition and Borrelia Detection in Ixodes scapularis Ticks at the Northwestern Edge of Their Range. Tropical Medicine and Infectious Disease. 2020; 5(4):173. https://doi.org/10.3390/tropicalmed5040173
Chicago/Turabian StyleSperling, Janet L. H., Daniel Fitzgerald, Felix A. H. Sperling, and Katharine E. Magor. 2020. "Microbiome Composition and Borrelia Detection in Ixodes scapularis Ticks at the Northwestern Edge of Their Range" Tropical Medicine and Infectious Disease 5, no. 4: 173. https://doi.org/10.3390/tropicalmed5040173
APA StyleSperling, J. L. H., Fitzgerald, D., Sperling, F. A. H., & Magor, K. E. (2020). Microbiome Composition and Borrelia Detection in Ixodes scapularis Ticks at the Northwestern Edge of Their Range. Tropical Medicine and Infectious Disease, 5(4), 173. https://doi.org/10.3390/tropicalmed5040173