Isomerization and Properties of Isomers of Carbocyanine Dyes


Abstract
:1. Introduction
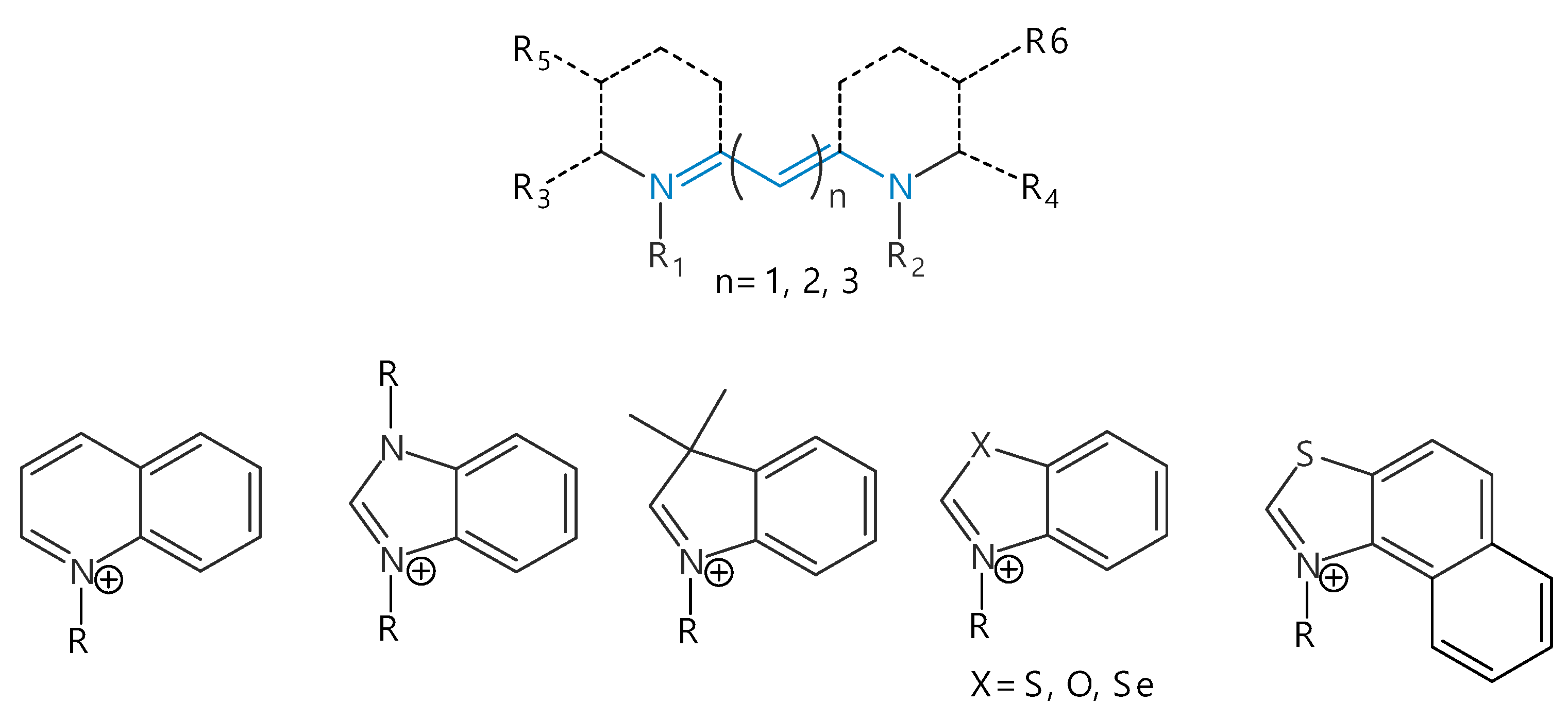
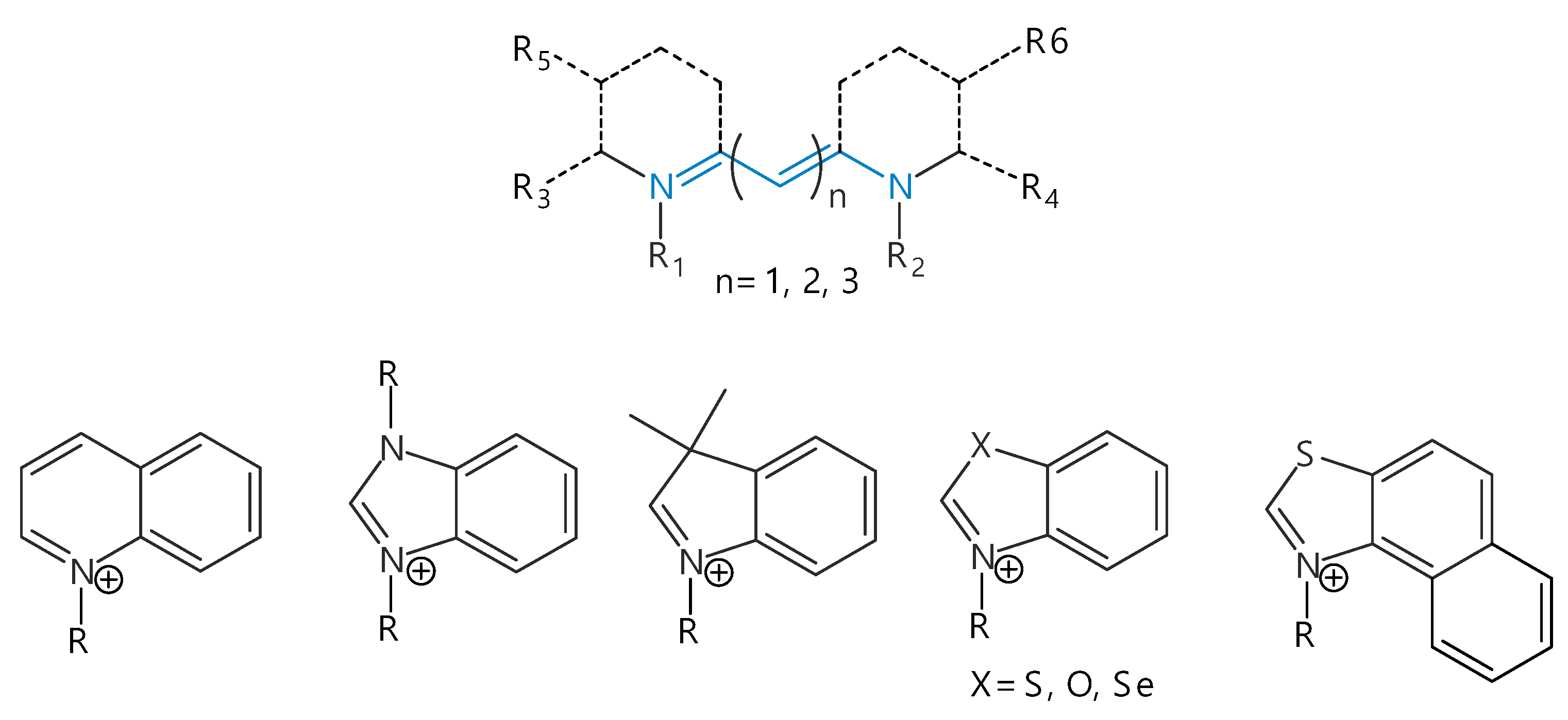
2. Structure and Spectral-Fluorescent Properties of Cyanine Dyes
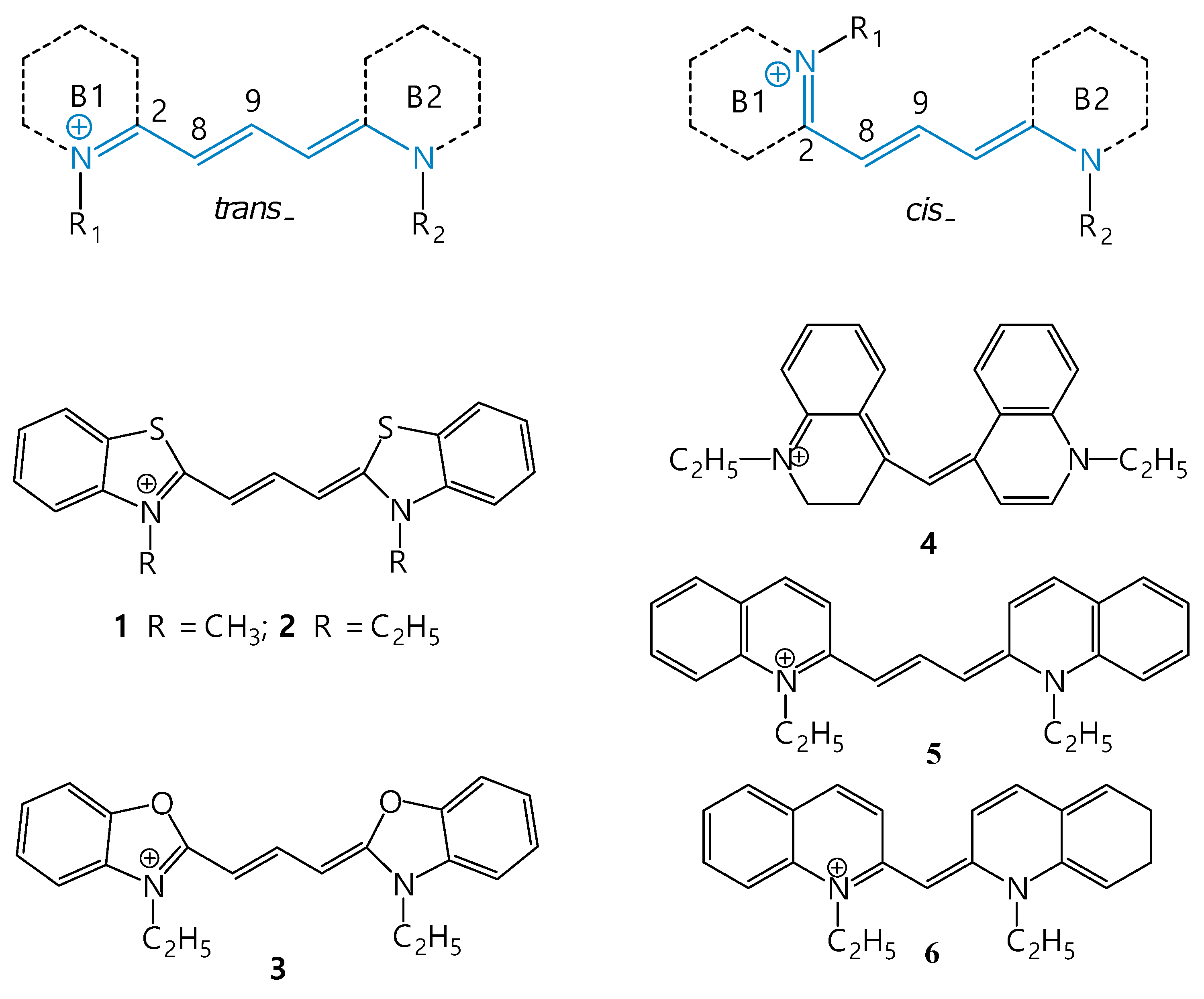
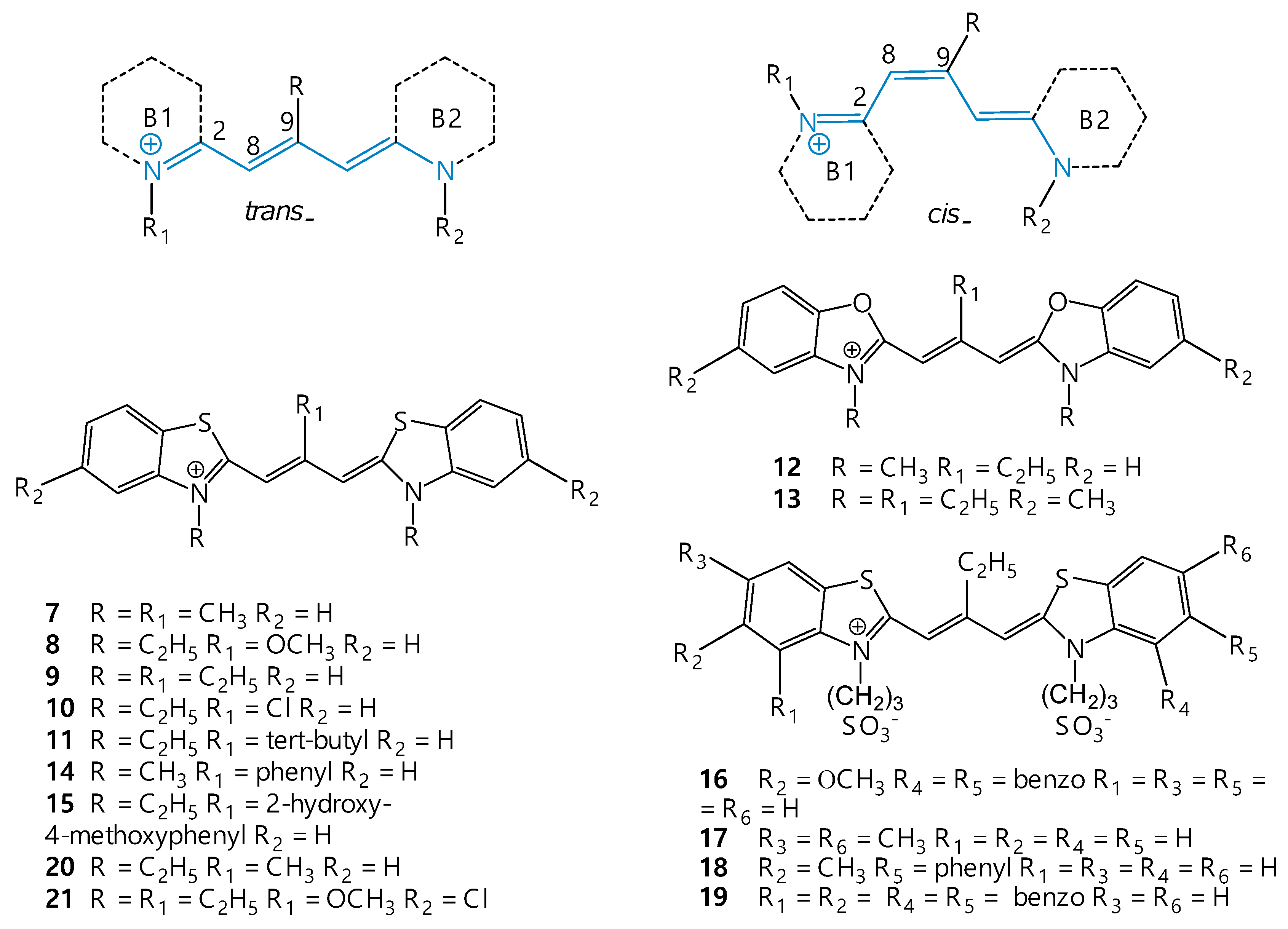
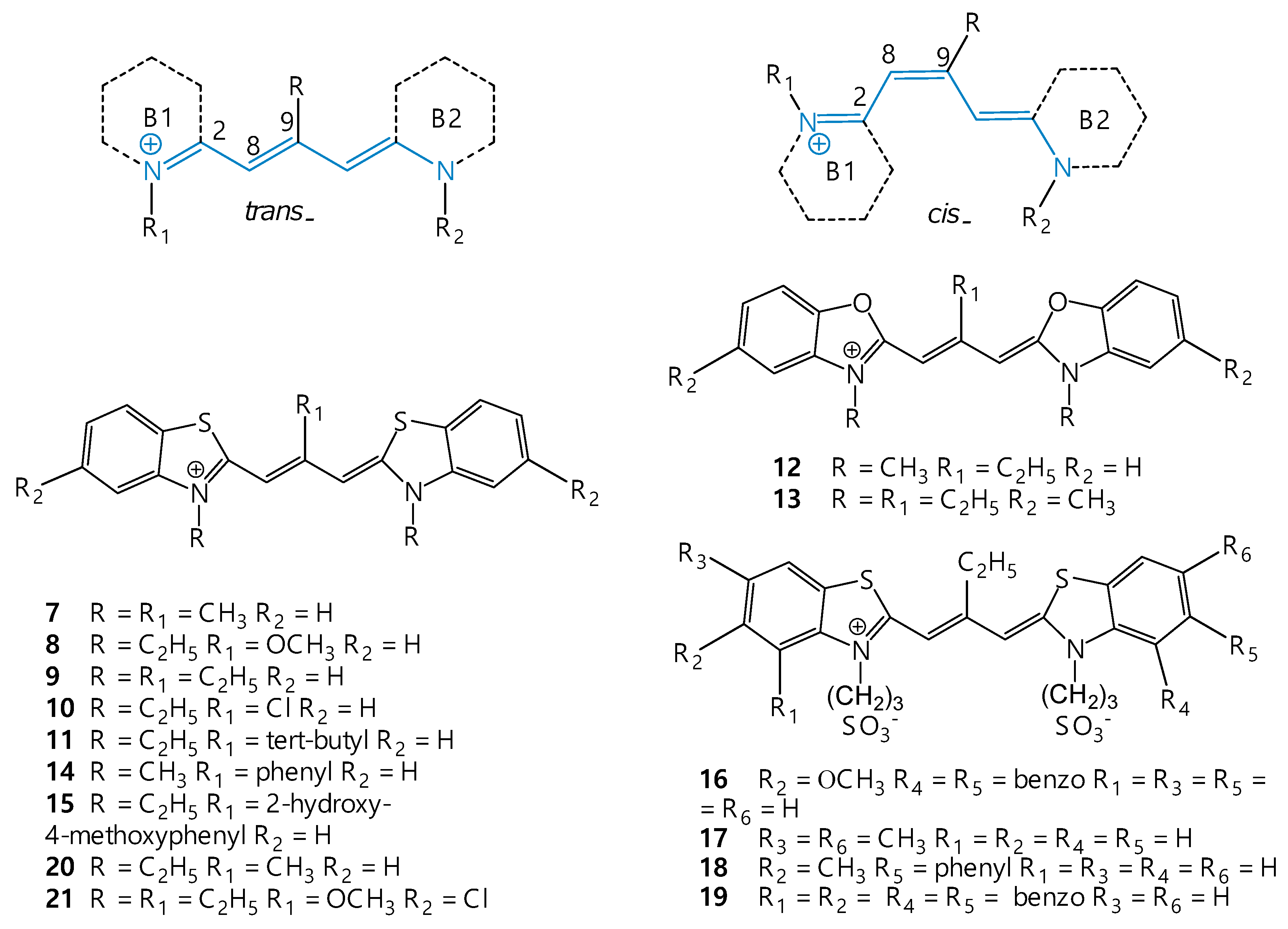
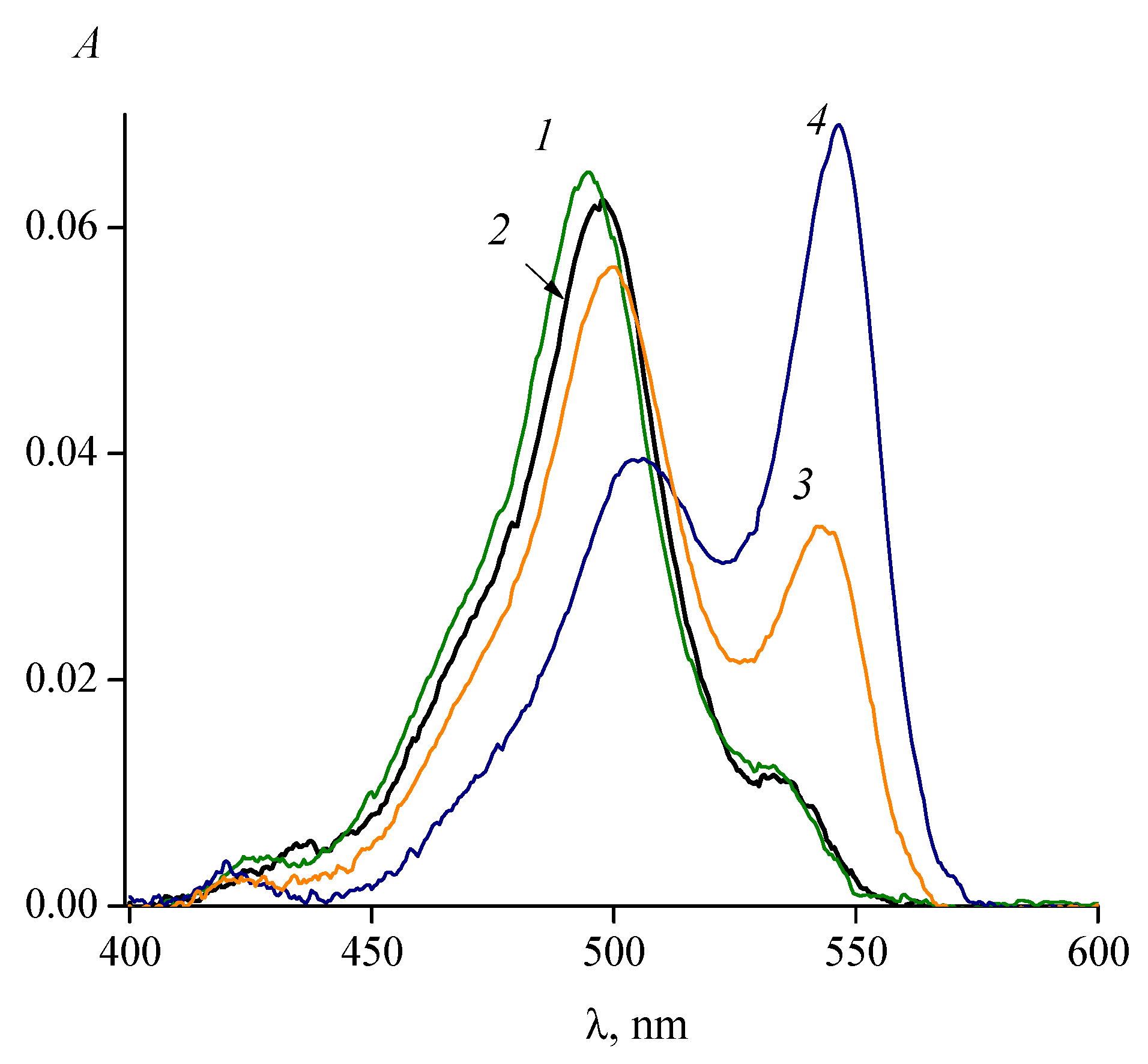
3. Isomerization of Carbocyanine Dyes
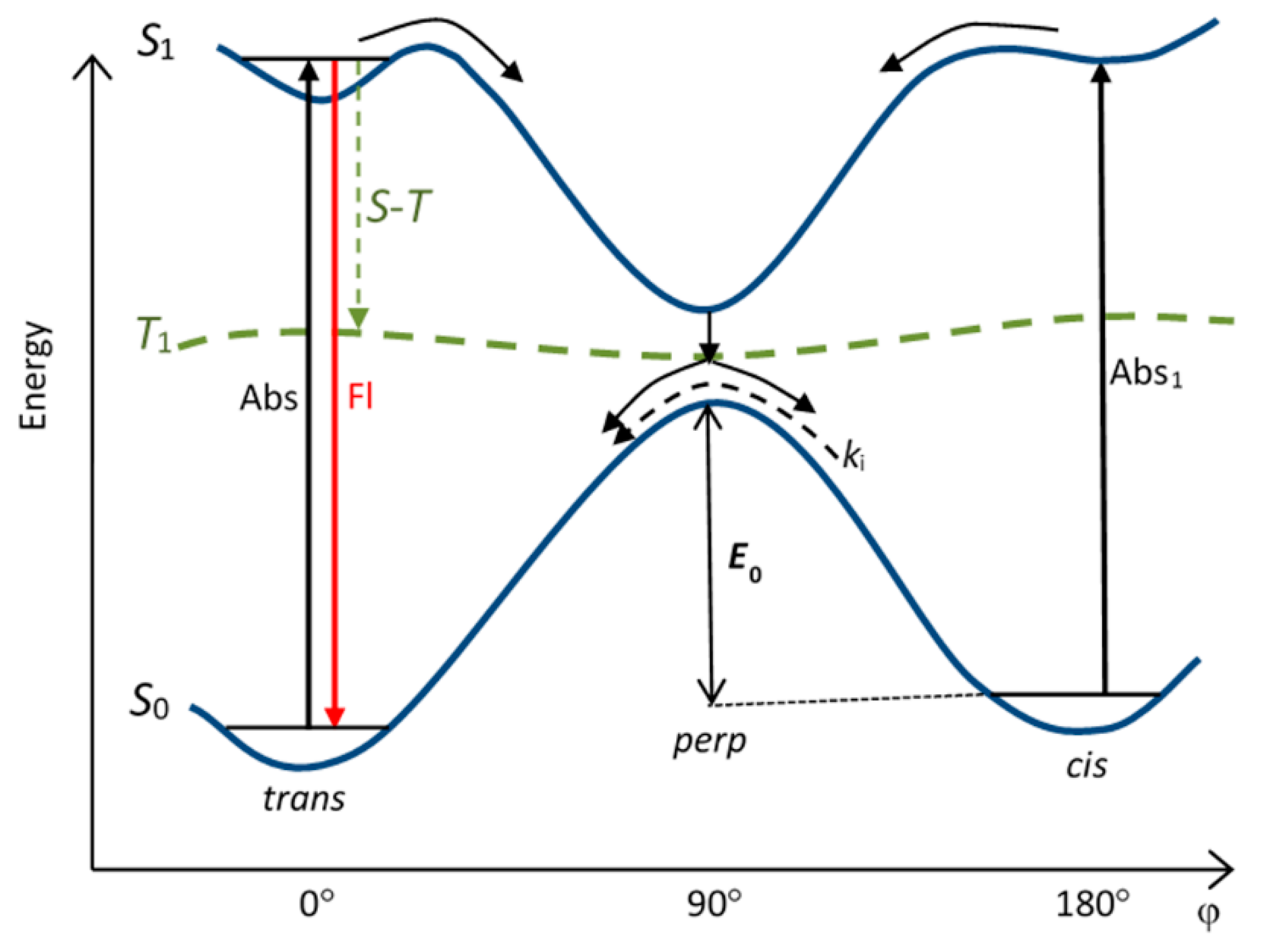
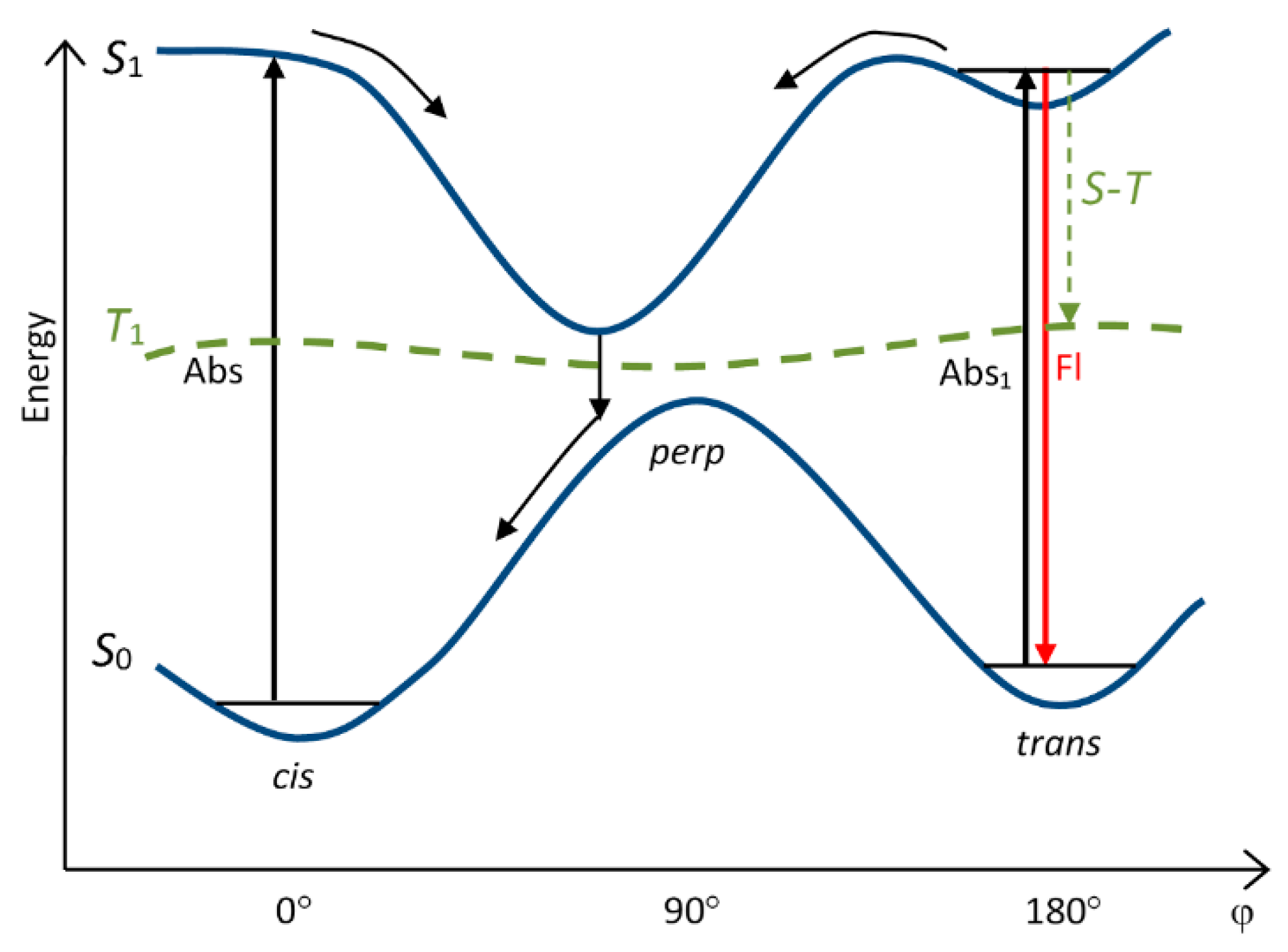
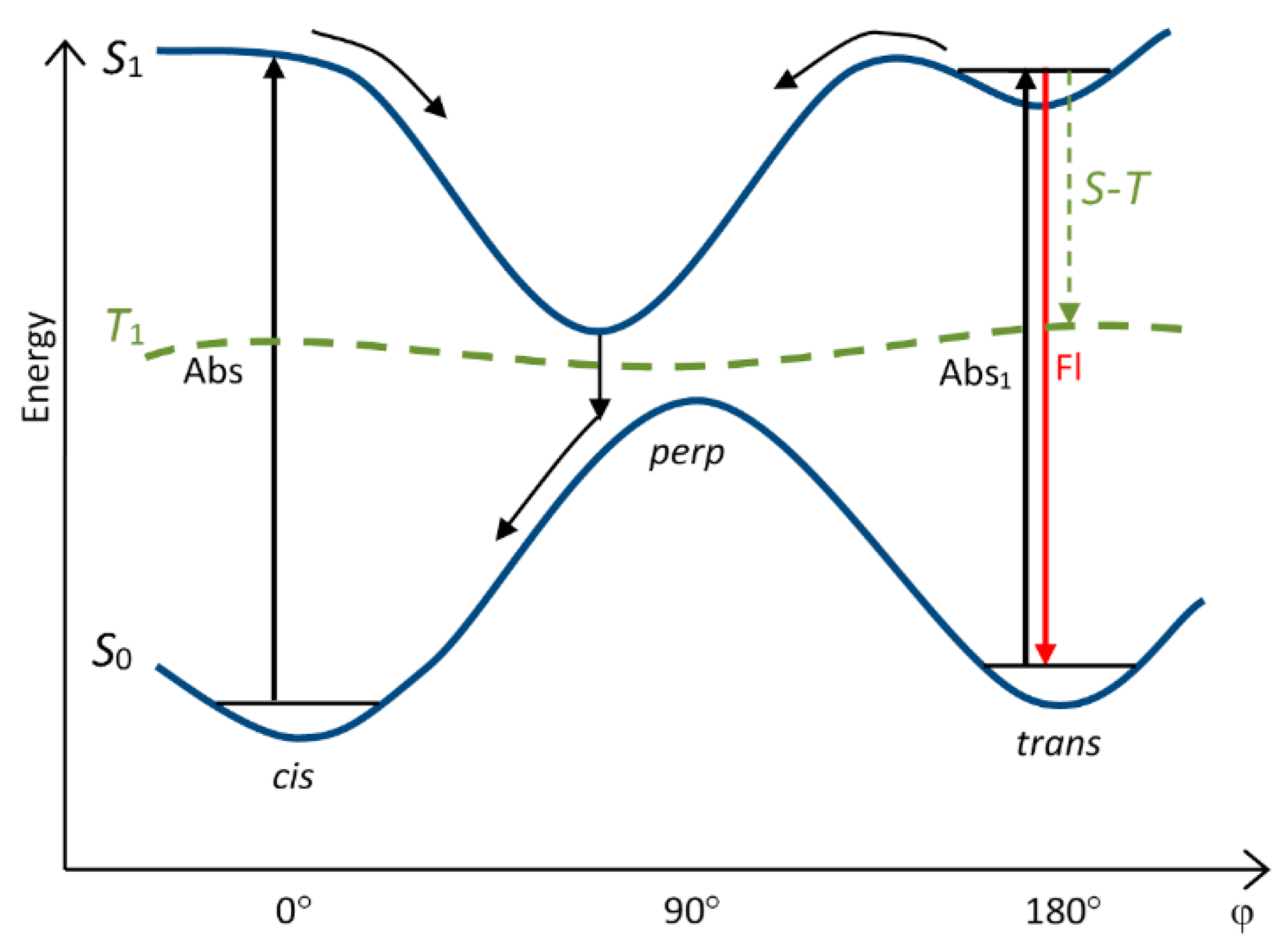
4. Potential Surfaces of Isomerization of Cyanine Dyes
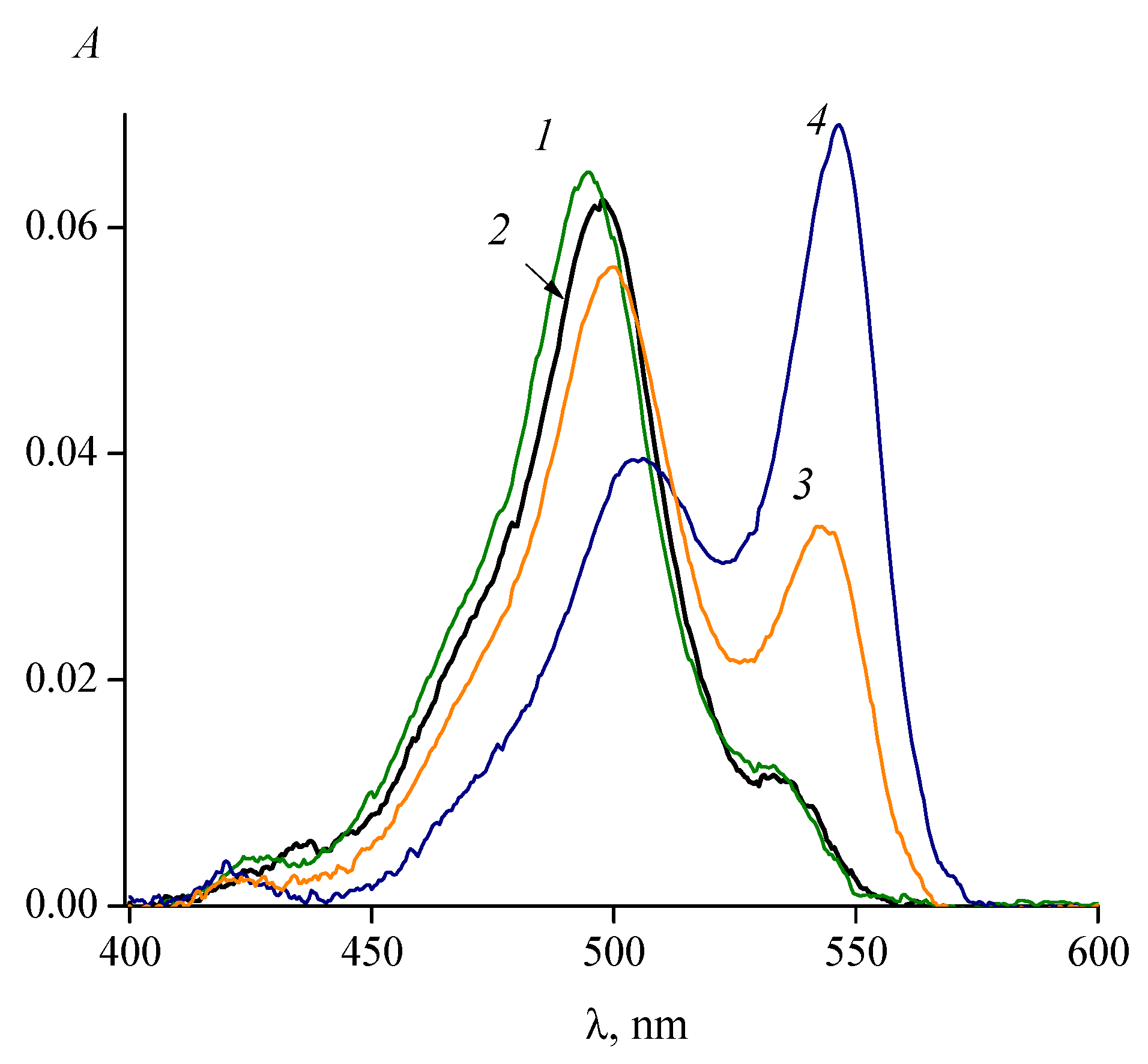
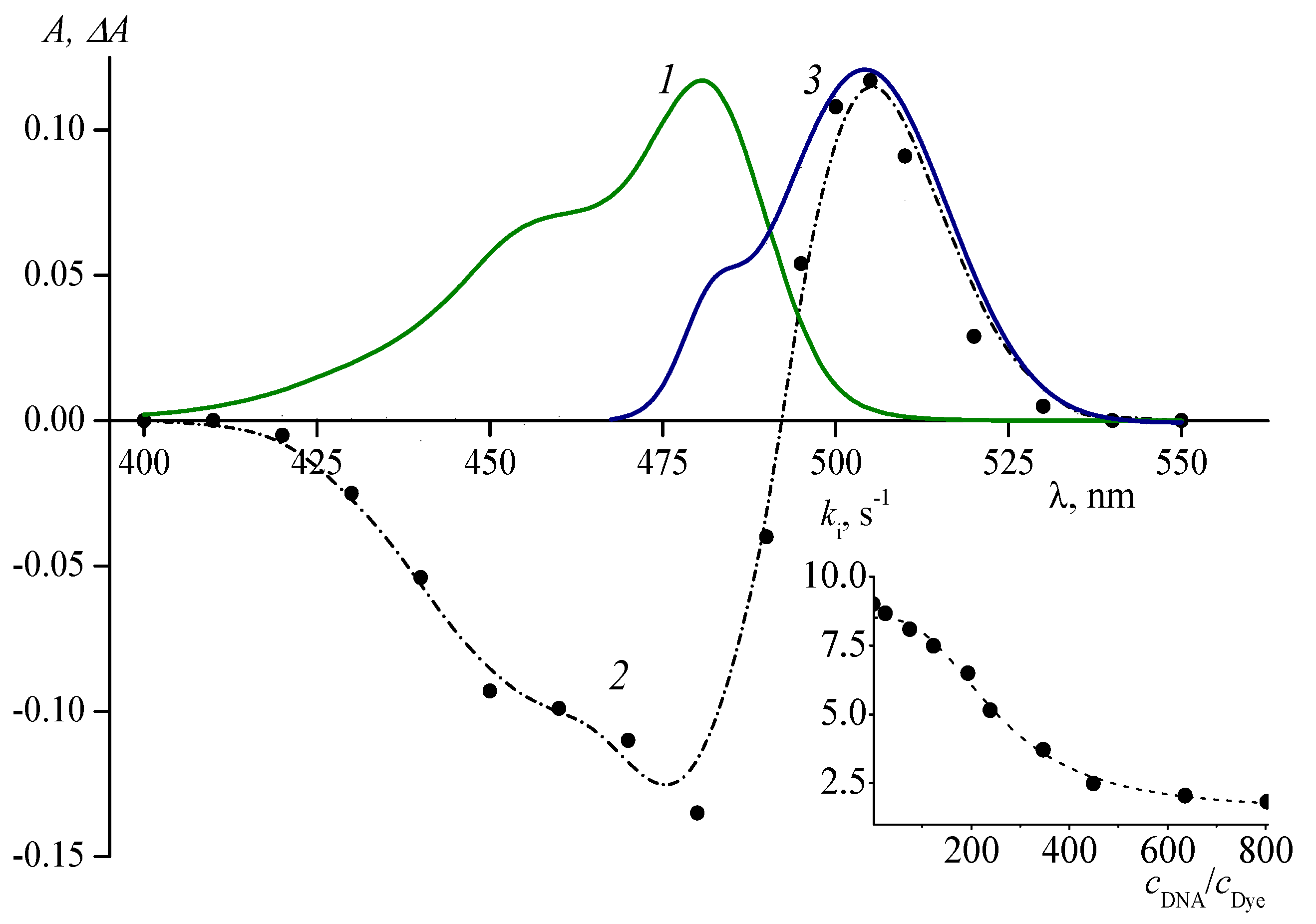
5. Effect of Solvent Viscosity on the Kinetics of Photoisomerization and Back (Thermal) Isomerization
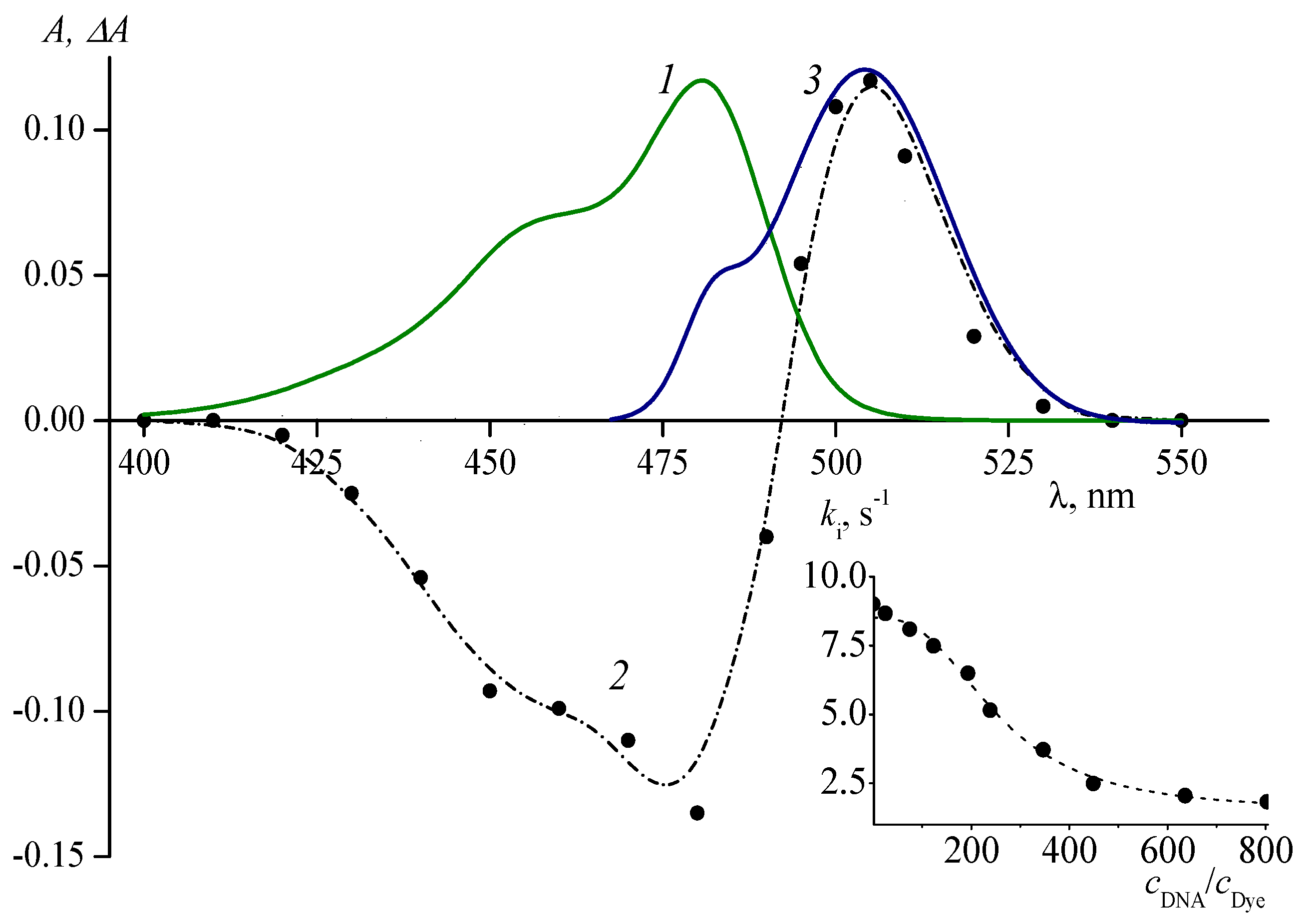
6. Effects of Ion Pair Formation on the Isomerization Processes of Cyanine Dyes
7. Isomerization of Meso-Substituted Carbocyanine Dyes
8. Isomerization of Cyanine Dyes in Structurally Organized Media
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, J.E.; Tauber, M.J.; Mathies, R.A. Wavelength dependent cis-trans isomerization in vision. Biochemistry 2001, 40, 13774–13778. [Google Scholar] [CrossRef]
- Kandori, H.; Shichida, Y.; Yoshizawa, T. Photoisomerization in rhodopsin. Biochemistry (Moscow) 2001, 66, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Zechmeister, L. Cis-trans isomerization and stereochemistry of carotenoids and diphenylpolyenes. Chem. Rev. 1944, 34, 267–344. [Google Scholar] [CrossRef]
- Herkstroeter, W.G. The Mechanism of Syn-Anti Isomerization of Azomethine Dyes. J. Am. Chem. Soc. 1973, 95, 8686–8691. [Google Scholar] [CrossRef]
- Liu, X.-M.; Jin, X.-Y.; Zhang, Z.-X.; Wang, J.; Bai, F.-Q. Theoretical study on the reaction mechanism of the thermal cis–trans isomerization of fluorine-substituted azobenzene derivatives. RSC Adv. 2018, 8, 11580–11588. [Google Scholar] [CrossRef]
- El-Shishtawy, R.M. Functional dyes, and some hi-tech applications. Int. J. Photoenergy 2009, 2009, 434897. [Google Scholar] [CrossRef]
- Pierce, B.M. Theoretical analysis of the third-order nonlinear optical properties of linear cyanines and polyenes. Proc. SPIE 1991, 1560, 148–161. [Google Scholar] [CrossRef]
- Pittman, M.; Plaza, P.; Martin, M.M.; Meyer, Y.H. Subpicosecond reverse saturable absorption in organic and organometallic solutions. Opt. Commun. 1998, 158, 201–212. [Google Scholar] [CrossRef]
- Levitus, M.; Ranjit, S. Cyanine dyes in biophysical research: The photophysics of polymethine fluorescent dyes in biomolecular environments. Q. Rev. Biophys. 2011, 44, 123–151. [Google Scholar] [CrossRef]
- Tatikolov, A.S. Polymethine dyes as spectral-fluorescent probes for biomacromolecules. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 55–90. [Google Scholar] [CrossRef]
- Zhang, Y.; Bi, J.; Xia, S.; Mazi, W.; Wan, S.; Mikesell, L.; Luck, R.L.; Liu, H. A near-infrared fluorescent probe based on a FRET rhodamine donor linked to a cyanine acceptor for sensitive detection of intracellular pH alternations. Molecules 2018, 23, 2679. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Massie, T.L.; Maeda, T.; Nakazumi, H.; Colyer, C.L. A long-wavelength fluorescent squarylium cyanine dye possessing boronic acid for sensing monosaccharides and glycoproteins with high enhancement in aqueous solution. Sensors 2012, 12, 5420–5431. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.K.; James, N.S.; Chen, Y.; Dobhal, M.P. Cyanine dye-based compounds for tumor imaging with and without photodynamic therapy. Top. Heterocycl. Chem. 2008, 14, 41–74. [Google Scholar] [CrossRef]
- James, N.S.; Chen, Y.; Joshi, P.; Ohulchanskyy, T.Y.; Ethirajan, M.; Henary, M.; Strekowski, L.; Pandey, R.K. Evaluation of polymethine dyes as potential probes for near infrared fluorescence imaging of tumors: Part 1. Theranostics 2013, 3, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhao, Y.; Zhang, H.; Chen, X.; Zhao, N.; Tan, D.; Zhang, H.; Shi, C. The application of heptamethine cyanine dye DZ-1 and indocyanine green for imaging and targeting in xenograft models of hepatocellular carcinoma. Int. J. Mol. Sci. 2017, 18, 1332. [Google Scholar] [CrossRef] [PubMed]
- Yen, T.-L.; Chang, C.-C.; Chung, C.-L.; Ko, W.-C.; Yang, C.-H.; Hsieh, C.-Y. Neuroprotective effects of platonin, a therapeutic immunomodulating medicine, on traumatic brain injury in mice after controlled cortical impact. Int. J. Mol. Sci. 2018, 19, 1100. [Google Scholar] [CrossRef]
- Ishchenko, A.A. Structure and spectral-luminescent properties of polymethine dyes. Russ. Chem. Rev. 1991, 60, 865–884. [Google Scholar] [CrossRef]
- Mishra, A.; Behera, R.K.; Behera, P.K.; Mishra, B.K.; Behera, G.B. Cyanines during the 1990s: A Review. Chem. Rev. 2000, 100, 1973–2011. [Google Scholar] [CrossRef]
- Hamer, F.M.; Heilbron, I.M.; Reade, J.H.; Walls, H.N. Cyanine dyes and related compounds. J. Chem. Soc. 1932, 251–260. [Google Scholar] [CrossRef]
- McCartin, P.J. Observation of metastable geometrical isomers of cyanines by flash photolysis. J. Chem. Phys. 1965, 42, 2980–2981. [Google Scholar] [CrossRef]
- Khimenko, V.; Chibisov, A.K.; Gorner, H. Effects of alkyl substituents in the polymethine chain on the photoprocesses in thiacarbocyanine dyes. J. Phys. Chem. A. 1997, 101, 7304–7310. [Google Scholar] [CrossRef]
- Ponterini, G.; Momicchioli, F. Trans-cis photoisomerization mechanism of carbocyanines: Experimental check of theoretical models. Chem. Phys. 1991, 151, 111–126. [Google Scholar] [CrossRef]
- Krieg, M.; Redmond, R.W. Photophysical properties of 3,3′-dialkylthiacarbocyanine dyes in homogeneous solution. Photochem. Photobiol. 1993, 57, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Caselli, M.; Latterini, L.; Ponterini, G. Consequences of H-dimerization on the photophysics and photochemistry of oxacarbocyanines. Phys. Chem. Chem. Phys. 2004, 6, 3857–3863. [Google Scholar] [CrossRef]
- Pronkin, P.G.; Tatikolov, A.S. Photochemical properties of oxacarbocyanine dyes in solutions and in complexes with DNA. High Energy Chem. 2015, 49, 368–371. [Google Scholar] [CrossRef]
- Henrichs, P.M.; Gross, S. Conformational analysis of carbocyanine dyes with variable-temperature proton Fourier transform nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 1976, 98, 7169–7175. [Google Scholar] [CrossRef]
- Aramendia, P.F.; Negri, R.M.; Roman, E.S. Temperature dependence of fluorescence and photoisomerization in symmetric carbocyanines. Influence of medium viscosity and molecular structure. J. Phys. Chem. 1994, 98, 3165–3173. [Google Scholar] [CrossRef]
- Rulliere, C. Laser action and photoisomerisation of 3,3′-diethyl oxadicarbocyanine iodide (DODCI): Influence of temperature and concentration. Chem. Phys. Lett. 1976, 43, 303–308. [Google Scholar] [CrossRef]
- Orlandi, G.; Siebrand, W. Model for the direct photo-isomerization of stilbene. Chem. Phys. Lett. 1975, 30, 352–354. [Google Scholar] [CrossRef]
- Momicchioli, F.; Baraldi, I.; Berthier, G. Theoretical study of trans-cis photoisomerism in polymethine cyanines. Chem. Phys. 1988, 123, 103–112. [Google Scholar] [CrossRef]
- Dietz, F.; Rentsch, S.K. On the mechanism of photoisomerization and the structure of the photoisomers of cyanine dyes. Chem. Phys. 1985, 96, 145–151. [Google Scholar] [CrossRef]
- Smedartchina, Z. The influence of a polar medium on the rate of fast photoprocesses. J. Photochem. 1985, 30, 13–23. [Google Scholar] [CrossRef]
- Sundstrh, V.; Gillbro, T. Viscosity-dependent isomerization yields of some cyanine dyes. A picosecond laser spectroscopy study. J. Phys. Chem. 1982, 86, 1788–1794. [Google Scholar] [CrossRef]
- Dietzek, B.; Tarnovsky, A.N.; Yartsev, A. Visualizing overdamped wavepacket motion: Excited-state isomerization of pseudocyanine in viscous solvents. Chem. Phys. 2009, 357, 54–62. [Google Scholar] [CrossRef]
- Åkesson, E.; Hakkarainen, A.; Laitinen, E.; Helenius, V.; Gillbro, T.; Korppi-Tommola, J.; Sundström, V. Analysis of microviscosity and reaction coordinate concepts in isomerization dynamics described by Kramers’ theory. J. Chem. Phys. 1991, 95, 6508–6523. [Google Scholar] [CrossRef]
- Kramers, H.A. Brownian motion in a field of force and the diffusion model of chemical reactions. Physica 1940, 7, 284–304. [Google Scholar] [CrossRef]
- Sumi, H.; Asano, T. Slow thermal isomerization in viscous solvents. J. Chem. Phys. 1995, 102, 9565–9573. [Google Scholar] [CrossRef]
- Murarka, R.K.; Bhattacharyya, S.; Biswas, R.; Bagchi, B. Isomerization dynamics in viscous liquids: Microscopic investigation of the coupling and decoupling of the rate to and from solvent viscosity and dependence on the intermolecular potential. J. Chem. Phys. 1999, 110, 7365–7375. [Google Scholar] [CrossRef]
- Grote, R.F.; Hynes, J.T. The stable states picture of chemical reactions. II. Rate constants for condensed and gas phase reaction models. J. Chem. Phys. 1980, 73, 2715–2732. [Google Scholar] [CrossRef]
- Sundstrom, V.; Gtllbro, T. Dynamics of the isomerization of trans-stilbene in n-alcohols studied by ultraviolet picosecond absorption recovery. Chem. Phys. Lett. 1984, 109, 538–543. [Google Scholar] [CrossRef]
- Åkesson, E.; Sundstrom, V.; Gillbro, T. Solvent-dependent barrier heights of excited-state photoisomerization reactions. Chem. Phys. Lett. 1985, 121, 513–522. [Google Scholar] [CrossRef]
- Ponterini, G.; Casselli, M. Photoisomerization dynamics of 3,3′-diethyloxacarbocyanine. Intramolecular and solvent viscosity effects. Ber. Bunsenges. Phys. Chem. 1992, 96, 564–573. [Google Scholar] [CrossRef]
- Chibisov, A.K.; Voznyak, D.A.; Petrov, N.K.; Alfimov, M.V. The specific feature of photochemical processes in molecules of 3,3′-dialkylthiacarbocyanines in binary solvent mixtures. High Energy Chem. 2009, 43, 38–43. [Google Scholar] [CrossRef]
- Tatikolov, A.S.; Dzhulibekov, K.S.; Shvedova, L.A.; Kuzmin, V.A. Influence of “inert” counterions on the photochemistry of some cationic polymethine dyes. J. Phys. Chem. 1995, 99, 6525–6529. [Google Scholar] [CrossRef]
- Kolesnikov, A.M.; Mikhailenko, F.A. The conformations of polymethine dyes. Russ. Chem. Rev. 1987, 56, 275–287. [Google Scholar] [CrossRef]
- West, W.; Pearce, S.; Grum, F. Stereoisomerism in cyanine dyes-meso-substituted thiacarbocyanines. J. Phys. Chem. 1967, 71, 1316–1326. [Google Scholar] [CrossRef]
- Tatikolov, A.S.; Akimkin, T.M.; Pronkin, P.G.; Yarmoluk, S.M. Enhancement of photoisomerization of polymethine dyes in complexes with biomacromolecules. Chem. Phys. Lett. 2013, 556, 287–291. [Google Scholar] [CrossRef]
- Allmann, R.; Anis, H.-J.; Benn, R.; Grahn, W.; Olejnek, S.; Waskowska, A. Konformationsanalyse von polymethinen i. erstmaliger nachweis von di-, tri- und all-cis-konformationen bei sterisch gehinderten trimethincyaninen (carbocyaninen) der indolin- und benzothiazolreihe. Angew. Chem. Suppl. 1983, 22, 1147–1175. [Google Scholar] [CrossRef]
- Pronkin, P.G.; Tatikolov, A.S.; Anikovskii, M.Y.; Kuzmin, V.A. The study of cis-trans equilibrium and complexation with DNA of meso-substituted carbocyanine dyes. High Energy Chem. 2005, 39, 237–243. [Google Scholar] [CrossRef]
- Pronkin, P.G.; Tatikolov, A.S.; Sklyarenko, V.I.; Kuzmin, V.A. Photochemical properties of meso-substituted thiacarbocyanine dyes in solutions and in complexes with DNA. High Energy Chem. 2006, 40, 252–258. [Google Scholar] [CrossRef]
- Tocho, J.O.; Duchowicz, R.; Scaffardi, L.; Blimes, G.M.; Dipaolo, R.; Murphy, M. Spectroscopic properties of isomerizable cyanine dyes. Trends Phys. Chem. 1992, 3, 31–47. [Google Scholar]
- Pronkin, P.G.; Tatikolov, A.S. Influence of the interaction with DNA on the spectral-fluorescent and photochemical properties of some meso-substituted polymethine dyes. Spectrochim. Acta Part A 2018, 202, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Anikovsky, M.Y.; Tatikolov, A.S.; Pronkin, P.G.; Levin, P.P.; Sklyarenko, V.I.; Kuzmin, V.A. DNA effect on cis–trans equilibrium and fluorescent properties of 3,3′-diethyl-9-thiomethylthiacarbocyanine iodide in aqueous solution. High Energy Chem. 2003, 37, 398–404. [Google Scholar] [CrossRef]
- Armitage, B.A. Cyanine dye–DNA interactions: Intercalation, groove binding, and aggregation. Top. Curr. Chem. 2005, 253, 55–76. [Google Scholar] [CrossRef]
- Atabekyan, L.S.; Chibisov, A.K. Photoprocesses in aqueous solutions of 9-ethylthiacarbocyanine dyes in the presence of surfactants. High Energy Chem. 2007, 41, 91–96. [Google Scholar] [CrossRef]
- Tatikolov, A.S.; Pronkin, P.G. Photochemical processes in molecules of polymethine dye probes in the presence of bile salts. J. Appl. Spectrosc. 2018, in press. [Google Scholar]
- Slavnova, T.D.; Chibisov, A.K.; Gorner, H. Photoprocesses of thiacarbocyanine monomers, dimers, and aggregates bound to polyanions. J. Phys. Chem. A 2002, 106, 10985–10990. [Google Scholar] [CrossRef]
- Anikovsky, M.Y.; Tatikolov, A.S.; Kuzmin, V.A. Complex formation between 3,3′-diethylthiacarbocyanine iodide and DNA and its investigation in aqueous solution. Int. J. Photoenergy 1999, 1, 35–39. [Google Scholar] [CrossRef]
- Chibisov, A.K.; Gorner, H. Photophysics of aggregated 9-methylthiacarbocyanine bound to polyanions. Chem. Phys. Lett. 2002, 357, 434–439. [Google Scholar] [CrossRef]
- Valandro, S.R.; Poli, A.L.; Correia, T.F.A.; Lombardo, P.C.; Schmitt, C.C. Photophysical behavior of isocyanine/clay hybrids in the solid state. Langmuir 2017, 33, 891–899. [Google Scholar] [CrossRef]
- Ghann, W.; Kang, H.; Emerson, E.; Oh, J.; Chavez-Gil, T.; Nesbitt, F.; Williams, R.; Uddin, J. Photophysical properties of near-IR cyanine dyes and their application as photosensitizers in dye sensitized solar cells. Inorg. Chim. Acta 2017, 467, 123–131. [Google Scholar] [CrossRef]
- Tahara, S.; Takeuchi, S.; Ohtani, H.; Tahara, T. Vibrational Wavepacket motion in ultrafast cyanine photoisomerization revealed by femtosecond stimulated Raman spectroscopy. In Proceedings of the International Conference on Ultrafast Phenomena, OSA, Santa Fe, NM, USA, 17–22 July 2016; Abstract Number UM2A.4. Optical Society of America: Washington, DC, USA, 2016. [Google Scholar] [CrossRef]
- Bleger, D.; Yu, Z.; Hecht, S. Toward optomechanics: Maximizing the photodeformation of individual molecules. Chem. Commun. 2011, 47, 12260–12266. [Google Scholar] [CrossRef]
- Kaiser, C.; Halbritter, T.; Heckel, A.; Wachtveitl, J. Thermal, photochromic and dynamic properties of water-soluble spiropyrans. ChemistrySelect 2017, 2, 4111–4123. [Google Scholar] [CrossRef]
- Zhang, Z.-X.; Bai, F.-Q.; Li, L.; Zhang, H.-X. Synthesis of novel sulfonamide azoles via C–N cleavage of sulfonamides by azole ring and relational antimicrobial study. New J. Chem. 2015, 39, 1634–1642. [Google Scholar] [CrossRef]
- Yao, H.-H.; Cheng, H.-H.; Cheng, C.-H.; Lin, C.-K.; Yang, J.-S.; Chen, I.C. Charge-transfer and isomerization reactions of trans-4-(N-arylamino)stilbenes. Phys. Chem. Chem. Phys. 2016, 18, 28164–28174. [Google Scholar] [CrossRef]
- Chmyrov, V.; Spielmann, T.; Hevekerl, H.; Widengren, J. Trans-cis isomerization of lipophilic dyes probes membrane microviscosity in biological membranes and in live cells. Anal. Chem. 2015, 87, 5690–5697. [Google Scholar] [CrossRef] [PubMed]
- Vinogradov, A.M.; Tatikolov, A.S.; Costa, S.M.B. The effect of anionic, cationic and neutral surfactants on the photophysics and isomerization of 3,3′-diethylthiacarbocyanine. Phys. Chem. Chem. Phys. 2001, 3, 4325–4332. [Google Scholar] [CrossRef]
- Henary, M.; Mojzych, M. Stability and reactivity of polymethine dyes in solution. In Heterocyclic Polymethine Dyes; Strekowski, L., Ed.; Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2008; Volume 14, pp. 221–238. [Google Scholar] [CrossRef]
- Bricks, J.L.; Kachkovskii, A.D.; Slominskii, Y.L.; Gerasov, A.O.; Popov, S.V. Molecular design of near infrared polymethine dyes: A review. Dyes Pigments 2015, 121, 238–255. [Google Scholar] [CrossRef]
- Yarmoluk, S.M.; Kovalska, V.B.; Lukashov, S.S.; Slominskii, Y.L. Interaction of cyanine dyes with nucleic acids. XII.β-substituted carbocyanines as possible fluorescent probes for nucleic acids detection. Bioorg. Med. Chem. Lett. 1999, 9, 1677–16789. [Google Scholar] [CrossRef]
- Armitage, B.A. Cyanine dye–nucleic acid interactions. In Heterocyclic Polymethine Dyes; Strekowski, L., Ed.; Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2008; Volume 14, pp. 11–29. [Google Scholar] [CrossRef]
- Panova, I.G.; Sharova, N.P.; Dmitrieva, S.B.; Poltavtseva, R.A.; Sukhikh, G.T.; Tatikolov, A.S. Use of a cyanine dye as a probe for albumin and collagen in the extracellular matrix. Anal. Biochem. 2007, 361, 183–189. [Google Scholar] [CrossRef]
- Tatikolov, A.S.; Akimkin, T.M.; Kashin, A.S.; Panova, I.G. Meso-substituted polymethine dyes as efficient spectral and fluorescent probes for biomacromolecules. High Energy Chem. 2010, 44, 224–227. [Google Scholar] [CrossRef]
- Akimkin, T.M.; Tatikolov, A.S.; Yarmoluk, S.M. Spectral and fluorescent study of the interaction of cyanine dyes Cyan 2 and Cyan 45 with DNA. High Energy Chem. 2011, 45, 222–228. [Google Scholar] [CrossRef]
- Bychkova, A.V.; Pronkin, P.G.; Sorokina, O.N.; Tatikolov, A.S.; Rosenfeld, M.A. Study of protein coatings cross-linked via the free-radical mechanism on magnetic nanoparticles by the method of spectral and fluorescent probes. Colloid J. 2014, 76, 387–394. [Google Scholar] [CrossRef]
- Gorobets, M.G.; Wasserman, L.A.; Vasilyeva, A.D.; Bychkova, A.V.; Pronkin, P.G.; Bugrova, A.E.; Indeykina, M.I.; Shilkina, N.G.; Konstantinova, M.L.; Kononikhin, A.S.; et al. Modification of human serum albumin under induced oxidation. Dokl. Biochem. Biophys. 2017, 474, 231–235. [Google Scholar] [CrossRef]
- Tatikolov, A.S.; Costa, S.M.B. Medium effects on the isomerization of an anionic polymethine dye. Chem. Phys. Lett. 2007, 440, 73–78. [Google Scholar] [CrossRef]
- Tatikolov, A.S.; Costa, S.M.B. Photophysical and aggregation properties of a long-chain squarylium indocyanine dye. J. Photochem. Photobiol. A Chem. 2001, 140, 147–156. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dye | R | Solvent | ki, s−1 | Ref. |
|---|---|---|---|---|
| Thiacarbocyanine dyes | ||||
| 2 | – | ethanol | 250 | [21] |
| 8 | OCH3 | isopropanol | 1.7 × 106 | [50] |
| 9 | C2H5 | dioxane | ~5 × 103 | |
| 10 | Cl | isopropanol | 1.6 × 103 | |
| 14 | C6H5 | ethanol | 340 | [52] |
| 15 | C7H6OH | 865 | ||
| 20 | SCH3 | isopropanol | 5 × 105 | [53] |
| Oxacarbocyanine dyes | ||||
| 3 | – | aqueous solutions | 9.0 | [25] |
| 11 | C2H5 | 1.5 × 104 | ||
| 12 | C2H5 | 1 × 104 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pronkin, P.; Tatikolov, A. Isomerization and Properties of Isomers of Carbocyanine Dyes. Sci 2019, 1, 19. https://doi.org/10.3390/sci1010019
Pronkin P, Tatikolov A. Isomerization and Properties of Isomers of Carbocyanine Dyes. Sci. 2019; 1(1):19. https://doi.org/10.3390/sci1010019
Chicago/Turabian StylePronkin, Pavel, and Alexander Tatikolov. 2019. "Isomerization and Properties of Isomers of Carbocyanine Dyes" Sci 1, no. 1: 19. https://doi.org/10.3390/sci1010019
APA StylePronkin, P., & Tatikolov, A. (2019). Isomerization and Properties of Isomers of Carbocyanine Dyes. Sci, 1(1), 19. https://doi.org/10.3390/sci1010019