Artemisinin-Based Antimalarial Drug Therapy: Molecular Pharmacology and Evolving Resistance

Abstract
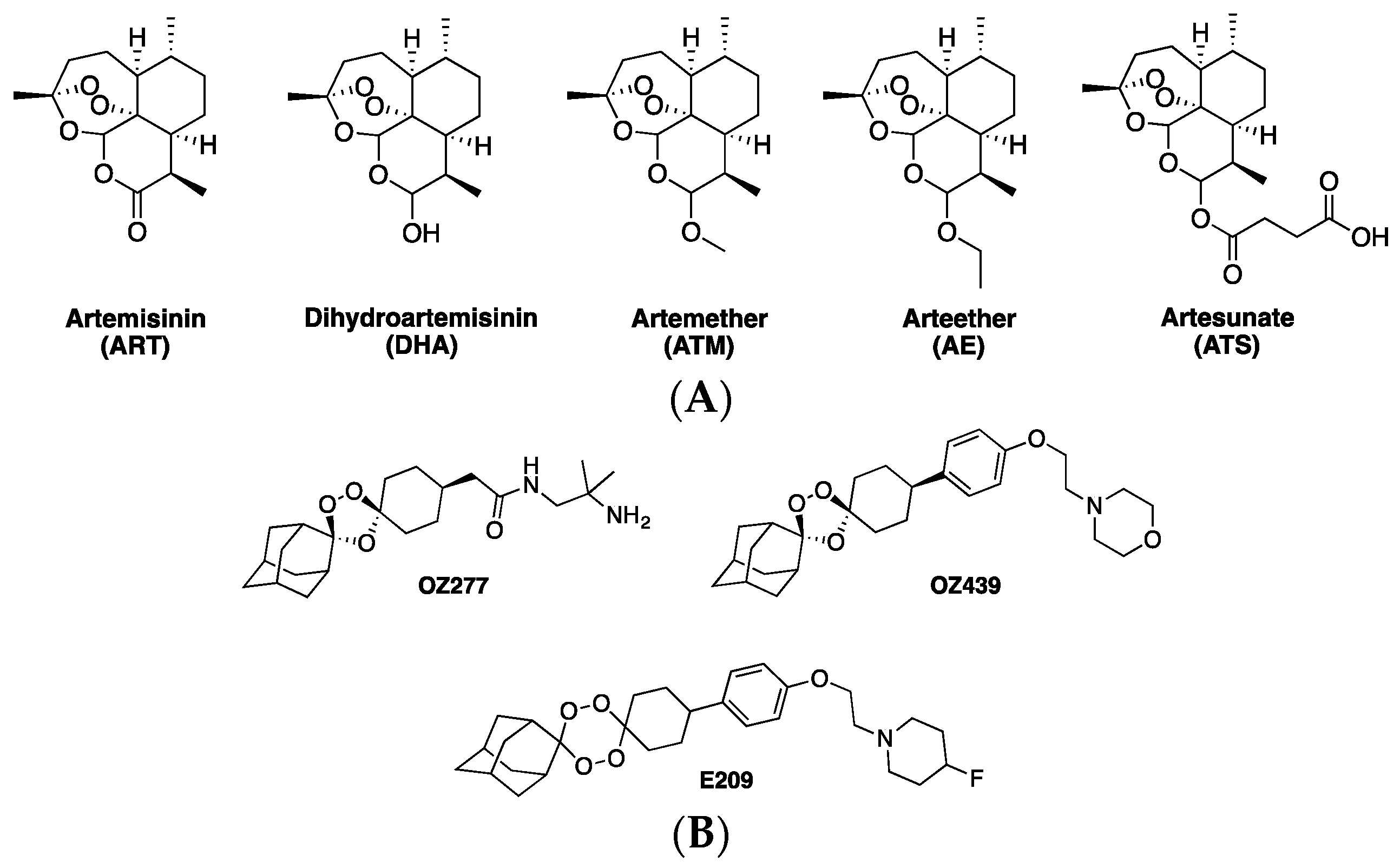
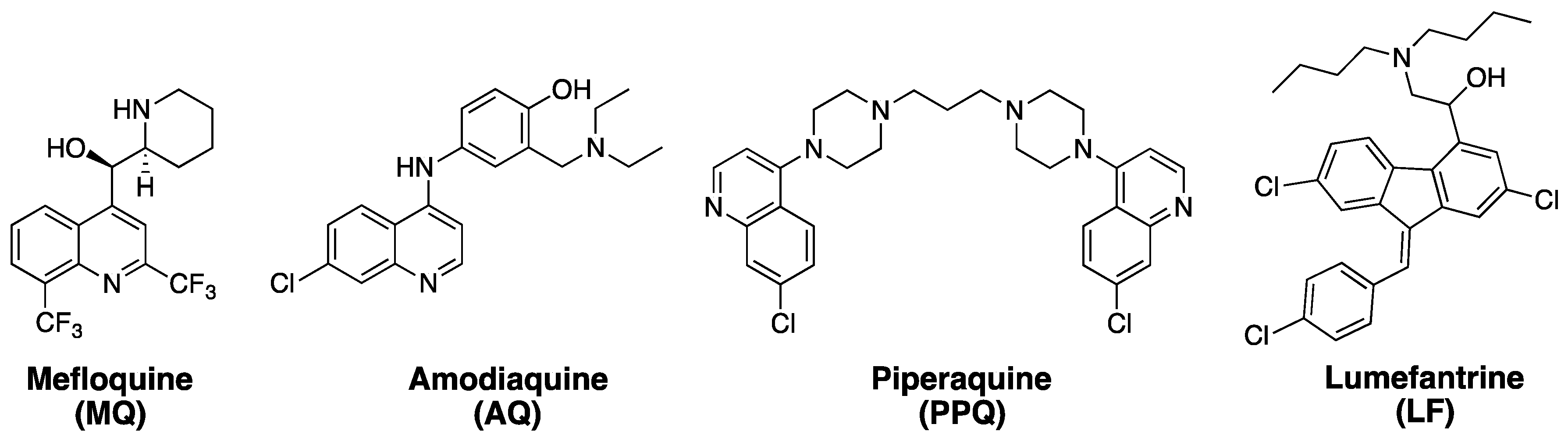
1. Artemisinins and ACTs
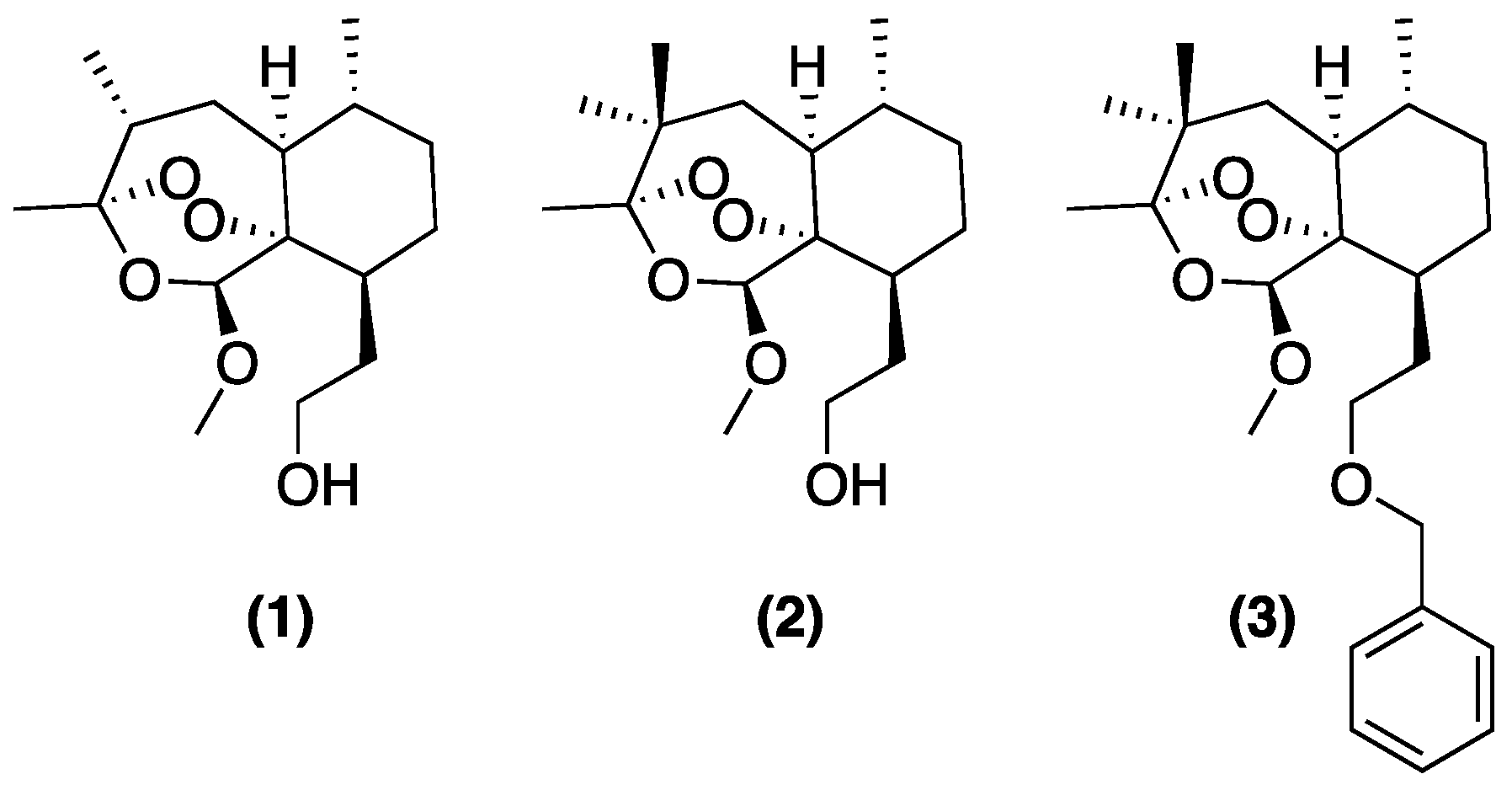
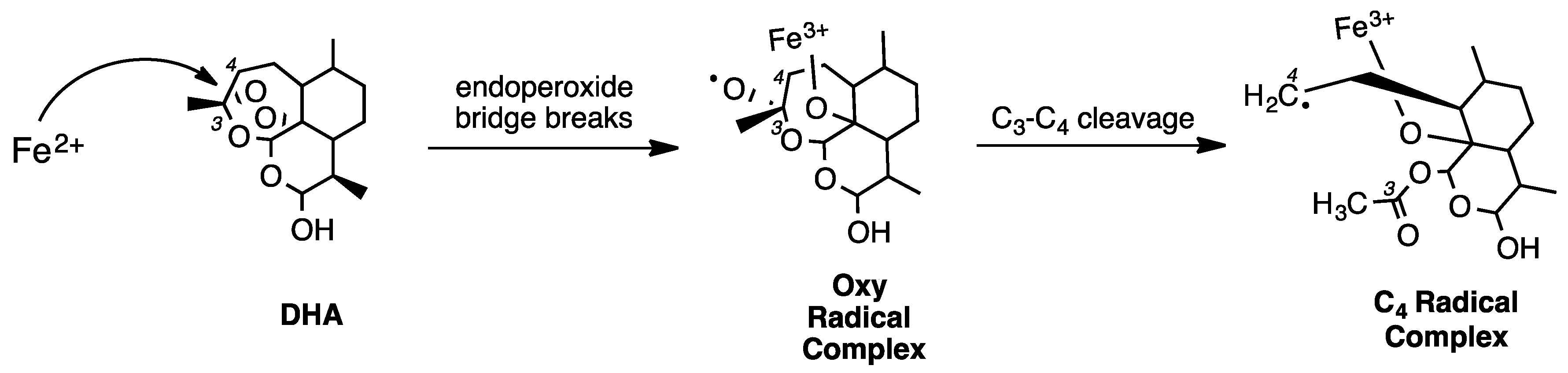
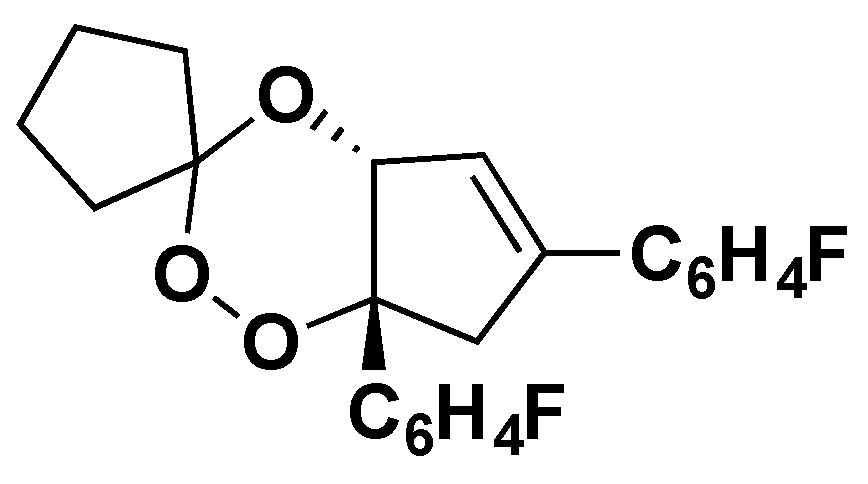
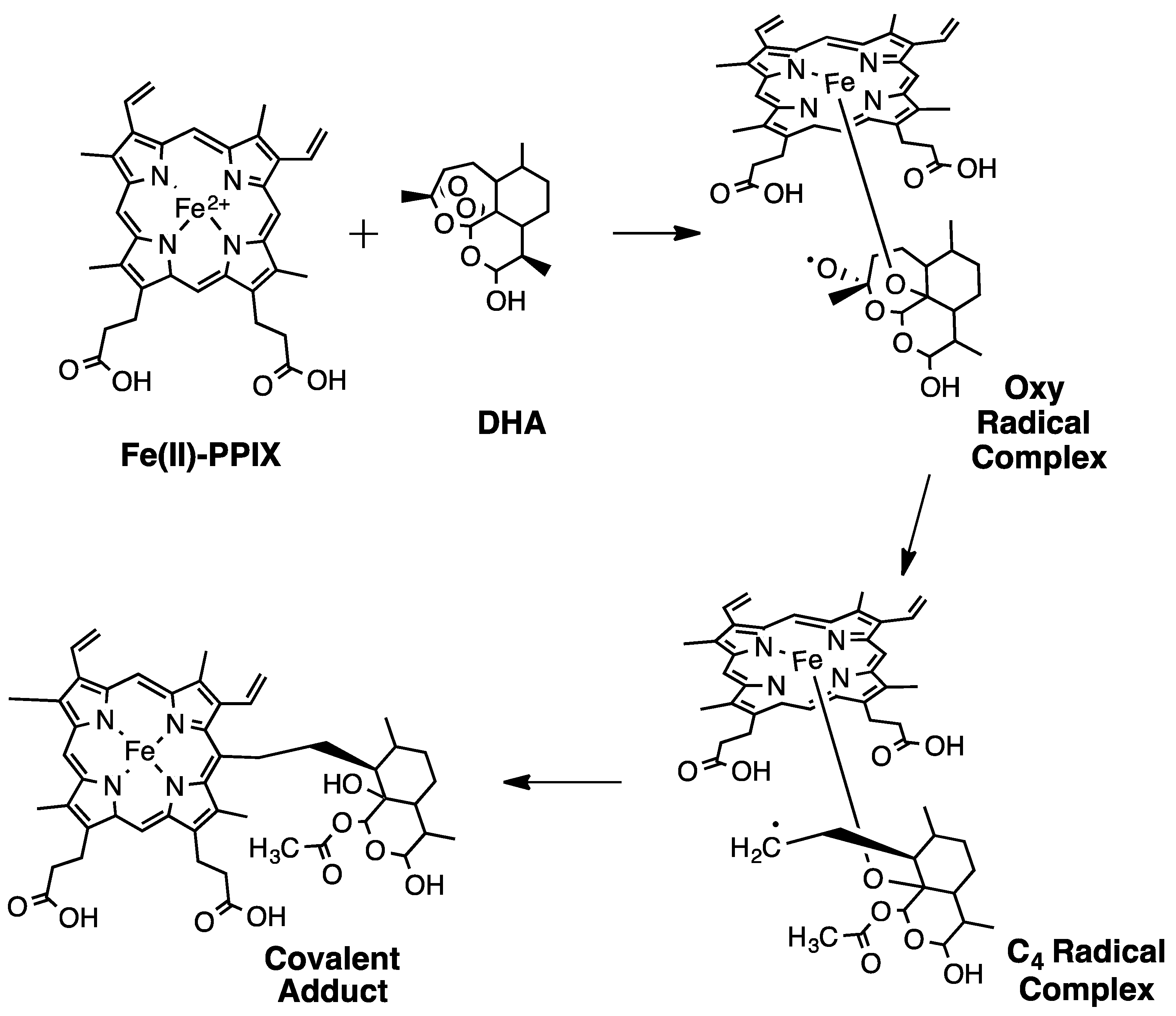
2. Early Investigations of ART Molecular Pharmacology
3. Proteomics Studies Suggest Multiple ART Drug Targets
4. Initial Evidence for Evolving ART Resistance (ARTR)
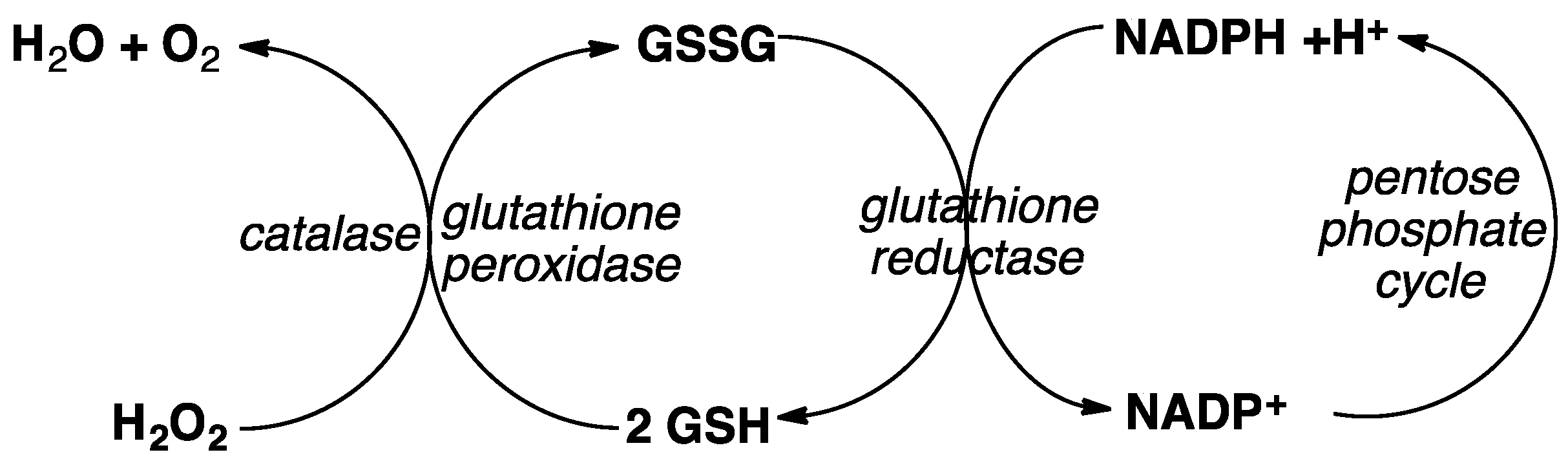
5. Hemoglobin (Hb) and Glutathione (GSH) Metabolism Versus ART Potency
6. Additional Evidence for Altered Drug-FPIX Interactions in ARTR Parasites
7. PfK13 Mutations Associated with ARTR
8. PfPI3K
9. ARTR without Associated PfK13 Mutations
10. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ACT | artemisinin combination therapy |
| ALLN | N-acetyl-L-leucyl-L-norleucinal |
| AQ | amodiaquine |
| ART | artemisinin |
| ARTR/S | artemisinin resistant/sensitive |
| ATM | artemether |
| ATS | artesunate |
| BiPY | 2,2′-bipyridyl |
| DCP | delayed clearance phenotype |
| DFO | Deferoxamine |
| DFP | Deferiprone |
| DHA | Dihydroartemisinin |
| DV | digestive vacuole |
| EPR | electron paramagnetic resonance |
| ER | endoplasmic reticulum |
| FPIX | ferriprotoporphyrin IX heme |
| ggcs, | gamma glutamylcysteine synthetase |
| GSH | glutathione |
| GSSG | glutathione disulfide |
| GWAS | genome-wide association study |
| Hb | hemoglobin |
| Hz | hemozoin |
| IC50 | half-maximal inhibitory concentration |
| iRBC | infected red blood cell |
| LD50 | half-maximal lethal concentration |
| LF | lumefantrine |
| MOA | mechanism of action |
| MQ | mefloquine |
| MRP | multidrug resistance protein |
| MS | mass spectrometry |
| PfPI3K | Plasmodium falciparum phosphatidyl-3′-kinase |
| PPQ | piperaquine |
| RSA | ring stage assay |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| SEA | Southeast Asia |
| SNP | single nucleotide polymorphism |
| QTL | quantitative trait locus |
References
- Klayman, D.L. Qinghaosu (artemisinin): An antimalarial drug from China. Science 1985, 228, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, X.; Kamaraj, S.; Bulbule, V.J.; Chiu, F.C.K.; Chollet, J.; Dhanasekaran, M.; Hein, C.D.; Papastogiannidis, P.; Morizzi, J.; et al. Structure-activity relationship of the antimalarial ozonide Artefenomel (OZ439). J. Med. Chem. 2017, 60, 2654–2668. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, P.M.; Amewu, R.K.; Charman, S.A.; Sabbani, S.; Gnadig, N.F.; Straimer, J.; Fidock, D.A.; Shore, E.R.; Roberts, N.L.; Wong, M.H.-L.; et al. A tetraoxane-based antimalarial drug candidate that overcomes PfK13-C580Y dependent artemisinin resistance. Nat. Commun. 2018, 8, 15159. [Google Scholar] [CrossRef] [PubMed]
- ter Kuile, F.; White, N.J.; Holloway, P.; Pasvol, G.; Krishna, S. Plasmodium falciparum: In vitro studies of the pharmacodynamic properties of drugs used for the treatment of severe malaria. Exp. Parasitol. 1993, 76, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, B.; Lelievre, J.; Barragan, M.J.L.; Laurent, V.; Su, X.Z.; Berry, A.; Benoit-Vical, F. Increased tolerance to artemisinin in plasmodium falciparum is mediated by a quiescence mechanism. Antimicrob. Agents Chemother. 2010, 54, 1872–1877. [Google Scholar] [CrossRef] [PubMed]
- Price, R.N.; Nosten, F.; Luxemburger, C.; ter Kuile, F.O.; Paiphun, L.; Chungsuphajaisiddji, T.; White, N.J. Effects of artemisinin derivatives on malaria transmissibility. Lancet 1996, 347, 1654–1658. [Google Scholar] [CrossRef]
- Meunier, B.; Robert, A. Heme as Trigger and Target for Trioxane-Containing Antimalarial Drugs. Acc. Chem. Res. 2010, 43, 1444–1451. [Google Scholar] [CrossRef]
- Noedl, H.; Se, Y.; Schaecher, K.; Smith, B.L.; Socheat, D.; Fukuda, M.M. Evidence of artemisinin-resistant malaria in western Cambodia. N. Engl. J. Med. 2008, 359, 2619–2620. [Google Scholar] [CrossRef]
- Noedl, H.; Socheat, D.; Satimai, W. Artemisinin-resistant malaria in Asia. N. Engl. J. Med. 2009, 361, 540–541. [Google Scholar] [CrossRef]
- Dondorp, A.M.; Nosten, F.; Yi, P.; Das, D.; Phyo, A.P.; Tarning, J.; Lwin, K.M.; Ariey, F.; Hanpithakpong, W.; Lee, S.J.; et al. Artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2009, 361, 455–467. [Google Scholar] [CrossRef]
- Meshnick, S.R.; Yang, Y.; Lima, V.; Kuypers, F.; Kamchonwongpaisan, S.; Yuthavong, Y. Iron-Dependent Free Radical Generation from the Antimalarial Agent Artemisinin (Qinghaosu). Antimicrob. Agents Chemother. 1993, 37, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Heller, L.E.; Goggins, E.; Roepe, P.D. Dihydroartemisinin-Ferriprotoporphyrin IX Adduct Abundance in Plasmodium falciparum Malarial Parasites and the Relationship to Emerging Artemisinin Resistance. Biochemistry 2018, 57, 6935–6945. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). 2018 Report. Available online: https://www.who.int/malaria/publications/world-malaria-report-2018/en/ (accessed on 6 March 2019).
- White, N.J. Antimalarial drug resistance. J. Clin. Invest. 2004, 113, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Carrara, V.I.; Zwang, J.; Ashley, E.A.; Price, R.N.; Stepniewska, K.; Barends, M.; Brockman, A.; Anderson, T.; McGready, R.; Phaiphun, L.; et al. Changes in the treatment re- sponses to artesunate-mefloquine on the northwestern border of Thailand during 13 years of continuous deployment. PLoS ONE 2009, 4, e4551. [Google Scholar] [CrossRef] [PubMed]
- Meshnick, S.R.; Thomas, A.; Ranz, A.; Xu, C.; Pan, H. Artemisinin (qinghaosu): The role of intracellular hemin in its mechanism of antimalarial action. Mol. Biochem. Parasitol. 1991, 49, 181–189. [Google Scholar] [CrossRef]
- Posner, G.H.; Oh, C.H. A Regiospecifically Oxygen-18 Labeled 1,2,4-Trioxane: A Simple Chemical Model System To Probe the Mechanism(s)for the Antimalarial Activity of Artemisinin (Qinghaosu). J. Am. Chem. Soc. 1992, 114, 8328–8329. [Google Scholar] [CrossRef]
- Zhang, F.; Gosser, D.K.; Meshnick, S.R. Hemin-catalyzed decomposition of artemisinin (qinghaosu). Biochem. Pharmacol. 1992, 43, 1805–1809. [Google Scholar] [CrossRef]
- Yang, Y.; Asawamahasakada, W.; Meshnick, S.R. Alkylation of human albumin by the antimalarial artemisinin. Biochem. Pharmacol. 1993, 46, 336–339. [Google Scholar] [CrossRef]
- Yang, Y.; Little, B.; Meshnick, S.R. Alkylation of proteins by Artemisinin: Effects of heme, pH, and drug structure. Biochem. Pharmacol. 1994, 48, 569–573. [Google Scholar] [CrossRef]
- Goldberg, D.E.; Slater, A.G.; Cerami, A.; Henderson, G.B. Hemoglobin degradation in the malaria parasite Plasmodium falciparum: An ordered process in a unique organelle. Proc. Natl. Acad. Sci. USA 1990, 87, 2931–2935. [Google Scholar] [CrossRef]
- Hong, Y.; Yang, Y.; Meshnick, S.R. The interaction of artemisinin with malarial hemozoin. Mol. Biochem. Parasitol. 1994, 63, 121–128. [Google Scholar] [CrossRef]
- Pagola, S.; Stephens, P.W.; Bohle, D.S.; Kosar, A.D.; Madsen, S.K. The structure of malaria pigment B-hematin. Nature 2000, 404, 307–310. [Google Scholar] [CrossRef] [PubMed]
- Posner, G.H.; Oh, C.H.; Wang, D.; Genera, L.; Milhous, W.K.; Meshnick, S.R.; Asawamahasakada, W. Mechanism-Based Design, Synthesis, and in Vitro Antimalarial Testing of New 4-Methylated Trioxanes Structurally Related to Artemisinin: The Importance of a Carbon-Centered Radical for Antimalarial Activity. J. Med. Chem. 1994, 37, 1256–1258. [Google Scholar] [CrossRef] [PubMed]
- Robert, A.; Meunier, B. Characterization of the First Covalent Adduct between Artemisinin and a Heme Model. J. Am. Chem. Soc. 1997, 119, 5968–5969. [Google Scholar] [CrossRef]
- Robert, A.; Meunier, B. Alkylating Properties of Antimalarial Artemisinin Derivatives and Synthetic Trioxanes when Activated by a Reduced Heme Model. Chem. Eur. J. 1998, 4, 1287–1296. [Google Scholar] [CrossRef]
- Robert, A.; Cazelles, J.; Meunier, B. Characterization of the alkylation product of heme by the antimalarial drug artemisinin. Angew. Chem. Int. Ed. 2001, 40, 1954–1957. [Google Scholar] [CrossRef]
- Robert, A.; Coppel, Y.; Meunier, B. Alkylation of heme by the antimalarial drug artemisinin. Chem. Commun. 2002, 5, 414–415. [Google Scholar] [CrossRef]
- Robert, A.; Dechy-Cabaret, O.; Cazelles, J.; Meunier, B. From mechanistic studies on artemisinin derivatives to new modular antimalarial drugs. Acc. Chem. Res. 2002, 35, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Cazelles, J.; Robert, A.; Meunier, R. Alkylating capacity and reaction products of antimalarial trioxanes after activation by a heme model. J. Org. Chem. 2002, 67, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Haynes, R.K. Reply to Comments on “Highly Antimalaria-Active Artemisinin Derivatives: Biological Activity Does Not Correlate with Chemical Reactivity”. Angew. Chem. Int. Ed. 2005, 44, 2064–2065. [Google Scholar] [CrossRef]
- Laurent, S.A.; Robert, A.; Meunier, B. C10-Modified artemisinin derivatives: Efficient heme-alkylating agents. Angew. Chem. Int. Ed. 2005, 44, 2060–2063. [Google Scholar] [CrossRef] [PubMed]
- Robert, A.; Benoit-Vical, F.; Claparols, C.; Meunier, B. The antimalarial drug artemisinin alkylates heme in infected mice. Proc. Natl. Acad. Sci. USA 2005, 103, 13676–13680. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.E.; Slater, A.F.G.; Beavis, R.; Chait, B.; Cerami, A.; Henderson, G.B. Hemoglobin Degradation in the Human Malaria Pathogen Plasmodium falciparum: A Catabolic Pathway Initiated by a Specific Aspartic Protease. J. Exp. Med. 1991, 173, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Francis, S.E.; Gluzman, I.Y.; Oksman, A.; Knickerbocker, A.; Mueller, R.; Bryant, M.L.; Sherman, D.R.; Russell, D.G.; Goldberg, D.E. Molecular characterization and inhibition of a Plasmodium falciparum aspartic hemoglobinase. EMBO J. 1994, 13, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Sijwali, P.S.; Shenai, B.R.; Gut, J.; Singh, J.; Rosenthal, P.J. Expression and characterization of the Plasmodium falciparum haemoglobinase falcipain-3. Biochem. J. 2001, 360, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, C.J.; Chia, W.N.; Loh, C.C.; Li, Z.; Lee, Y.M.; He, Y.; Yuan, L.X.; Lim, T.K.; Liu, M.; et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat. Commun. 2015, 6, 10111. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.; Barton, V.; Phanchana, M.; Charoensutthivarakul, S.; Wong, M.H.; Hemingway, J.; Biagini, G.A.; O’Neill, P.M.; Ward, S.A. Artemisinin activity-based probes identify multiple molecular targets within the asexual stage of the malaria parasites Plasmodium falciparum 3D7. Proc. Natl. Acad. Sci. USA 2016, 113, 2080–2085. [Google Scholar] [CrossRef] [PubMed]
- Ismail, H.M.; Barton, V.E.; Panchana, M.; Charoensutthivarakul, S.; Biagini, G.A.; Ward, S.A.; O’Neill, P.M. A Click Chemistry-Based Proteomic Approach Reveals that 1,2,4-Trioxolane and Artemisinin Antimalarials Share a Common Protein Alkylation Profile. Angew. Chem. 2016, 128, 6511–6515. [Google Scholar] [CrossRef]
- Heller, L.E.; Roepe, P.D. Quantification of Free Ferriprotoporphyrin IX Heme and Hemozoin for Artemisinin Sensitive vs Delayed Clearance Phenotype Plasmodium falciparum Malarial Parasites. Biochemistry 2018, 57, 6927–6934. [Google Scholar] [CrossRef]
- Bopp, S.; Magistrado, P.; Wong, W.; Schaffner, S.F.; Mukherjee, A.; Lim, P.; Dhorda, M.; Amaratunga, C.; Woodrow, C.J.; Ashley, E.A.; et al. Plasmepsin II-III copy number accounts for bimodal Piperaquine resistance among Cambodian Plasmodium falciparum. Nat. Commun. 2018, 9, 1769. [Google Scholar] [CrossRef]
- World Health Organization (WHO). 2008 Report. Available online: https://www.who.int/malaria/publications/atoz/9789241563697/en/ (accessed on 6 March 2019).
- Denis, M.B.; Tsuyuoka, R.; Lim, P.; Lindegardh, N.; Yi, P.; Top, S.N.; Socheat, D.; Fandeur, T.; Annerberg, A.; Christophel, E.M.; et al. Efficacy of artemether-lumefantrine for the treatment of uncomplicated falciparum malaria in northwest Cambodia. Trop. Med. Int. Health 2006, 11, 1800–1807. [Google Scholar] [CrossRef] [PubMed]
- Vijaykadga, S.; Rojanawatsirivej, C.; Cholpol, S.; Phoungmanee, D.; Nakavej, A.; Wongsrichanalai, C. In vivo sensitivity monitoring of mefloquine monotherapy and artesunate-mefloquine combinations for the treatment of uncomplicated falciparum malaria in Thailand in 2003. Trop. Med. Int. Health 2006, 11, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Nosten, F.; Imvithaya, S.; Vincenti, M.; Delmas, G.; Lebihan, G.; Hausler, B.; White, N. Malaria on the Thai-Burmese border: Treatment of 5192 patients with mefloquine-sulfadoxine-pyrimethamine. Bull. World Health Organ. 1987, 65, 891–896. [Google Scholar] [PubMed]
- Nosten, F.; ter Kuile, F.; Chongsuphajaisiddhi, T.; Luxemburger, C.; Webster, H.K.; Edstein, M.; Phaipun, L.; Thew, K.L.; White, N.J. Mefloquine-resistant falciparum malaria on the Thai-Burmese border. Lancet 1991, 337, 1140–1143. [Google Scholar] [CrossRef]
- Denis, M.B.; Tsuyuoka, R.; Poravuth, Y.; Poravuth, Y.; Narann, T.S.; Seila, S.; Lim, C.; Incardona, S.; Lim, P.; Sem, R.; et al. Surveillance of the efficacy of artesunate and mefloquine combination for the treatment of uncomplicated falciparum malaria in Cambodia. Trop. Med. Int. Health 2006, 11, 1360–1366. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A.; Dhorda, M.; Fairhurst, R.M.; Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; Anderson, J.M.; Mao, S.; Sam, B.; et al. Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med. 2014, 371, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Amaratunga, C.; Lim, P.; Suon, S.; Sreng, S.; Mao, S.; Sopha, C.; Sam, B.; Dek, D.; Try, V.; Amato, R.; et al. Dihydroartemisinin–piperaquine resistance in Plasmodium falciparum malaria in Cambodia: A multisite prospective cohort study. Lancet Infect. Dis. 2016, 16, 357–365. [Google Scholar] [CrossRef]
- Amaratunga, C.; Sreng, S.; Suon, S.; Phelps, E.S.; Stepniewska, K.; Lim, P.; Zhou, C.; Mao, S.; Anderson, J.M.; Lindegardh, N.; et al. Artemisinin-resistant Plasmodium falciparum in Pursat province, western Cambodia: A parasite clearance rate study. Lancet Infect. Dis. 2012, 12, 851–858. [Google Scholar] [CrossRef]
- Miotto, O.; Almagro-Garcia, J.; Manske, M.; Macinnis, B.; Campino, S.; Rockett, K.A.; Amaratunga, C.; Lim, P.; Sreng, S.; Anderson, J.M.; et al. Multiple populations of artemisinin-resistant Plasmodium falciparum in Cambodia. Nat Genet. 2013, 45, 648–655. [Google Scholar] [CrossRef]
- Cheeseman, I.H.; Miller, B.A.; Nair, S.; Nkhoma, S.; Tan, A.; Tan, J.C.; Al Saai, S.; Phyo, A.P.; Moo, C.L.; Lwin, K.M.; et al. A major genome region underlying artemisinin resistance in malaria. Science 2012, 336, 79–82. [Google Scholar] [CrossRef]
- Ariey, F.; Witkowski, B.; Amaratunga, C.; Beghain, J.; Langlois, A.C.; Khim, N.; Kim, S.; Duru, V.; Bouchier, C.; Ma, L.; et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Menard, D.; Khim, N.; Beghain, J.; Adegnika, A.A.; Shafiul-Alam, M.; Amodu, O.; Rahim-Awab, G.; Barnadas, C.; Berry, A.; Boum, Y.; et al. A worldwide map of Plasmodium falciparum K13-propeller polymorphisms. N. Engl. J. Med. 2016, 374, 2453–2464. [Google Scholar] [CrossRef] [PubMed]
- MalariaGEN Plasmodium falciparum Community Project. Genomic epidemiology of artemisinin resistant malaria. eLife 2016, 5, e08714. [Google Scholar] [CrossRef] [PubMed]
- Woodrow, C.J.; White, N.J. The clinical impact of artemisinin resistance in southeast asia and the potential for future spread. FEMS Microbiol. Rev. 2017, 41, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Asawamabasakda, W.; Ittarat, I.; Chang, C.; McElroy, P.; Meshnick, S.R. Effects of antimalarials and protease inhibitors on plasmodial hemozoin production. Mol. Biochem. Parasitol. 1994, 67, 183–191. [Google Scholar] [CrossRef]
- Zhang, J.; Krugliak, M.; Ginsburg, H. The fate of ferriprotoporphyrin IX in malaria infected erythrocytes in conjunction with the mode of action of antimalarial drugs. Mol. Biochem. Parasitol. 1999, 99, 129–141. [Google Scholar] [CrossRef]
- Gligorijevic, B.; McAllister, R.; Urbach, J.S.; Roepe, P.D. Spinning disk confocal microscopy of live, intraerythrocytic malarial parasites. 1. Quantification of hemozoin development for drug sensitive versus resistant malaria. Biochemistry 2006, 45, 12400–12410. [Google Scholar] [CrossRef]
- Klonis, N.; Crespo-Ortiz, M.P.; Bottova, I.; Abu-Bakar, N.; Kenny, S.; Rosenthal, P.; Tilley, L. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc. Natl. Acad. Sci. USA 2011, 108, 11405–11410. [Google Scholar] [CrossRef]
- Hott, A.; Casandra, D.; Sparks, K.N.; Morton, L.C.; Castanares, G.G.; Rutter, A.; Kyle, D.E. Artemisinin-resistant Plasmodium falciparum parasites exhibit altered patterns of development in infected erythrocytes. Antimicrob. Agents Chemother. 2015, 59, 3156–3167. [Google Scholar] [CrossRef]
- Xie, S.C.; Dogovski, C.; Hanssen, E.; Chiu, F.; Yang, T.; Crespo, M.P.; Stafford, C.; Batinovic, S.; Teguh, S.; Charman, S.; et al. Haemoglobin degradation underpins the sensitivity of early ring stage Plasmodium falciparum to artemisinins. J. Cell. Sci. 2016, 129, 406–416. [Google Scholar] [CrossRef]
- Perez-Rosado, J.; Gervais, G.W.; Ferrer-Rodriguez, I.; Peters, W.; Serrano, A.E. Plasmodium berghei: Analysis of the gamma-glutamylcysteine synthetase gene in drug-resistant lines. Exp. Parasitol. 2002, 101, 175–182. [Google Scholar] [CrossRef]
- Vega-Rodriguez, J.; Pastrana-Mena, R.; Crespo-Llado, K.; Ortiz, J.G.; Ferrer-Rodriguez, I.; Serrano, A.E. Implications of Glutathione Levels in the Plasmodium berghei Response to Chloroquine and Artemisinin. PLoS ONE 2015, 10, e0128212. [Google Scholar] [CrossRef] [PubMed]
- Mok, S.; Imwong, M.; Mackinnon, M.J.; Sim, J.; Ramadoss, R.; Yi, P.; Mayxay, M.; Chotivanich, K.; Liong, K.; Russell, B.; et al. Artemisinin resistance in Plasmodium falciparum is associated with an altered temporal pattern of transcription. BMC Genomics 2011, 12, 391. [Google Scholar] [CrossRef] [PubMed]
- Rocamora, D.; Zhu, L.; Liong, K.Y.; Dondorp, A.; Miotto, O.; Mok, S.; Bozdech, Z. Oxidative stress and protein damage responses mediate artemisinin resistance in malaria parasites. PLoS Pathog. 2018, 14, e1006930. [Google Scholar] [CrossRef] [PubMed]
- Roepe, P.D. To kill or not to kill, that is the question: Cytocidal antimalarial drug resistance. Trends Parasitol. 2014, 30, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, B.; Amaratunga, C.; Khim, N.; Sreng, S.; Chim, P.; Kim, S.; Lim, P.; Mao, S.; Sopha, C.; Sam, B.; et al. Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: In-vitro and ex-vivo drug-response studies. Lancet Infect. Dis. 2013, 13, 1043–1049. [Google Scholar] [CrossRef]
- Siriwardana, A.; Iyengar, K.; Roepe, P.D. Endoperoxide drug cross resistance patterns for plasmodium falciparum exhibiting an artemisinin delayed clearance phenotype. Antimicrob. Agents Chemother. 2016, 60, 6952–6956. [Google Scholar] [CrossRef] [PubMed]
- Straimer, J.; Gnädig, N.F.; Witkowski, B.; Amaratunga, C.; Duru, V.; Ramadani, A.P.; Dacheux, M.; Khim, N.; Zhang, L.; Lam, S.; et al. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 2015, 347, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.J.; Lansdell, P.; Sanders, M.; Muwanguzi, J.; van Schalkwyk, D.A.; Kaur, H.; Nolder, D.; Tucker, J.; Bennett, H.M.; Otto, T.D.; et al. Pfk13-Independent Treatment failure in four imported cases of Plasmodium falciparum malaria treated with artemether-lumefantrine in the United Kingdom. Antimicrob. Agents Chemother. 2017, 61, e02382-16. [Google Scholar] [CrossRef] [PubMed]
- Demas, A.R.; Sharma, A.I.; Wong, W.; Early, A.M.; Redmond, A.M.; Bopp, S.; Neafsey, D.E.; Volkman, S.K.; Hartl, D.L.; Wirth, D.F. Mutations in Plasmodium falciparum actin-binding protein coronin confer reduced artemisinin susceptibility. Proc. Natl. Acad. Sci. USA 2018, 115, 12799–12804. [Google Scholar] [CrossRef] [PubMed]
- Mbengue, A.; Bhattacharjee, S.; Pandharkar, T.; Liu, H.; Estiu, G.; Stahelin, R.V.; Rizk, S.S.; Njimoh, D.L.; Ryan, Y.; Chotivanich, K.; et al. A molecular mechanism of artemisinin resistance in Plasmodium falciparum malaria. Nature 2015, 520, 683–687. [Google Scholar] [CrossRef]
- Hassett, M.R.; Sternberg, A.R.; Riegel, B.E.; Thomas, C.J.; Roepe, P.D. Heterologous expression, purification, and functional analysis of plasmodium falciparum phosphatidylinositol 3’-kinase. Biochemistry 2017, 56, 4335–4345. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Coppens, I.; Mbengue, A.; Suresh, N.; Ghorbal, M.; Slouka, Z.; Safeukui, I.; Tang, H.; Speicher, D.W.; Stahelin, R.V.; et al. Remodeling of the malaria parasite and host human red cell by vesicle amplification that induces artemisinin resistance. Blood 2018, 131, 1234–1247. [Google Scholar] [CrossRef]
- Hassett, M.R.; Roepe, P.D. PIK-ing new malaria chemotherapy. Trends Parasitol. 2018, 34, 925–927. [Google Scholar] [CrossRef]
- Vaid, A.; Ranjan, R.; Smythe, W.A.; Hoppe, H.C.; Sharma, P. PfPI3K, a phosphatidylinositol-3 kinase from Plasmodium falciparum, is exported to the host erythrocyte and is involved in hemoglobin trafficking. Blood 2010, 115, 2500–2507. [Google Scholar] [CrossRef] [PubMed]
- Tawk, L.; Chicanne, G.; Dubremetz, J.F.; Richard, V.; Payrastre, B.; Vial, H.J.; Roy, C.; Wengelnik, K. Phosphatidylinositol 3-monophosphate is involved in toxoplasma apicoplast biogenesis. Eukaryot. Cell. 2010, 9, 1519–1530. [Google Scholar] [CrossRef] [PubMed]
- Hassett, M.R.; Sternberg, A.R.; Roepe, P.D. Inhibition of human class I vs class III phosphatidylinositol 3′ kinases. Biochemistry 2017, 56, 4326–4334. [Google Scholar] [CrossRef] [PubMed]
- Gaviria, D.; Paguio, M.; Turnbull, L.; Tan, A.; Siriwardana, A.; Ghosh, D.; Ferdig, M.; Sinai, A.; Roepe, P. A Process Similar to Autophagy Is Associated with Cytocidal Chloroquine Resistance in Plasmodium falciparum. PLoS ONE 2013, 8, e79059. [Google Scholar] [CrossRef]
- Hain, A.U.; Bosch, J. Autophagy in Plasmodium, a multifunctional pathway? Comput. Struct. Biotechnol. J. 2013, 8, e201308002. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cabrera, M.; Yang, J.; Yuan, L.; Gupta, B.; Liang, X.; Kemirembe, K.; Shrestha, S.; Brashear, A.; Li, X.; et al. Genome-wide association analysis identifies genetic loci associated with resistance to multiple antimalarials in plasmodium falciparum china-myanmar border. Nat. Sci. Rep. 2016, 6, 33891. [Google Scholar] [CrossRef]
- Mott, B.T.; Eastman, R.T.; Guha, R.; Sherlach, K.S.; Siriwardana, A.; Shinn, P.; McKnight, C.; Michael, S.; Lacerda-Queiroz, N.; Patel, P.R.; et al. High-throughput matrix screening identifies synergistic and antagonistic antimalarial drug combinations. Nat. Sci. Rep. 2015, 5, 13891. [Google Scholar] [CrossRef] [PubMed]
- Brennand, A.; Gualdrón-López, M.; Coppens, I.; Rigden, D.J.; Ginger, M.L.; Michels, P.A. Autophagy in parasitic protists: Unique features and drug targets. Mol. Biochem. Parasitol. 2011, 177, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Breglio, K.F.; Rahman, R.S.; Sa, J.M.; Hott, A.; Roberts, D.J.; Wellems, T.E. Kelch mutations in Plasmodium falciparum Protein K13 do not modulate dormancy after artemisinin exposure and sorbitol selection in vitro. Antimicrob. Agents Chemother. 2018, 62, e02256-17. [Google Scholar] [CrossRef]
- Sa, J.M.; Kaslow, S.R.; Krause, M.A.; Melendez-Muniz, V.A.; Salzman, R.E.; Kite, W.A.; Zhang, M.; Moraes Barros, R.R.; Mu, J.; Han, P.K.; et al. Artemisinin resistance phenotypes and K13 inheritance in a Plasmodium falciparum cross and Aotus model. Proc. Natl. Acad. Sci. USA 2018, 115, 12513–12518. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein (Gene) ID | Prtotein ART Targets Found in All 3 Studies | Putative Location |
|---|---|---|
| PF3D7_0102200 | Ring-infected erythrocyte surface antigen | Dense Granules (Merozoites) |
| PF3D7_0322900 | 40S ribosomal protein S3A, putative | Ribosome |
| PF3D7_0523000 | Multidrug resistance protein | DV |
| PF3D7_0608800 | Ornithine aminotransferase | Cytosol |
| PF3D7_0624000 | Hexokinase | Cytosol |
| PF3D7_0708400 | Heat shock protein 90 | Cytoplasm |
| PF3D7_0818900 | Heat shock protein 70 | Nucleus |
| PF3D7_0903700 | Alpha tubulin 1 | Microtubule |
| PF3D7_0930300 | Merozoite surface protein 1 | Plasma Membrane |
| PF3D7_1008700 | Tubulin beta chain | Microtubule |
| PF3D7_1012400 | Hypoxanthine-guanine phosphoribosyltransferase | Cytosol |
| PF3D7_1015900 | Enolase | DV |
| PF3D7_1246200 | Actin I | Actin Filament/Cytoskeleton |
| PF3D7_1311900 | Vacuolar ATP synthase subunit a | DV |
| PF3D7_1324900 | L-lactate dehydrogenase | Cytosol |
| PF3D7_1357100 | Elongation factor 1-alpha | Cytosol |
| PF3D7_1407900 | Plasmepsin I | DV |
| PF3D7_1408000 | Plasmepsin II | DV |
| PF3D7_1444800 | Fructose-bisphosphate aldolase | Cytosol |
| Gene ID | Found in All 3 Studies | Putative Location of Encoded Protein | Ring (9 HPI) Expression Value (Log2 Ratio) in 3D7 | Trophozoite (28 HPI) Expression Value (Log2 Ratio) in 3D7 | Essential? |
|---|---|---|---|---|---|
| PF3D7_0102200 | Ring-infected erythrocyte surface antigen | Dense Granules (Merozoites) | 0.8 | −2.52 | Unknown |
| PF3D7_0608800 | Ornithine aminotransferase | Cytoplasm | −1.74 | 0.7 | No |
| PF3D7_0903700 | Alpha tubulin 1 | Microtubule | −1.3 | −0.08 | No |
| PF3D7_0930300 | Merozoite surface protein 1 | Plasma Membrane | −0.59 | −1.52 | Yes |
| PF3D7_1008700 | Tubulin beta chain | Microtubule | −1.53 | −0.02 | Yes |
| PF3D7_1246200 | Actin I | Actin Filament/Cytoskeleton | −0.55 | −1.05 | Yes |
| PF3D7_1324900 | L-lactate dehydrogenase | Cytosol | −0.76 | 0.49 | Yes |
| PF3D7_1407900 | Plasmepsin I | DV | 1.17 | −1.01 | Yes |
| Gene ID | Up-Regulated Gene Description | FDR |
|---|---|---|
| PF3D7_0512200 | glutathione synthetase | 0.20 |
| PF3D7_1224600 | cytochrome c heme lyase, putative | 0.23 |
| PF3D7_0825600 | cytochrome c oxidase assembly protein, putative | 0.19 |
| PF3D7_1311700 | cytochrome c2 precursor, putative | 0.21 |
| Gene ID | Down-Regulated Gene Description | FDR |
|---|---|---|
| PF3D7_0727200 | cysteine desulfurase, putative | 0.19 |
| PF3D7_1438900 | thioredoxin peroxidase 1 | 0.24 |
| PF3D7_1457200 | thioredoxin 1 | 0.12 |
| PF3D7_1455400 | hemolysin, putative | 0.22 |
| PF3D7_1419800 | glutathione reductase | 0.16 |
| PF3D7_1012300 | ubiquitinol-cytochrome c reductase complex | 0.19 |
| PF3D7_1352500 | thioredoxin-related protein, putative | 0.24 |
| PF3D7_1011900 | heme oxygenase | 0.24 |
| PF3D7_1458000 | cysteine proteinase falcipain 1 | 0.16 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heller, L.E.; Roepe, P.D. Artemisinin-Based Antimalarial Drug Therapy: Molecular Pharmacology and Evolving Resistance. Trop. Med. Infect. Dis. 2019, 4, 89. https://doi.org/10.3390/tropicalmed4020089
Heller LE, Roepe PD. Artemisinin-Based Antimalarial Drug Therapy: Molecular Pharmacology and Evolving Resistance. Tropical Medicine and Infectious Disease. 2019; 4(2):89. https://doi.org/10.3390/tropicalmed4020089
Chicago/Turabian StyleHeller, Laura E., and Paul D. Roepe. 2019. "Artemisinin-Based Antimalarial Drug Therapy: Molecular Pharmacology and Evolving Resistance" Tropical Medicine and Infectious Disease 4, no. 2: 89. https://doi.org/10.3390/tropicalmed4020089
APA StyleHeller, L. E., & Roepe, P. D. (2019). Artemisinin-Based Antimalarial Drug Therapy: Molecular Pharmacology and Evolving Resistance. Tropical Medicine and Infectious Disease, 4(2), 89. https://doi.org/10.3390/tropicalmed4020089