
Combating Malaria: Targeting the Ubiquitin-Proteasome System to Conquer Drug Resistance


Abstract
1. Introduction
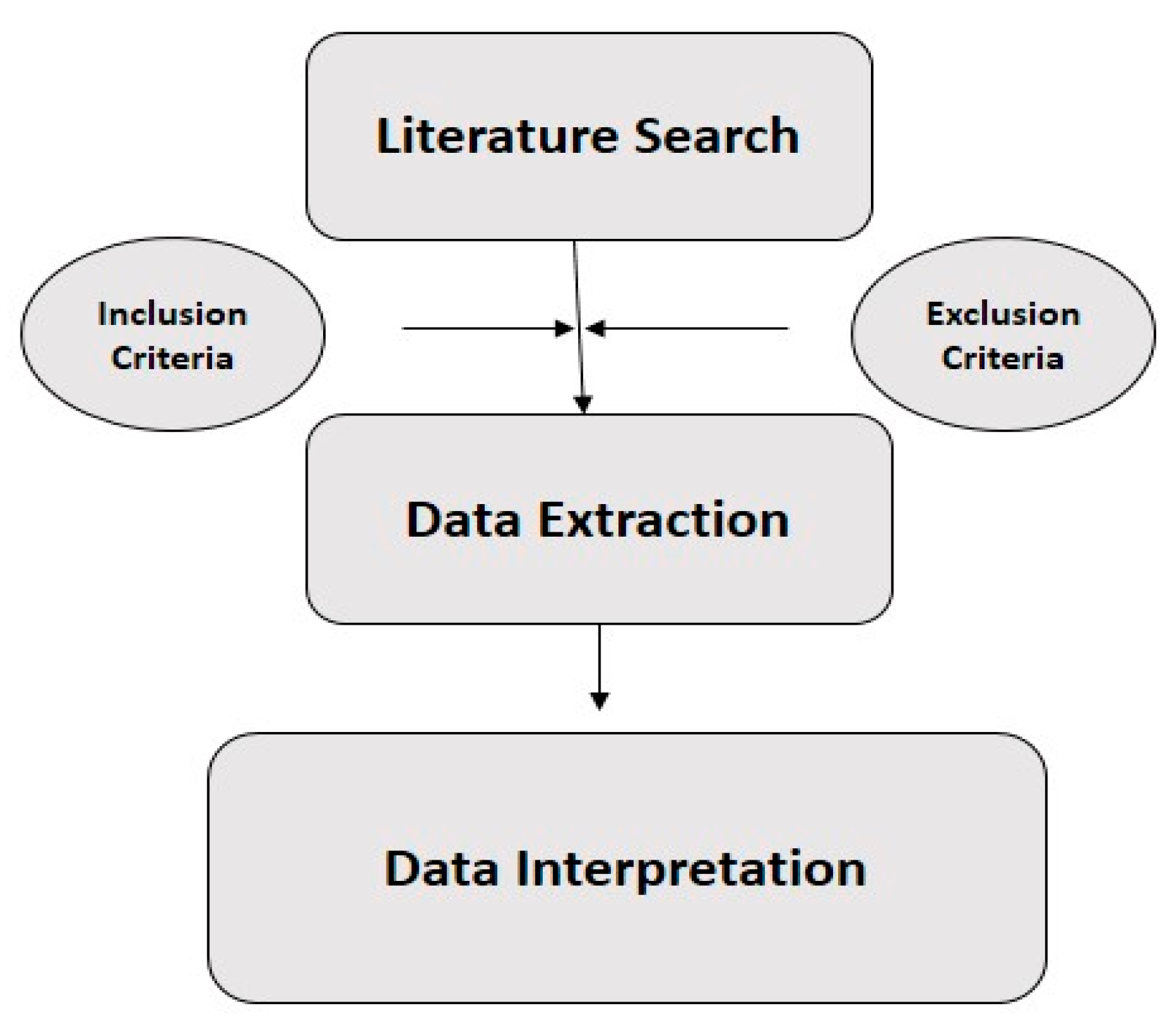
2. Methodology
2.1. Literature Search
2.2. Inclusion and Exclusion Criteria
2.3. Data Extraction
2.4. Interpretation
3. Results Interpretation
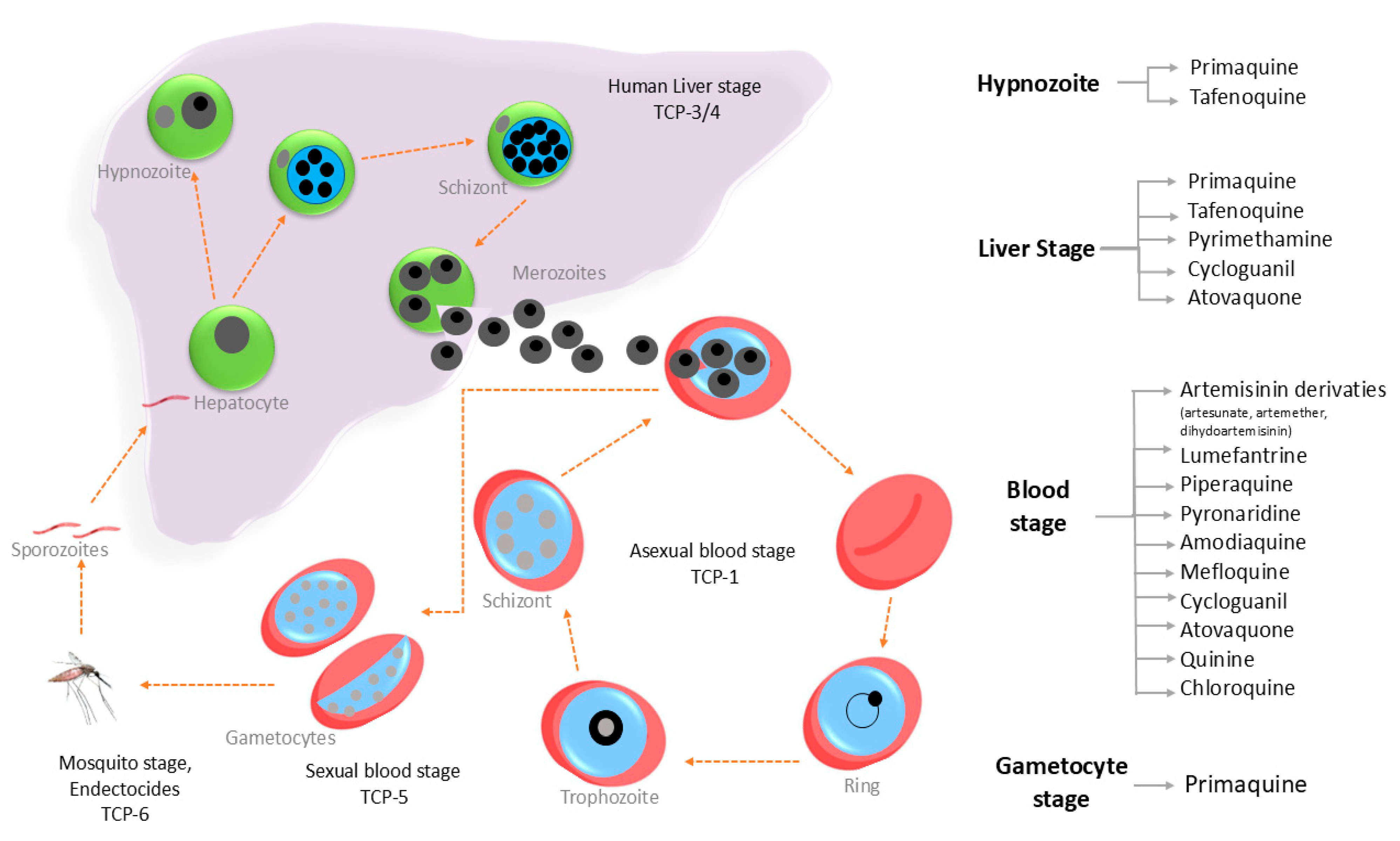
3.1. Current Antimalarial Drug Targets
3.2. Possible Next-Generation Therapies
3.3. Emergence of Drug Resistance Against Antimalarial Drugs
Resistance to Artemisinin and Other Therapeutics
3.4. Ubiquitin Proteasome System (UPS) as an Antimalarial Drug Target
3.4.1. Structure and Composition of Plasmodium UPS
3.4.2. Ubiquitination of Subjected Proteins
3.4.3. Protein Deubiquitination
3.5. Advancements in Proteasome Inhibitor Design for Malaria
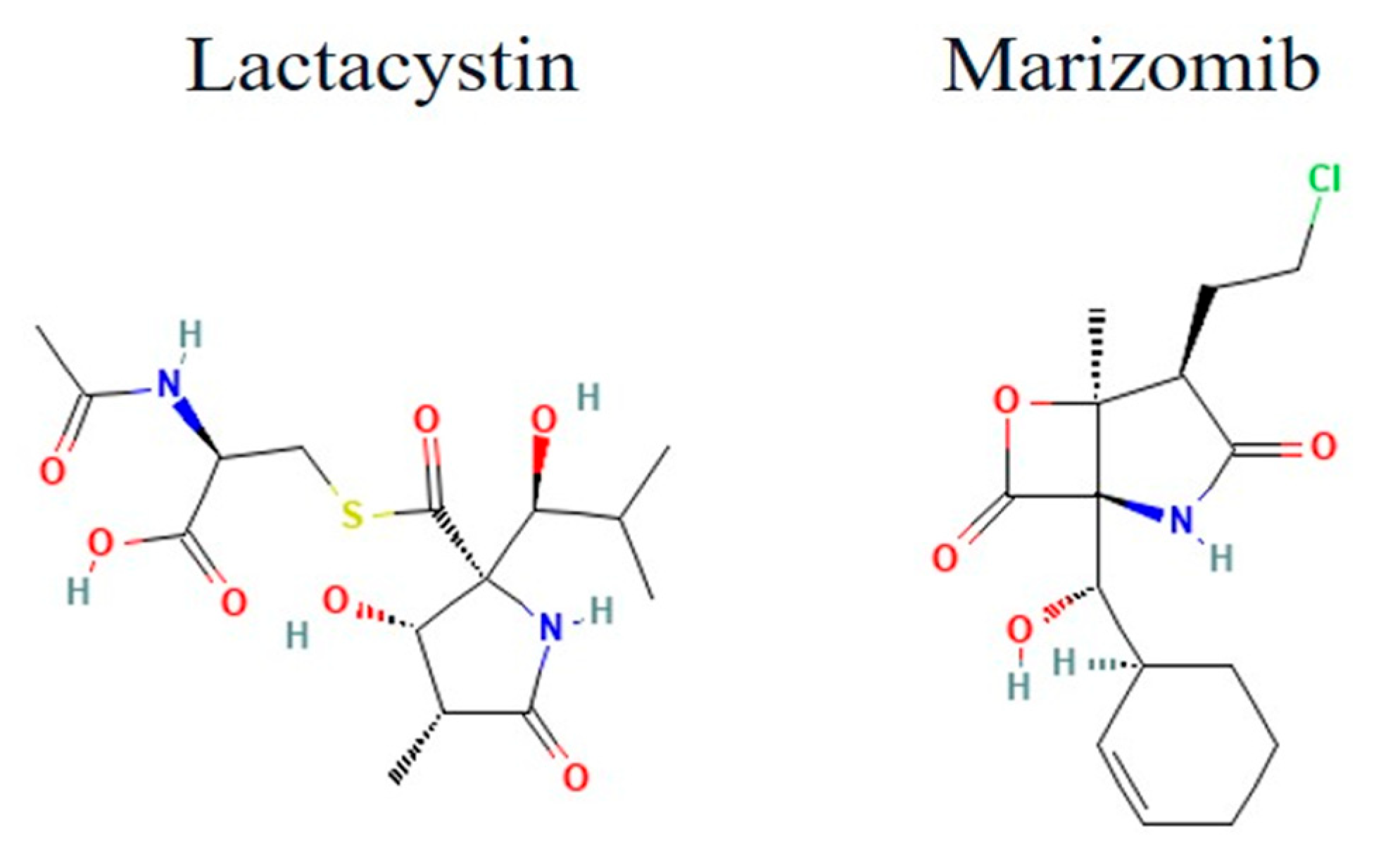
3.5.1. Lactacystins and Salinosporamides
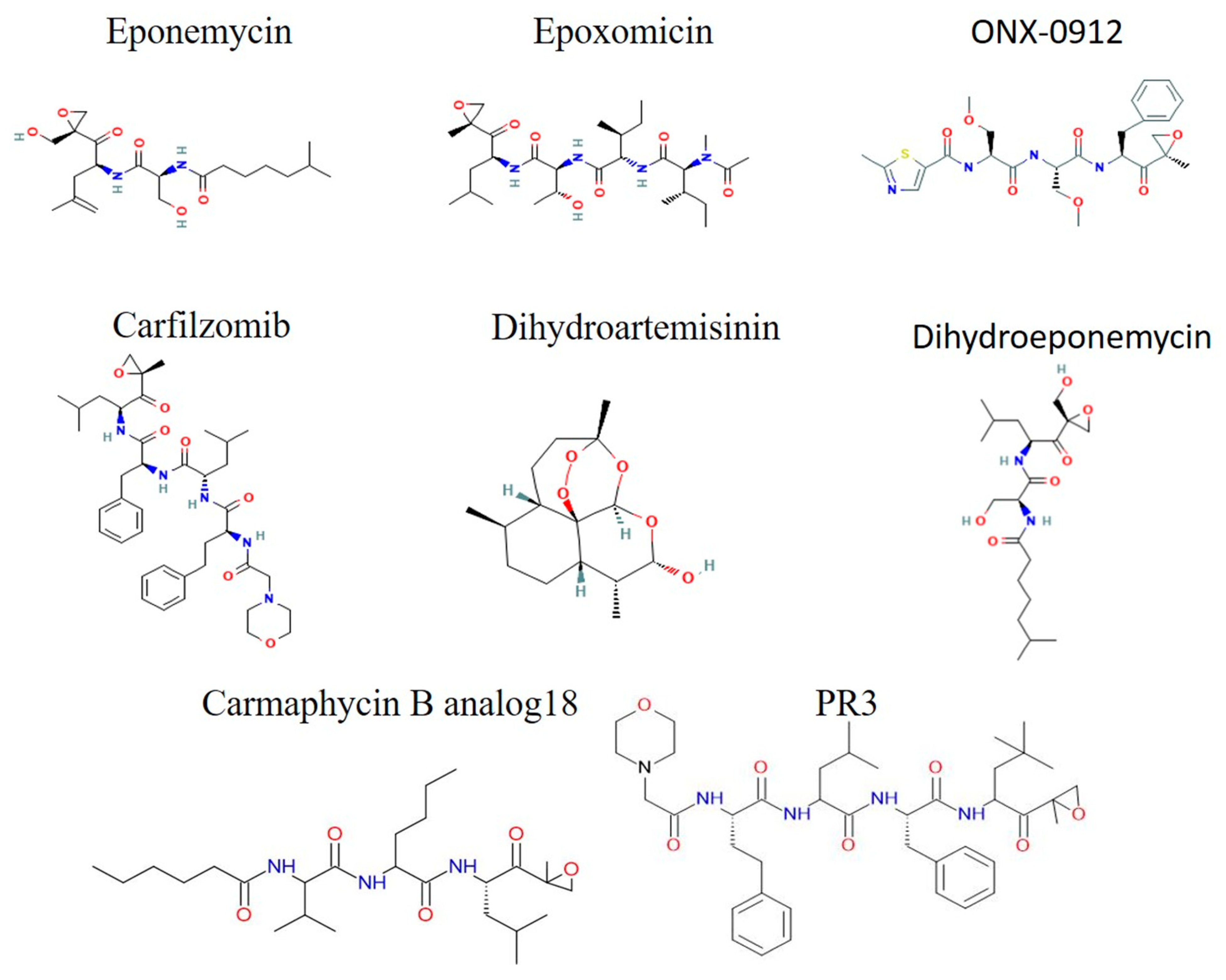
3.5.2. Epoxyketone Derivatives
3.5.3. Sulfonyl Fluorides
3.5.4. Peptide Aldehydes and Boronic Acids (Non-Peptide Small Molecules)
3.5.5. Cyclic Peptides
3.5.6. AsnEDA and Derivative
4. Future Directions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zareen, S.; Rehman, H.U.; Gul, N.; Zareen, H.; Hisham, M.; Ullah, I.; Rehman, M.U.; Bibi, S.; Bakht, A.; Khan, J. Malaria is still a life threatening disease review. J. Entomol. Zool. Stud. 2016, 105, 105–112. [Google Scholar]
- Cowman, A.F.; Healer, J.; Marapana, D.; Marsh, K. Malaria: Biology and disease. Cell 2016, 167, 610–624. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, H.; Bhalerao, P.; Singh, S.; Arya, H.; Alotaibi, B.S.; Rashid, S.; Hasan, M.R.; Bhatt, T.K. Malaria therapeutics: Are we close enough? Parasit Vectors 2023, 16, 130. [Google Scholar] [CrossRef]
- Maitland, K. Severe malaria in African children—The need for continuing investment. N. Engl. J. Med. 2016, 375, 2416–2417. [Google Scholar] [CrossRef]
- Nnamonu, E.I.; Ndukwe-Ani, P.A.; Imakwu, C.A.; Okenyi, C.I.; Ugwu, F.J.; Aniekwe, M.I.; Odo, S.I.; Ezenwosu, S.U. Malaria: Trend of burden and impact of control strategies. Int. J. Trop. Dis. Health 2020, 41, 18–30. [Google Scholar] [CrossRef]
- Rosenthal, P.J.; John, C.C.; Rabinovich, N.R. Malaria: How are we doing and how can we do better? Am. J. Trop. Med. Hyg. 2019, 100, 239–241. [Google Scholar] [CrossRef]
- Abanyam, N.L. Malaria and Socio-Economic Development in Nigeria. Lead City J. Soc. Sci. 2020, 5, 81. [Google Scholar]
- Haldar, K.; Bhattacharjee, S.; Safeukui, I. Drug resistance in Plasmodium. Nat. Rev. Microbiol. 2018, 16, 156–170. [Google Scholar] [CrossRef]
- Ehrlich, H.Y.; Somé, A.F.; Bazié, T.; Ebou, C.N.; Dembélé, E.L.; Balma, R.; Goodwin, J.; Wade, M.; Bei, A.K.; Ouédraogo, J.-B.; et al. Tracking antimalarial drug resistance using mosquito blood meals: A cross-sectional study. Lancet Microbe 2023, 4, e461–e469. [Google Scholar] [CrossRef]
- Fontinha, D.; Moules, I.; Prudêncio, M. Repurposing drugs to fight hepatic malaria parasites. Molecules 2020, 25, 3409. [Google Scholar] [CrossRef]
- Poonam; Gupta, Y.; Gupta, N.; Singh, S.; Wu, L.; Chhikara, B.S.; Rawat, M.; Rathi, B. Multistage inhibitors of the malaria parasite: Emerging hope for chemoprotection and malaria eradication. Med. Res. Rev. 2018, 38, 1511–1535. [Google Scholar] [PubMed]
- Mata-Cantero, L.; Chaparro, M.J.; Colmenarejo, G.; Cid, C.; Cabrera, A.C.; Rodriguez, M.M.S.; Martín, J.J.; Gamo, F.J.; Gomez-Lorenzo, M.G. Identification of small molecules disrupting the ubiquitin proteasome system in Malaria. ACS Infect. Dis. 2019, 5, 2105–2117. [Google Scholar] [PubMed]
- Sinha, A.; Sarkar, S. Ubiquitin-Proteasome System—A target to control pathogenic protozoa. Microb. Pathog. Combat. Sci. Technol. Educ. 2013, 1, 764–773. [Google Scholar]
- Bridgford, J.L.; Xie, S.C.; Cobbold, S.A.; Pasaje, C.F.A.; Herrmann, S.; Yang, T.; Gillett, D.L.; Dick, L.R.; Ralph, S.A.; Dogovski, C.; et al. Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat. Commun. 2018, 9, 3801. [Google Scholar]
- Arora, P.; Narwal, M.; Thakur, V.; Mukhtar, O.; Malhotra, P.; Mohmmed, A. A Plasmodium falciparum ubiquitin-specific protease (PfUSP) is essential for parasite survival and its disruption enhances artemisinin efficacy. Biochem. J. 2023, 480, 25–39. [Google Scholar]
- Mata-Cantero, L.; Azkargorta, M.; Aillet, F.; Xolalpa, W.; LaFuente, M.J.; Elortza, F.; Carvalho, A.S.; Martin-Plaza, J.; Matthiesen, R.; Rodriguez, M.S. New insights into host-parasite ubiquitin proteome dynamics in P. falciparum infected red blood cells using a TUBEs-MS approach. J. Proteom. 2016, 139, 45–59. [Google Scholar]
- Shibeshi, M.A.; Kifle, Z.D.; Atnafie, S.A. Antimalarial drug resistance and novel targets for antimalarial drug discovery. Infect. Drug Resist. 2020, 13, 4047–4060. [Google Scholar]
- Simwela, N.V.; Hughes, K.R.; Rennie, M.T.; Barrett, M.P.; Waters, A.P. Mammalian deubiquitinating enzyme inhibitors display in vitro and in vivo activity against malaria parasites and potentiate artemisinin action. ACS Infect. Dis. 2021, 7, 333–346. [Google Scholar]
- Krishnan, K.M.; Williamson, K.C. The proteasome as a target to combat malaria: Hits and misses. Transl. Res. 2018, 198, 40–47. [Google Scholar]
- Chung, D.-W.D.; Le Roch, K.G. Targeting the Plasmodium ubiquitin/proteasome system with anti-malarial compounds: Promises for the future. Infect. Disord. Drug Targets 2010, 10, 158–164. [Google Scholar]
- Hamilton, M.J.; Lee, M.; Le Roch, K.G. The ubiquitin system: An essential component to unlocking the secrets of malaria parasite biology. Mol. BioSystems 2014, 10, 715–723. [Google Scholar] [CrossRef]
- Zhan, W.; Zhang, H.; Ginn, J.; Leung, A.; Liu, Y.J.; Michino, M.; Toita, A.; Okamoto, R.; Wong, T.; Imaeda, T.; et al. Development of a highly selective Plasmodium falciparum proteasome inhibitor with anti-malaria activity in humanized mice. Angew. Chem. 2021, 133, 9365–9369. [Google Scholar] [CrossRef]
- Silva, M.J.F. Defining Molecular Pathways in Antimalarial Drug Resistance; Universidade do Minho: Braga, Portugal, 2022. [Google Scholar]
- Mathews, E.S.; John, A.R.O. Tackling resistance: Emerging antimalarials and new parasite targets in the era of elimination. F1000Research 2018, 7, 1170. [Google Scholar] [CrossRef] [PubMed]
- Egwu, C.O.; Pério, P.; Augereau, J.-M.; Tsamesidis, I.; Benoit-Vical, F.; Reybier, K. Resistance to artemisinin in falciparum malaria parasites: A redox-mediated phenomenon. Free Radic. Biol. Med. 2022, 179, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Renar, K.S.; Iskra, J.; Križaj, I. Understanding malarial toxins. Toxicon 2016, 119, 319–329. [Google Scholar] [CrossRef]
- Plucinski, M.M.; Dimbu, P.R.; Macaia, A.P.; Ferreira, C.M.; Samutondo, C.; Quivinja, J.; Afonso, M.; Kiniffo, R.; Mbounga, E.; Kelley, J.S.; et al. Efficacy of artemether-lumefantrine, artesunate–amodiaquine, and dihydroartemisinin–piperaquine for treatment of uncomplicated Plasmodium falciparum malaria in Angola, 2015. Malar. J. 2017, 16, 1–10. [Google Scholar] [CrossRef]
- Wicht, K.J.; Mok, S.; Fidock, D.A. Molecular mechanisms of drug resistance in Plasmodium falciparum malaria. Annu. Rev. Microbiol. 2020, 74, 431–454. [Google Scholar] [CrossRef]
- Agomo, P.U.; Meremikwu, M.M.; Watila, I.M.; Omalu, I.J.; Odey, F.A.; Oguche, S.; Ezeiru, V.I.; Aina, O.O. Efficacy, safety and tolerability of artesunate-mefloquine in the treatment of uncomplicated Plasmodium falciparum malaria in four geographic zones of Nigeria. Malar. J. 2008, 7, 172. [Google Scholar] [CrossRef]
- Burrows, J.N.; van Huijsduijnen, R.H.; Möhrle, J.J.; Oeuvray, C.; Wells, T.N. Designing the next generation of medicines for malaria control and eradication. Malar. J. 2013, 12, 187. [Google Scholar] [CrossRef]
- Siqueira-Neto, J.L.; Wicht, K.J.; Chibale, K.; Burrows, J.N.; Fidock, D.A.; Winzeler, E.A. Antimalarial drug discovery: Progress and approaches. Nat. Rev. Drug Discov. 2023, 22, 807–826. [Google Scholar] [CrossRef]
- Burrows, J.N.; Duparc, S.; Gutteridge, W.E.; van Huijsduijnen, R.H.; Kaszubska, W.; Macintyre, F.; Mazzuri, S.; Möhrle, J.J.; Wells, T.N.C. New developments in anti-malarial target candidate and product profiles. Malar. J. 2017, 16, 1–29. [Google Scholar]
- Alaithan, H.; Kumar, N.; Islam, M.Z.; Liappis, A.P.; Nava, V.E. Novel therapeutics for malaria. Pharmaceutics 2023, 15, 1800. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.-Q.; Ye, Q.; Huang, H.; Zheng, W.-Y. An overview of available antimalarials: Discovery, mode of action and drug resistance. Curr. Mol. Med. 2020, 20, 583–592. [Google Scholar] [PubMed]
- Reyburn, H. New WHO guidelines for the treatment of malaria. BMJ 2010, 340, c2637. [Google Scholar]
- Kumar, S.; Bhardwaj, T.; Prasad, D.; Singh, R.K. Drug targets for resistant malaria: Historic to future perspectives. Biomed. Pharmacother. 2018, 104, 8–27. [Google Scholar]
- Paget-McNicol, S.; Saul, A. Mutation rates in the dihydrofolate reductase gene of Plasmodium falciparum. Parasitology 2001, 122, 497–505. [Google Scholar]
- Müller, O. Malaria in Africa: Challenges for Control and Elimination in the 21st Century; Peter Lang: Frankfurt am Main, Germany, 2011. [Google Scholar]
- Newton, P.N.; Green, M.D.; Mildenhall, D.C.; Plançon, A.; Nettey, H.; Nyadong, L.; Hostetler, D.M.; Swamidoss, I.; Harris, G.A.; Powell, K.; et al. Poor quality vital anti-malarials in Africa-an urgent neglected public health priority. Malar. J. 2011, 10, 1–22. [Google Scholar]
- Hall, K.A.; Newton, P.N.; Green, M.D.; De Veij, M.; Vandenabeele, P.; Pizzanelli, D.; Mayxay, M.; Dondorp, A.; Fernandez, F.M. Characterization of counterfeit artesunate antimalarial tablets from southeast Asia. Am. J. Trop. Med. Hyg. 2006, 75, 804–811. [Google Scholar]
- Sidhu, A.B.S.; Uhlemann, A.-C.; Valderramos, S.G.; Valderramos, J.-C.; Krishna, S.; Fidock, D.A. Decreasing pfmdr1 copy number in Plasmodium falciparum malaria heightens susceptibility to mefloquine, lumefantrine, halofantrine, quinine, and artemisinin. J. Infect. Dis. 2006, 194, 528–535. [Google Scholar]
- Goldberg, D.E.; Siliciano, R.F.; Jacobs, W.R. Outwitting evolution: Fighting drug-resistant TB, malaria, and HIV. Cell 2012, 148, 1271–1283. [Google Scholar] [CrossRef]
- Bray, P.G.; Ward, S.A.; O’neill, P.M. Quinolines and artemisinin: Chemistry, biology and history. Curr. Top Microbiol. Immunol. 2005, 295, 3–38. [Google Scholar] [PubMed]
- Morris, C.A.; Duparc, S.; Borghini-Fuhrer, I.; Jung, D.; Shin, C.-S.; Fleckenstein, L. Review of the clinical pharmacokinetics of artesunate and its active metabolite dihydroartemisinin following intravenous, intramuscular, oral or rectal administration. Malar. J. 2011, 10, 1–17. [Google Scholar]
- Yeung, S.; Socheat, D.; Moorthy, V.S.; Mills, A.J. Artemisinin resistance on the Thai–Cambodian border. Lancet 2009, 374, 1418–1419. [Google Scholar] [PubMed]
- White, N.J. Counter perspective: Artemisinin resistance: Facts, fears, and fables. Am. J. Trop. Med. Hyg. 2012, 87, 785. [Google Scholar]
- Baker, D.A.; Drought, L.G.; Flueck, C.; Nofal, S.D.; Patel, A.; Penzo, M.; Walker, E.M. Cyclic nucleotide signalling in malaria parasites. Open Biol. 2017, 7, 170213. [Google Scholar]
- Balikagala, B.; Fukuda, N.; Ikeda, M.; Katuro, O.T.; Tachibana, S.-I.; Yamauchi, M.; Opio, W.; Emoto, S.; Anywar, D.A.; Kimura, E.; et al. Evidence of artemisinin-resistant malaria in Africa. N. Engl. J. Med. 2021, 385, 1163–1171. [Google Scholar]
- Berzosa, P.; de la Fuente, I.M.; Ta-Tang, T.-H.; González, V.; García, L.; Rodríguez-Galet, A.; Díaz-Regañón, R.; Galán, R.; Cerrada-Gálvez, L.; Ncogo, P.; et al. Temporal evolution of the resistance genotypes of Plasmodium falciparum in isolates from Equatorial Guinea during 20 years (1999 to 2019). Malar. J. 2021, 20, 1–17. [Google Scholar]
- Egwu, C.O.; Augereau, J.-M.; Reybier, K.; Benoit-Vical, F. Reactive oxygen species as the brainbox in malaria treatment. Antioxidants 2021, 10, 1872. [Google Scholar] [CrossRef]
- Heller, L.E. Investigating the Mechanism of Action of Artemisinin Antimalarials and the Role of Ferriprotoporphyrin IX Heme; Georgetown University: Washington, DC, USA, 2019. [Google Scholar]
- Kong, X.; Feng, J.; Xu, Y.; Yan, G.; Zhou, S. Molecular surveillance of artemisinin resistance-related Pfk13 and pfcrt polymorphisms in imported Plasmodium falciparum isolates reported in eastern China from 2015 to 2019. Malar. J. 2022, 21, 369. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Stahelin, R.V.; Haldar, K. Host targeting of virulence determinants and phosphoinositides in blood stage malaria parasites. Trends Parasitol. 2012, 28, 555–562. [Google Scholar]
- Carter, T.E.; Boulter, A.; Existe, A.; Romain, J.R.; Victor, J.Y.S.; Mulligan, C.J.; Okech, B.A. Artemisinin resistance-associated polymorphisms at the K13-propeller locus are absent in Plasmodium falciparum isolates from Haiti. Am. J. Trop. Med. Hyg. 2015, 92, 552–554. [Google Scholar] [PubMed]
- Lu, F.; Culleton, R.; Zhang, M.; Ramaprasad, A.; von Seidlein, L.; Zhou, H.; Zhu, G.; Tang, J.; Liu, Y.; Wang, W.; et al. Emergence of indigenous artemisinin-resistant Plasmodium falciparum in Africa. N. Engl. J. Med. 2017, 376, 991–993. [Google Scholar] [PubMed]
- Berger, F.; Gomez, G.M.; Sanchez, C.P.; Posch, B.; Planelles, G.; Sohraby, F.; Nunes-Alves, A.; Lanzer, M. pH-dependence of the Plasmodium falciparum chloroquine resistance transporter is linked to the transport cycle. Nat. Commun. 2023, 14, 4234. [Google Scholar] [PubMed]
- Amusengeri, A.; Tata, R.B.; Bishop, Ö.T. Understanding the Pyrimethamine Drug Resistance Mechanism via Combined Molecular Dynamics and Dynamic Residue Network Analysis. Molecules 2020, 25, 904. [Google Scholar] [CrossRef]
- Rasmussen, C.; Alonso, P.; Ringwald, P. Current and emerging strategies to combat antimalarial resistance. Expert Rev. Anti- Infect. Ther. 2022, 20, 353–372. [Google Scholar]
- Belete, T.M. Recent progress in the development of new antimalarial drugs with novel targets. Drug Des. Dev. Ther. 2020, 14, 3875–3889. [Google Scholar]
- Pandey, S.K.; Anand, U.; Siddiqui, W.A.; Tripathi, R. Drug Development Strategies for Malaria: With the Hope for New Antimalarial Drug Discovery—An Update. Adv. Med. 2023, 2023, 5060665. [Google Scholar]
- Zhao, L.; Zhao, J.; Zhong, K.; Tong, A.; Jia, D. Targeted protein degradation: Mechanisms, strategies and application. Signal Transduct. Target. Ther. 2022, 7, 113. [Google Scholar]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar]
- Karpiyevich, M.; Artavanis-Tsakonas, K. Ubiquitin-like modifiers: Emerging regulators of protozoan parasites. Biomolecules 2020, 10, 1403. [Google Scholar] [CrossRef]
- Hyer, M.L.; Milhollen, M.A.; Ciavarri, J.; Fleming, P.; Traore, T.; Sappal, D.; Huck, J.; Shi, J.; Gavin, J.; Brownell, J.; et al. A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat. Med. 2018, 24, 186–193. [Google Scholar] [PubMed]
- Cano, L.H. Decoding Ubiquitination in the Fight Against Malaria: A Network-Based Exploration of E1-E2-E3 Enzyme Triples in Plasmodium Falciparum; Universidade de São Paulo: São Paulo, Brazil, 2023. [Google Scholar]
- Wang, D.; Ma, L.; Wang, B.; Liu, J.; Wei, W. E3 ubiquitin ligases in cancer and implications for therapies. Cancer Metastasis Rev. 2017, 36, 683–702. [Google Scholar] [PubMed]
- Luth, M.R.; Gupta, P.; Ottilie, S.; Winzeler, E.A. Using in vitro evolution and whole genome analysis to discover next generation targets for antimalarial drug discovery. ACS Infect. Dis. 2018, 4, 301–314. [Google Scholar]
- Raedler, L. Velcade (Bortezomib) receives 2 new FDA indications: For retreatment of patients with multiple myeloma and for first-line treatment of patients with mantle-cell lymphoma. Am. Health Drug Benefits 2015, 8, 135. [Google Scholar]
- Kumar, P.; Kumar, P.; Mandal, D.; Velayutham, R. The emerging role of Deubiquitinases (DUBs) in parasites: A foresight review. Front. Cell. Infect. Microbiol. 2022, 12, 985178. [Google Scholar]
- Qin, B.; Chen, X.; Wang, F.; Wang, Y. DUBs in Alzheimer’s disease: Mechanisms and therapeutic implications. Cell Death Discov. 2024, 10, 475. [Google Scholar]
- Pereira, P.H.S.; Curra, C.; Garcia, C.R.S. Ubiquitin proteasome system as a potential drug target for malaria. Curr. Top. Med. Chem. 2018, 18, 315–320. [Google Scholar]
- Jin, J.-O.; Puranik, N.; Bui, Q.T.; Yadav, D.; Lee, P.C.-W. The ubiquitin system: An emerging therapeutic target for lung cancer. Int. J. Mol. Sci. 2021, 22, 9629. [Google Scholar] [CrossRef]
- Yang, Q.; Zhao, J.; Chen, D.; Wang, Y. E3 ubiquitin ligases: Styles, structures and functions. Mol. Biomed. 2021, 2, 1–17. [Google Scholar]
- Mevissen, T.E.; Komander, D. Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar]
- Sun, M.; Zhang, X. Current methodologies in protein ubiquitination characterization: From ubiquitinated protein to ubiquitin chain architecture. Cell Biosci. 2022, 12, 126. [Google Scholar]
- Budenholzer, L.; Cheng, C.L.; Li, Y.; Hochstrasser, M. Proteasome structure and assembly. J. Mol. Biol. 2017, 429, 3500–3524. [Google Scholar] [PubMed]
- Li, H.; Bogyo, M.; da Fonseca, P.C.A. The cryo-EM structure of the Plasmodium falciparum 20S proteasome and its use in the fight against malaria. FEBS J. 2016, 283, 4238–4243. [Google Scholar]
- Sijwali, P.; Rosenthal, P. The Proteolytic Repertoire of Malaria Parasites. In Advances in Malaria Research; Wiley-Blackwell: Hoboken, NJ, USA, 2016; pp. 325–352. [Google Scholar]
- LaMonte, G.M.; Almaliti, J.; Bibo-Verdugo, B.; Keller, L.; Zou, B.Y.; Yang, J.; Antonova-Koch, Y.; Orjuela-Sanchez, P.; Boyle, C.A.; Vigil, E.; et al. Development of a potent inhibitor of the Plasmodium proteasome with reduced mammalian toxicity. J. Med. Chem. 2017, 60, 6721–6732. [Google Scholar]
- Ignatz-Hoover, J.J.; Murphy, E.V.; Driscoll, J.J. Targeting proteasomes in cancer and infectious disease: A parallel strategy to treat malignancies and microbes. Front. Cell. Infect. Microbiol. 2022, 12, 925804. [Google Scholar]
- Xie, S.C.; Dick, L.R.; Gould, A.; Brand, S.; Tilley, L. The proteasome as a target for protozoan parasites. Expert Opin. Ther. Targets 2019, 23, 903–914. [Google Scholar]
- Rao, S.P.; Manjunatha, U.H.; Mikolajczak, S.; Ashigbie, P.G.; Diagana, T.T. Drug discovery for parasitic diseases: Powered by technology, enabled by pharmacology, informed by clinical science. Trends Parasitol. 2023, 39, 260–271. [Google Scholar]
- Reynolds, J.M.; El Bissati, K.; Brandenburg, J.; Günzl, A.; Mamoun, C.B. Antimalarial activity of the anticancer and proteasome inhibitor bortezomib and its analog ZL3B. BMC Clin. Pharmacol. 2007, 7, 13. [Google Scholar]
- Li, H.; Ponder, E.L.; Verdoes, M.; Asbjornsdottir, K.H.; Deu, E.; Edgington, L.E.; Lee, J.T.; Kirk, C.J.; Demo, S.D.; Williamson, K.C.; et al. Validation of the proteasome as a therapeutic target in Plasmodium using an epoxyketone inhibitor with parasite-specific toxicity. Chem. Biol. 2012, 19, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Mardhiyyah, K.; Arif, M.H.; Fitri, L.E.; Winarsih, S.; Chandradikusuma, D.; Nurseta, T. Dihydroeponemycin inhibits ubiquitin proteasome system of Plasmodium falciparum in silico. J. Phys. Conf. Ser. 2019, 1374, 012040. [Google Scholar] [CrossRef]
- Czesny, B.; Goshu, S.; Cook, J.L.; Williamson, K.C. The proteasome inhibitor epoxomicin has potent Plasmodium falciparum gametocytocidal activity. Antimicrob. Agents Chemother. 2009, 53, 4080–4085. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Winzeler, E.A. New targets for antimalarial drug discovery. Curr. Opin. Microbiol. 2022, 70, 102220. [Google Scholar] [CrossRef] [PubMed]
- Hatabu, T.; Hagiwara, M.; Taguchi, N.; Kiyozawa, M.; Suzuki, M.; Kano, S.; Sato, K. Plasmodium falciparum: The fungal metabolite gliotoxin inhibits proteasome proteolytic activity and exerts a plasmodicidal effect on P. falciparum. Exp. Parasitol. 2006, 112, 179–183. [Google Scholar] [PubMed]
- Staszczak, M. Fungal secondary metabolites as inhibitors of the ubiquitin–proteasome system. Int. J. Mol. Sci. 2021, 22, 13309. [Google Scholar] [CrossRef]
- Gantt, S.M.; Myung, J.M.; Briones, M.R.; Li, W.D.; Corey, E.; Omura, S.; Nussenzweig, V.; Sinnis, P. Proteasome inhibitors block development of Plasmodium spp. Antimicrob. Agents Chemother. 1998, 42, 2731–2738. [Google Scholar]
- Aminake, M.N.; Arndt, H.-D.; Pradel, G. The proteasome of malaria parasites: A multi-stage drug target for chemotherapeutic intervention? Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 1–10. [Google Scholar]
- Prudhomme, J.; McDaniel, E.; Ponts, N.; Bertani, S.; Fenical, W.; Jensen, P.; Le Roch, K. Marine actinomycetes: A new source of compounds against the human malaria parasite. PLoS ONE 2008, 3, e2335. [Google Scholar]
- Lindenthal, C.; Weich, N.; Chia, Y.-S.; Heussler, V.; Klinkert, M.-Q. The proteasome inhibitor MLN-273 blocks exoerythrocytic and erythrocytic development of Plasmodium parasites. Parasitology 2005, 131, 37–44. [Google Scholar] [CrossRef]
- Tschan, S.; Mordmüller, B.; Kun, J.F. Threonine peptidases as drug targets against malaria. Expert Opin. Ther. Targets 2011, 15, 365–378. [Google Scholar]
- Kreidenweiss, A.; Kremsner, P.G.; Mordmüller, B. Comprehensive study of proteasome inhibitors against Plasmodium falciparum laboratory strains and field isolates from Gabon. Malar. J. 2008, 7, 178. [Google Scholar]
- Prasad, R.; Atul, N.; Kolla, V.K.; Legac, J.; Singhal, N.; Navale, R.; Rosenthal, P.J.; Sijwali, P.S. Blocking Plasmodium falciparum development via dual inhibition of hemoglobin degradation and the ubiquitin proteasome system by MG132. PLoS ONE 2013, 8, e73530. [Google Scholar] [CrossRef] [PubMed]
- Muchamuel, T.; Basler, M.; Aujay, M.A.; Suzuki, E.; Kalim, K.W.; Lauer, C.; Sylvain, C.; Ring, E.R.; Shields, J.; Jiang, J.; et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009, 15, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Huber, E.M.; Heinemeyer, W.; Groll, M. Bortezomib-resistant mutant proteasomes: Structural and biochemical evaluation with carfilzomib and ONX 0914. Structure 2015, 23, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.-C.; Li, D.; Zhan, W.; Ye, J.; Liu, Y.J.; Leung, A.; Qin, J.; Crespo, B.; Gamo, F.-J.; Zhang, H.; et al. Structures revealing mechanisms of resistance and collateral sensitivity of Plasmodium falciparum to proteasome inhibitors. Nat. Commun. 2023, 14, 8302. [Google Scholar] [CrossRef]
- Tallorin, L.; Durrant, J.D.; Nguyen, Q.G.; McCammon, J.A.; Burkart, M.D. Celastrol inhibits Plasmodium falciparum enoyl-acyl carrier protein reductase. Bioorganic Med. Chem. 2014, 22, 6053–6061. [Google Scholar] [CrossRef]
- Ng, J.P.L.; Han, Y.; Yang, L.J.; Birkholtz, L.-M.; Coertzen, D.; Wong, H.N.; Haynes, R.K.; Coghi, P.; Wong, V.K.W. Antimalarial and antitumour activities of the steroidal quinone-methide celastrol and its combinations with artemiside, artemisone and methylene blue. Front. Pharmacol. 2022, 13, 988748. [Google Scholar] [CrossRef]
- Stokes, B.H.; Yoo, E.; Murithi, J.M.; Luth, M.R.; Afanasyev, P.; da Fonseca, P.C.A.; Winzeler, E.A.; Ng, C.L.; Bogyo, M.; Fidock, D.A. Covalent Plasmodium falciparum-selective proteasome inhibitors exhibit a low propensity for generating resistance in vitro and synergize with multiple antimalarial agents. PLoS Pathog. 2019, 15, e1007722. [Google Scholar] [CrossRef]
- Li, H.; O’Donoghue, A.J.; Van Der Linden, W.A.; Xie, S.C.; Yoo, E.; Foe, I.T.; Tilley, L.; Craik, C.S.; Da Fonseca, P.C.A.; Bogyo, M. Structure-and function-based design of Plasmodium-selective proteasome inhibitors. Nature 2016, 530, 233–236. [Google Scholar] [CrossRef]
- Martinez-Fonts, K.; Davis, C.; Tomita, T.; Elsasser, S.; Nager, A.R.; Shi, Y.; Finley, D.; Matouschek, A. The proteasome 19S cap and its ubiquitin receptors provide a versatile recognition platform for substrates. Nat. Commun. 2020, 11, 477. [Google Scholar] [CrossRef]
- Yao, H.; Xu, J. Regulation of cancer immune checkpoint: Mono-and poly-ubiquitination: Tags for fate. Adv. Exp. Med. Biol. 2020, 1248, 295–324. [Google Scholar]
- Horrocks, P.; Newbold, C.I. Intraerythrocytic polyubiquitin expression in Plasmodium falciparum is subjected to developmental and heat-hock control. Mol. Biochem. Parasitol. 2000, 105, 115–125. [Google Scholar] [PubMed]
- Kumar, T.; Maitra, S.; Rahman, A.; Bhattacharjee, S. A conserved guided entry of tail-anchored pathway is involved in the trafficking of a subset of membrane proteins in Plasmodium falciparum. PLoS Pathog. 2021, 17, e1009595. [Google Scholar]
- Bozdech, Z.; Llinás, M.; Pulliam, B.L.; Wong, E.D.; Zhu, J.; DeRisi, J.L. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biol. 2003, 1, e5. [Google Scholar] [CrossRef]
- Le Roch, K.G.; Zhou, Y.; Blair, P.L.; Grainger, M.; Moch, J.K.; Haynes, J.D.; De la Vega, P.; Holder, A.A.; Batalov, S.; Carucci, D.J.; et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science 2003, 301, 1503–1508. [Google Scholar]
- Ponts, N.; Yang, J.; Chung, D.-W.D.; Prudhomme, J.; Girke, T.; Horrocks, P.; Le Roch, K.G. Deciphering the ubiquitin-mediated pathway in apicomplexan parasites: A potential strategy to interfere with parasite virulence. PLoS ONE 2008, 3, e2386. [Google Scholar]
- Jain, J.; Jain, S.K.; Walker, L.A.; Tekwani, B.L. Inhibitors of ubiquitin E3 ligase as potential new antimalarial drug leads. BMC Pharmacol. Toxicol. 2017, 18, 40. [Google Scholar] [CrossRef]
- Shi, D.; Grossman, S.R. Ubiquitin becomes ubiquitous in cancer: Emerging roles of ubiquitin ligases and deubiquitinases in tumorigenesis and as therapeutic targets. Cancer Biol. Ther. 2010, 10, 737–747. [Google Scholar]
- Philip, N.; Haystead, T.A. Characterization of a UBC13 kinase in Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2007, 104, 7845–7850. [Google Scholar] [CrossRef]
- Canner, J.A.; Sobo, M.; Ball, S.; Hutzen, B.; DeAngelis, S.; Willis, W.; Studebaker, A.W.; Ding, K.; Wang, S.; Yang, D.; et al. MI-63: A novel small-molecule inhibitor targets MDM2 and induces apoptosis in embryonal and alveolar rhabdomyosarcoma cells with wild-type p53. Br. J. Cancer 2009, 101, 774–781. [Google Scholar] [CrossRef]
- Hartmann-Petersen, R.; Gordon, C. Protein degradation: Recognition of ubiquitinylated substrates. Curr. Biol. 2004, 14, R754–R756. [Google Scholar]
- Madura, K. Rad23 and Rpn10: Perennial wallflowers join the mêlée. Trends Biochem. Sci. 2004, 29, 637–640. [Google Scholar] [PubMed]
- Welchman, R.L.; Gordon, C.; Mayer, R.J. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat. Rev. Mol. Cell Biol. 2005, 6, 599–609. [Google Scholar] [PubMed]
- Bedford, L.; Paine, S.; Sheppard, P.W.; Mayer, R.J.; Roelofs, J. Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 2010, 20, 391–401. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Yamashita, K.; Tane, K.; Kizu, R.; Ohta, T.; Matsunaga, S.; Fusetani, N.; Kawahara, H.; Yokosawa, H. Girolline, an antitumor compound isolated from a sponge, induces G2/M cell cycle arrest and accumulation of polyubiquitinated p53. Biol. Pharm. Bull. 2004, 27, 699–701. [Google Scholar]
- Benoit-Vical, F.; Saléry, M.; Soh, P.N.; Ahond, A.; Poupat, C. Girolline: A potential lead structure for antiplasmodial drug research. Planta Med. 2008, 74, 438–444. [Google Scholar] [CrossRef]
- Gademann, K.; Kobylinska, J. Antimalarial natural products of marine and freshwater origin. Chem. Rec. 2009, 9, 187–198. [Google Scholar]
- Babu, S.A.; Padmavathi, R.; Aslam, N.A.; Rajkumar, V. Recent developments on the synthesis and applications of natural products-inspired spirooxindole frameworks. Stud. Nat. Prod. Chem. 2015, 46, 227–339. [Google Scholar]
- Boulet, C.; Gaynor, T.L.; Carvalho, T.G. Eryptosis and malaria: New experimental guidelines and re-evaluation of the antimalarial potential of eryptosis inducers. Front. Cell. Infect. Microbiol. 2021, 11, 630812. [Google Scholar]
- Bansal, M.; Upadhyay, C.; Poonam; Kumar, S.; Rathi, B. Phthalimide analogs for antimalarial drug discovery. RSC Med. Chem. 2021, 12, 1854–1867. [Google Scholar]
- Lee, J.; Lee, Y.; Jung, Y.M.; Park, J.H.; Yoo, H.S.; Park, J. Discovery of E3 ligase ligands for target protein degradation. Molecules 2022, 27, 6515. [Google Scholar] [CrossRef]
- Bedford, L.; Lowe, J.; Dick, L.R.; Mayer, R.J.; Brownell, J.E. Ubiquitin-like protein conjugation and the ubiquitin–proteasome system as drug targets. Nat. Rev. Drug Discov. 2011, 10, 29–46. [Google Scholar] [PubMed]
- Xie, Y. Structure, assembly and homeostatic regulation of the 26S proteasome. J. Mol. Cell Biol. 2010, 2, 308–317. [Google Scholar]
- Kraut, D.A.; Prakash, S.; Matouschek, A. To degrade or release: Ubiquitin-chain remodeling. Trends Cell Biol. 2007, 17, 419–421. [Google Scholar] [PubMed]
- Ponder, E.L.; Bogyo, M. Ubiquitin-like modifiers and their deconjugating enzymes in medically important parasitic protozoa. Eukaryot. Cell 2007, 6, 1943–1952. [Google Scholar]
- Artavanis-Tsakonas, K.; Misaghi, S.; Comeaux, C.A.; Catic, A.; Spooner, E.; Duraisingh, M.T.; Ploegh, H.L. Identification by functional proteomics of a deubiquitinating/deNeddylating enzyme in Plasmodium falciparum. Mol. Microbiol. 2006, 61, 1187–1195. [Google Scholar] [CrossRef]
- Artavanis-Tsakonas, K.; Weihofen, W.A.; Antos, J.M.; Coleman, B.I.; Comeaux, C.A.; Duraisingh, M.T.; Gaudet, R.; Ploegh, H.L. Characterization and structural studies of the Plasmodium falciparum ubiquitin and Nedd8 hydrolase UCHL3. J. Biol. Chem. 2010, 285, 6857–6866. [Google Scholar]
- Frickel, E.-M.; Quesada, V.; Muething, L.; Gubbels, M.-J.; Spooner, E.; Ploegh, H.; Artavanis-Tsakonas, K. Apicomplexan UCHL3 retains dual specificity for ubiquitin and Nedd8 throughout evolution. Cell. Microbiol. 2007, 9, 1601–1610. [Google Scholar]
- Rosenthal, P.J. Falcipains and other cysteine proteases of malaria parasites. Adv. Exp. Med. Biol. 2011, 712, 30–48. [Google Scholar]
- Daviet, L.; Colland, F. Targeting ubiquitin specific proteases for drug discovery. Biochimie 2008, 90, 270–283. [Google Scholar]
- Aurrecoechea, C.; Brestelli, J.; Brunk, B.P.; Dommer, J.; Fischer, S.; Gajria, B.; Gao, X.; Gingle, A.; Grant, G.; Harb, O.S.; et al. PlasmoDB: A functional genomic database for malaria parasites. Nucleic Acids Res. 2009, 37, D539–D543. [Google Scholar]
- Ngwa, C.J.; Scheuermayer, M.; Mair, G.R.; Kern, S.; Brügl, T.; Wirth, C.C.; Aminake, M.N.; Wiesner, J.; Fischer, R.; Vilcinskas, A.; et al. Changes in the transcriptome of the malaria parasite Plasmodium falciparum during the initial phase of transmission from the human to the mosquito. BMC Genom. 2013, 14, 256. [Google Scholar] [CrossRef] [PubMed]
- Fenteany, G.; Standaert, R.F.; Lane, W.S.; Choi, S.; Corey, E.J.; Schreiber, S.L. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 1995, 268, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Gulder, T.A.M.; Moore, B.S. Salinosporamide natural products: Potent 20 S proteasome inhibitors as promising cancer chemotherapeutics. Angew. Chem. Int. Ed. Engl. 2010, 49, 9346–9367. [Google Scholar] [CrossRef] [PubMed]
- Fitri, L.E.; Cahyono, A.W.; Nugraha, R.Y.B.; Alkarimah, A.; Ramadhani, N.N.; Laksmi, D.A.; Trianty, L.; Noviyanti, R. Analysis of eponemycin (α’β’epoxyketone) analog compound from Streptomyces hygroscopicus subsp hygroscopicus extracts and its antiplasmodial activity during plasmodium berghei infection. Biomed. Res. 2017, 28, 164–172. [Google Scholar]
- Hanada, M.; Sugawara, K.; Kaneta, K.; Toda, S.; Nishiyama, Y.; Tomita, K.; Yamamoto, H.; Konishi, M.; Oki, T. Epoxomicin, a new antitumor agent of microbial origin. J. Antibiot. 1992, 45, 1746–1752. [Google Scholar] [CrossRef]
- Bibo-Verdugo, B.; Jiang, Z.; Caffrey, C.R.; O’Donoghue, A.J. Targeting proteasomes in infectious organisms to combat disease. FEBS J. 2017, 284, 1503–1517. [Google Scholar] [CrossRef]
- Dogovski, C.; Xie, S.C.; Burgio, G.; Bridgford, J.; Mok, S.; McCaw, J.M.; Chotivanich, K.; Kenny, S.; Gnädig, N.; Straimer, J.; et al. Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS Biol. 2015, 13, e1002132. [Google Scholar] [CrossRef]
- Rosenthal, M.; Ng, C. Parasite proteostasis and artemisinin resistance. Res. Sq. 2023. [Google Scholar] [CrossRef]
- Bozdech, Z.; Ferreira, P.E.; Mok, S. A crucial piece in the puzzle of the artemisinin resistance mechanism in Plasmodium falciparum. Trends Parasitol. 2015, 31, 345–346. [Google Scholar] [CrossRef]
- BoáKim, K. From epoxomicin to carfilzomib: Chemistry, biology, and medical outcomes. Nat. Prod. Rep. 2013, 30, 600–604. [Google Scholar]
- Paul, G. Ubiquitin–Proteasome System as a Potential Drug Target for Malaria. In Drug Targets for Plasmodium Falciparum: Historic to Future Perspectives; Springer: Berlin/Heidelberg, Germany, 2024; pp. 167–182. [Google Scholar]
- Tan, L.T.; Phyo, M.Y. Marine cyanobacteria: A source of lead compounds and their clinically-relevant molecular targets. Molecules 2020, 25, 2197. [Google Scholar] [CrossRef] [PubMed]
- Palmer, J.T.; Rasnick, D.; Klaus, J.L.; Bromme, D. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J. Med. Chem. 1995, 38, 3193–3196. [Google Scholar]
- Bogyo, M.; McMaster, J.S.; Gaczynska, M.; Tortorella, D.; Goldberg, A.L.; Ploegh, H. Covalent modification of the active site threonine of proteasomal β subunits and the Escherichia coli homolog HslV by a new class of inhibitors. Proc. Natl. Acad. Sci. USA 1997, 94, 6629–6634. [Google Scholar] [PubMed]
- Tschan, S.; Brouwer, A.J.; Werkhoven, P.R.; Jonker, A.M.; Wagner, L.; Knittel, S.; Aminake, M.N.; Pradel, G.; Joanny, F.; Liskamp, R.M.J.; et al. Broad-spectrum antimalarial activity of peptido sulfonyl fluorides, a new class of proteasome inhibitors. Antimicrob. Agents Chemother. 2013, 57, 3576–3584. [Google Scholar] [PubMed]
- Vinitsky, A.; Michaud, C.; Powers, J.C.; Orlowski, M. Inhibition of the chymotrypsin-like activity of the pituitary multicatalytic proteinase complex. Biochemistry 1992, 31, 9421–9428. [Google Scholar]
- Tsubuki, S.; Kawasaki, H.; Saito, Y.; Miyashita, N.; Inomata, M.; Kawashima, S. Purification and characterization of a Z-Leu-Leu-Leu-MCA degrading protease expected to regulate neurite formation: A novel catalytic activity in proteasome. Biochem. Biophys. Res. Commun. 1993, 196, 1195–1201. [Google Scholar]
- Rock, K.L.; Gramm, C.; Rothstein, L.; Clark, K.; Stein, R.; Dick, L.; Hwang, D.; Goldberg, A.L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994, 78, 761–771. [Google Scholar]
- Coux, O.; Tanaka, K.; Goldberg, A.L. Structure and functions of the 20S and 26S proteasomes. Annu. Rev. Biochem. 1996, 65, 801–847. [Google Scholar]
- Oerlemans, R.; Franke, N.E.; Assaraf, Y.G.; Cloos, J.; van Zantwijk, I.; Berkers, C.R.; Scheffer, G.L.; Debipersad, K.; Vojtekova, K.; Lemos, C.; et al. Molecular basis of bortezomib resistance: Proteasome subunit β5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood 2008, 112, 2489–2499. [Google Scholar] [CrossRef]
- Ge, Y.; Li, A.; Wu, J.; Feng, H.; Wang, L.; Liu, H.; Xu, Y.; Xu, Q.; Zhao, L.; Li, Y. Design, synthesis and biological evaluation of novel non-peptide boronic acid derivatives as proteasome inhibitors. Eur. J. Med. Chem. 2017, 128, 180–191. [Google Scholar]
- de Wilt, L.H.; Jansen, G.; Assaraf, Y.G.; van Meerloo, J.; Cloos, J.; Schimmer, A.D.; Chan, E.T.; Kirk, C.J.; Peters, G.J.; Kruyt, F.A. Proteasome-based mechanisms of intrinsic and acquired bortezomib resistance in non-small cell lung cancer. Biochem. Pharmacol. 2012, 83, 207–217. [Google Scholar] [PubMed]
- Moore, B.S.; Eustáquio, A.S.; McGlinchey, R.P. Advances in and applications of proteasome inhibitors. Curr. Opin. Chem. Biol. 2008, 12, 434–440. [Google Scholar] [PubMed]
- Chauhan, D.; Hideshima, T.; Anderson, K.C. Proteasome inhibition in multiple myeloma: Therapeutic implication. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 465–476. [Google Scholar] [PubMed]
- Richardson, P.G.; Mitsiades, C.; Hideshima, T.; Anderson, K.C. Bortezomib: Proteasome inhibition as an effective anticancer therapy. Annu. Rev. Med. 2006, 57, 33–47. [Google Scholar]
- Chauhan, D.; Catley, L.; Li, G.; Podar, K.; Hideshima, T.; Velankar, M.; Mitsiades, C.; Mitsiades, N.; Yasui, H.; Letai, A.; et al. A novel orally active proteasome inhibitor induces apoptosis in multiple myeloma cells with mechanisms distinct from Bortezomib. Cancer cell 2005, 8, 407–419. [Google Scholar]
- Teixeira, C.; Vale, N.; Perez, B.; Gomes, A.; Gomes, J.R.; Gomes, P. “Recycling” classical drugs for malaria. Chem. Rev. 2014, 114, 11164–11220. [Google Scholar]
- Schwegmann, A.; Brombacher, F. Host-directed drug targeting of factors hijacked by pathogens. Sci. Signal. 2008, 1, re8. [Google Scholar]
- Dolloff, N.G. Emerging therapeutic strategies for overcoming proteasome inhibitor resistance. Adv. Cancer Res. 2015, 127, 191–226. [Google Scholar]
- Koguchi, Y.; Kohno, J.; Nishio, M.; Takahashi, K.; Okuda, T.; Ohnuki, T.; Komatsubara, S. TMC-95A, B, C, and D, novel proteasome inhibitors produced by Apiospora montagnei Sacc. TC 1093. Taxonomy, Production, Isolation, and Biological Activities. J. Antibiot. 2000, 53, 105–109. [Google Scholar]
- Li, H.; Tsu, C.; Blackburn, C.; Li, G.; Hales, P.; Dick, L.; Bogyo, M. Identification of potent and selective non-covalent inhibitors of the Plasmodium falciparum proteasome. J. Am. Chem. Soc. 2014, 136, 13562–13565. [Google Scholar]
- Wilson, D.L.; Meininger, I.; Strater, Z.; Steiner, S.; Tomlin, F.; Wu, J.; Jamali, H.; Krappmann, D.; Götz, M.G. Synthesis and evaluation of macrocyclic peptide aldehydes as potent and selective inhibitors of the 20S proteasome. ACS Med. Chem. Lett. 2016, 7, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Zhan, W.; Visone, J.; Ouellette, T.; Harris, J.C.; Wang, R.; Zhang, H.; Singh, P.K.; Ginn, J.; Sukenick, G.; Wong, T.-T.; et al. Improvement of asparagine ethylenediamines as anti-malarial Plasmodium-selective proteasome inhibitors. J. Med. Chem. 2019, 62, 6137–6145. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, L.A.; Zhan, W.; Visone, J.; Dziedziech, A.; Singh, P.K.; Fan, H.; Tong, X.; Bruzual, I.; Hara, R.; Kawasaki, M.; et al. Antimalarial proteasome inhibitor reveals collateral sensitivity from intersubunit interactions and fitness cost of resistance. Proc. Natl. Acad. Sci. USA 2018, 115, E6863–E6870. [Google Scholar] [CrossRef]
- Xin, B.-T.; De Bruin, G.; Huber, E.M.; Besse, A.; Florea, B.I.; Filippov, D.V.; Van Der Marel, G.A.; Kisselev, A.F.; Van Der Stelt, M.; Driessen, C.; et al. Structure-based design of β5c selective inhibitors of human constitutive proteasomes. J. Med. Chem. 2016, 59, 7177–7187. [Google Scholar] [CrossRef]
- Guedes, R.A.; Serra, P.; Salvador, J.A.; Guedes, R.C. Computational approaches for the discovery of human proteasome inhibitors: An overview. Molecules 2016, 21, 927. [Google Scholar] [CrossRef]
- Reamtong, O.; Srimuang, K.; Saralamba, N.; Sangvanich, P.; Day, N.P.; White, N.J.; Imwong, M. Protein profiling of mefloquine resistant Plasmodium falciparum using mass spectrometry-based proteomics. Int. J. Mass Spectrom. 2015, 391, 82–92. [Google Scholar] [CrossRef]
- Green, J.L.; Wu, Y.; Encheva, V.; Lasonder, E.; Prommaban, A.; Kunzelmann, S.; Christodoulou, E.; Grainger, M.; Truongvan, N.; Bothe, S.; et al. Ubiquitin activation is essential for schizont maturation in Plasmodium falciparum blood-stage development. PLoS Pathog. 2020, 16, e1008640. [Google Scholar] [CrossRef]
- Das, A.; Rajkhowa, S.; Sinha, S.; Zaki, M.E. Unveiling potential repurposed drug candidates for Plasmodium falciparum through in silico evaluation: A synergy of structure-based approaches, structure prediction, and molecular dynamics simulations. Comput. Biol. Chem. 2024, 110, 108048. [Google Scholar] [CrossRef]
- Tyagi, R.; Srivastava, M.; Jain, P.; Pandey, R.P.; Asthana, S.; Kumar, D.; Raj, V.S. Development of potential proteasome inhibitors against Mycobacterium tuberculosis. J. Biomol. Struct. Dyn. 2020, 40, 2189–2203. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Component | Target in Parasites (Antimalarial) | Target in Humans (Diseases) | Challenges | Ref. |
|---|---|---|---|---|
| Ubiquitin | Disrupt ubiquitin tagging to impair parasite protein homeostasis. | Target ubiquitin-activating enzymes in cancer therapy. | High conservation limits selectivity. | [63,64] |
| E3 Ligases | Inhibit parasite-specific E3 ligases to block protein degradation. | Target E3 ligases (e.g., MDM2) in cancer and inflammation. | Functional redundancy complicates specificity. | [65,66] |
| Proteasome | Use proteasome inhibitors (e.g., Epoxyketones) selective for Plasmodium. | FDA-approved inhibitors (e.g., Bortezomib) for cancer therapy. | Toxicity due to effects on normal human cells. | [67,68] |
| DUBs | Inhibit Plasmodium-specific DUBs to induce proteotoxic stress. | Target DUBs (e.g., USP7, USP14) for cancer and neurodegeneration. | High conservation; off-target effects must be minimized. | [69,70] |
| Compounds | IC50 (µM, In Vitro) | Pharmacokinetics | Class | Water Solubility | Ref | ||
|---|---|---|---|---|---|---|---|
| GI Abs | BBB Perm | Sol | Class | ||||
| Bortezomib | 0.031–0.043 (Pf3D7) | High | No | Peptide boronate | 7.53 × 10−1 | Soluble | [83] |
| Carfilzomib | 0.025 (Pf) | Low | No | Epoxyketone | 1.03 × 10−3 | Moderately soluble | [79,84] |
| Dihydroeponemycin | In silico study | High | No | Epoxyketone | 3.53 × 100 | Soluble | [85] |
| Epoxomicin | 0.054 (gametocyte) 0.041 (asexual stages) | Low | No | Epoxyketone | 2.04 × 10−1 | Soluble | [86,87] |
| Gliotoxin | 2.17 (Pf) | High | No | Non-covalent, reversible | 1.48 × 101 | Very soluble | [88,89] |
| Lactacystin | 1.2–1.5 (Pf blood stages) | Low | No | Β-lactone | 1.47 × 101 | Very soluble | [90,91] |
| Marizomib, NPI-0052, Salinosporamide A | 0.0114 (Pf) | High | No | Β-lactone-γ-lactam | 6.84 × 10−1 | Soluble | [92,93,94] |
| MG-115 | 0.0975 (Pf3D7) | High | No | Peptide aldehyde | 1.78 × 10−2 | Moderately soluble | [95] |
| MG-132 | 0.0476 (Pf) | High | No | Peptide aldehyde | 8.00 × 10−3 | Moderately soluble | [96] |
| ONX-0914 (PR-957) | n.d | High | No | β-lactone | 1.07 × 10−1 | Soluble | [97,98] |
| TDI-8304 | 3.1 (hypnozoite stage) | High | No | Vinyl sulfone | 2.17 × 10−2 | Moderately soluble | [22,99] |
| Tripterin (celastrol) | 0.50–0.82µM (asexual blood stage Pf) | Low | No | Triterpenoid | 2.21 × 10−4 | Poorly soluble | [100,101] |
| WLL_vs | 0.011–0.013 (against diverse strains) | Low | No | Vinyl sulfone | 2.18 × 10−2 | Moderately soluble | [102,103] |
| YU-101 | 0.0245 (Pf3D7) | n.d | n.d | Epoxyketone | n.d | n.d | [95] |
| ZL3B | 0.04 (Pf3D7) | High | No | Peptide boronate | 5.50 × 10−3 | Moderately soluble | [83] |
| Compounds | IC50 (µM, In Vitro) | Pharmacokinetics | Note | Water Solubility | Ref. | ||
|---|---|---|---|---|---|---|---|
| GI Abs | BBB Perm | Sol | Class | ||||
| HLI 373 | 2.36 (CQ-S), 3.47 (CQ-R) | High | Yes | HDM2 inhibitor | 1.75 × 10−2 | Moderately soluble | [111] |
| JNJ-26854165 | 2.17 (CQ-S), 1.86 (CQ-R) | High | Yes | Modulate ubiquitin proteasome pathway | 1.06 × 10−2 | Moderately soluble | [111] |
| MI-219 | n.d | High | No | MDM2 inhibitor | 3.35 × 10−3 | Moderately soluble | [122] |
| MI-63 | 0.58 | High | No | MDM2 inhibitor | 8.94 × 10−4 | Moderately soluble | [91] |
| Nutlin-3A | 12.76 (CQ-S), 18.56 (CQ-R) | High | No | MDM2 inhibitor | 1.73 × 10−4 | Poorly soluble | [111] |
| Oridonin | 2 | High | No | Activate E3 ubiquitin ligase | 2.58 × 100 | Soluble | [123] |
| SMER 3 | 12.06 (CQ-S), 20.58 (CQ-R) | High | Yes | Inhibitor of a yeast SCF family E3 ubiquitin ligase | 5.08 × 10−1 | Soluble | [111] |
| Thalidomide | n.d | High | No | Immunomodulatory | 3.94 × 100 | Very soluble | [111,124,125] |
| Proteasome Subunits | H. sapiens (GenBank) | P. falciparum (PlasmoDB) | Seq Identity (%) | E-Value | ||
|---|---|---|---|---|---|---|
| Accession No. | Seq Length (AA) | Accession No. | Seq Length (AA) | |||
| CP subunits | ||||||
| α type 1 | P25786 | 263 | PF14_0716 | 254 | 44.0 | 7 × 10−71 |
| α type 2 | P25787 | 234 | PFF0420c | 235 | 57.7 | 6 × 10−95 |
| α type 3 | P25788 | 255 | PFC0745c | 252 | 36.2 | 1 × 10−56 |
| α type 4 | P25789 | 261 | PF13_0282 | 246 | 53.4 | 8 × 10−88 |
| α type 5 | P28066 | 241 | PF07_0112 | 256 | 54.4 | 4 × 10−90 |
| α type 6 | P60900 | 246 | MAL8P1.128 | 260 | 44.2 | 4 × 10−76 |
| α type 7 | O14818 | 248 | MAL13P1.270 | 241 | 51.1 | 1 × 10−81 |
| β type 1 | P20618 | 241 | PFE0915c | 240 | 43.2 | 1 × 10−59 |
| β type 2 | P49721 | 201 | PF14_0676 | 195 | 40.2 | 9 × 10−46 |
| β type 3 | P49720 | 205 | PFA0400c | 218 | 44.0 | 4 × 10−65 |
| β type 4 | P28070 | 264 | MAL8P1.142 | 265 | 36.3 | 7 × 10−49 |
| β type 5 | P28074 | 263 | PF10_0111 | 271 | 53.5 | 7 × 10−78 |
| β type 6 | P28072 | 239 | PFI1545c | 282 | 28.5 | 6 × 10−36 |
| β type 7 | Q99436 | 277 | PF13_0156 | 270 | 55.8 | 7 × 10−95 |
| RP subunits (base) | ||||||
| RS 4 or RPT2 | P62191 | 440 | PF10_0081 | 448 | 76.6 | |
| RS 6A or RPT5 | P17980 | 439 | PF11_0314 | 439 | 71.3 | |
| RS 6B or RPT3 | P43686 | 418 | PFD0665c | 392 | 68.6 | |
| RS 7 or RPT1 | P35998 | 433 | PF13_0063 | 420 | 74.7 | |
| RS 8 or RPT6 | P62195 | 406 | PFL2345c | 435 | 77.3 | |
| RS 10B or RPT4 | P62333 | 389 | PF13_0033 | 393 | 68.9 | |
| Non-ATPase RS 1 or RPN2 | Q99460 | 953 | PF14_0632 | 1172 | 38.5 | |
| Non-ATPase RS 2 or RPN1 | Q13200 | 908 | PFB0260w | 966 | 36.1 | |
| Non-ATPase RS 4 or RPN10 | P55036 | 377 | PF08_0109 | 481 | 40.9 | 7 × 10−35 |
| Non-ATPase RS RPN13 | Q16186 | 407 | PF14_0138 | 253 | 33.8 | 1 × 10−17 |
| RP subunits (lid) | ||||||
| Non-ATPase RS 3 or RPN3 | O43242 | 534 | MAL13P1.190 | 503 | 38.8 | 1 × 10−100 |
| Non-ATPase RS 4 or RPN6 | O00231 | 422 | PF14_0025 | 666 | 35.9 | 2 × 10−28 |
| Non-ATPase RS 6 or RPN 7 | Q15008 | 389 | PF11_0303 | 393 | 38.3 | 1 × 10−102 |
| Non-ATPase RS 7 or RPN8 | P51665 | 324 | PFI0630w | 338 | 44.7 | 3 × 10−74 |
| Non-ATPase RS 8 or RPN 12 | P48556 | 350 | PFC0520w | 304 | 35.0 | 1 × 10−33 |
| Non-ATPase RS 12 or RPN5 | O00232 | 456 | PF10_0174 | 467 | 38.6 | 8 × 10−101 |
| Non-ATPase RS 13 or RPN9 | Q9UNM6 | 376 | PF10_0298 | 393 | 26.3 | 8 × 10−37 |
| Non-ATPase RS 14 or RPN 11 | O00487 | 310 | MAL13P1.343 | 311 | 63.0 | 2 × 10−138 |
| Non-ATPase RS RPN 15 or DSS1 | P60896 | 70 | MAL7P1.117 | 106 | 61.0 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanaullah, B.; Truong, N.V.; Nguyen, T.-K.; Han, E.-T. Combating Malaria: Targeting the Ubiquitin-Proteasome System to Conquer Drug Resistance. Trop. Med. Infect. Dis. 2025, 10, 94. https://doi.org/10.3390/tropicalmed10040094
Sanaullah B, Truong NV, Nguyen T-K, Han E-T. Combating Malaria: Targeting the Ubiquitin-Proteasome System to Conquer Drug Resistance. Tropical Medicine and Infectious Disease. 2025; 10(4):94. https://doi.org/10.3390/tropicalmed10040094
Chicago/Turabian StyleSanaullah, Bazgha, Nguyen Van Truong, Tuyet-Kha Nguyen, and Eun-Taek Han. 2025. "Combating Malaria: Targeting the Ubiquitin-Proteasome System to Conquer Drug Resistance" Tropical Medicine and Infectious Disease 10, no. 4: 94. https://doi.org/10.3390/tropicalmed10040094
APA StyleSanaullah, B., Truong, N. V., Nguyen, T.-K., & Han, E.-T. (2025). Combating Malaria: Targeting the Ubiquitin-Proteasome System to Conquer Drug Resistance. Tropical Medicine and Infectious Disease, 10(4), 94. https://doi.org/10.3390/tropicalmed10040094