2.1. Ultrafast Dissociation of Core Ionized Deoxyribose
In a previous paper [
17], we have modeled the dynamics induced by the removal of oxygen K-shell electrons from a deoxyribose molecule embedded in liquid water (direct effect). The only investigated initial configuration (atomic positions and velocities) is shown in
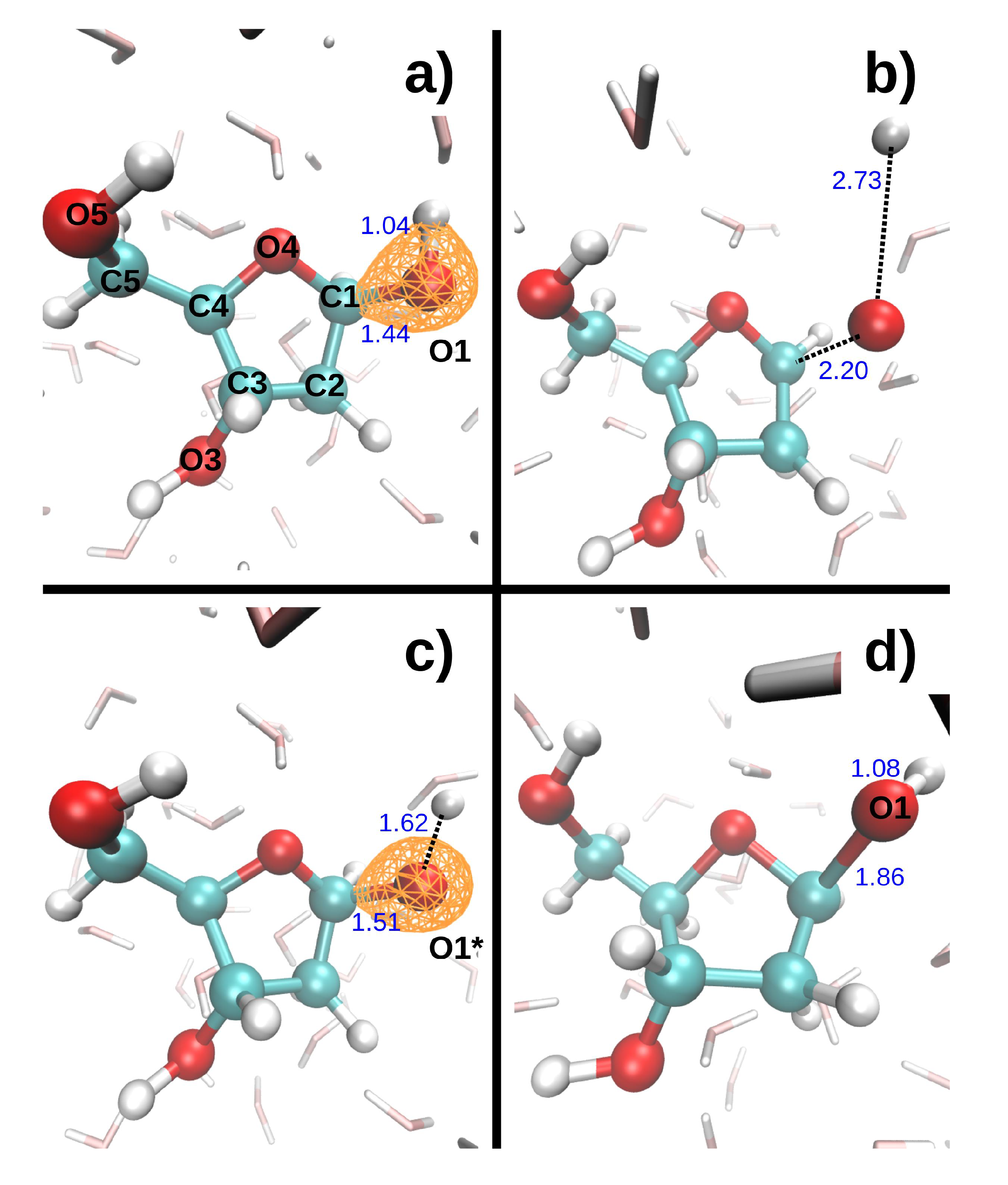
Figure 1. The C-O* bonds length increased by 0.1–0.2 Å during the 9.7 fs dynamics (the asterisk will always indicate the core-ionized atom). The hydroxyl groups, however, displayed very different bond elongations, from 0.05 Å for O3*-H3 to 0.78 Å for O5*-H5. To study indirect effects, two additional MD were performed, in which the core hole was localized on a water molecule donating an hydrogen bond (HB) either to O1 or O4. In both cases, one of the O*-H bonds broke during the 9.7 fs dynamics, and the proton was transferred either to another water molecule or to O4, respectively.
In the present paper, we have completed this study by investigating five additional initial configurations, thus allowing a statistical analysis of the results. Core holes were localized on the different carbon and oxygen atoms of the sugar (direct effect). The results will be compared to simulations of the isolated molecule [
20] to infer the role of the environment on the dissociation patterns. Core holes were also localized on the water molecules forming an HB with the sugar (indirect effect).
2.1.1. Hydration of the Sugar
We present first the DR hydration during the 1.2 ps dynamics of the neutral system, along which the six initial configurations were selected. Such simulation time is short for a full analysis of the hydration but our purpose here is to describe the starting point for the ensuing nonequilibrium dynamics. The radial distribution functions (RDF) between the different DR oxygen atoms and the water hydrogen atoms exhibited a first minimum around 2.4 Å, as did the RDF between the different hydrogen atoms belonging to the DR hydroxyl groups and the water oxygen atoms (see
Supplementary Materials, Figure S1). The number of water molecules in the first coordination sphere of the different DR atoms are shown in
Table 1.
2.1.2. Direct Effect
We have studied the consequence of carbon or oxygen K-shell ionizations in a deoxyribose molecule embedded in liquid water. Carbon K-shell vacancies did not induce any significant bond elongation during the 9.7 fs MD. On the contrary, when the core hole is localized on the oxygen atoms, the length of the CO* and O*H bonds systematically increases, as shown in
Table 2. The elongation of the CO* bonds was always below 0.34 Å at the end of all the 9.7 fs core hole MD. These results are similar to those obtained in the gas phase [
20]. Thus, the CO* bonds elongation does not seem to be influenced much by the DR environment. In particular, the largest average bond elongation is induced by core vacancies on O1 in both phases.
On the contrary, although O*H bonds are not much affected by the presence of the K-shell vacancy on O* in the gas phase [
20], these bonds may undergo very large elongations in liquid water, as shown in
Table 2. In fact, the elongation of the O*H bonds could reach 0.93 Å at the end of the 9.7 fs core hole dynamics. Considering OH bonds to be dissociated when the OH distance is larger than 1.5 Å, we find that the O3-H3 bond breaks in two configurations out of six, and the O1-H1 and O5-H5 bonds in four configurations out of six. The released proton is always transferred to a neighboring water molecule to form an hydronium ion (H
3O
+).
The dissociation of O*H bonds appears to depend strongly on the local environment of the hydroxyl groups, and, more specifically, on the presence of a water molecule (labeled H
2O
w) accepting an hydrogen bond from the sugar (O*H…O
w). When the initial distance between the DR hydrogen atom and O
w is less than ~1.8 Å, respectively, greater than ~1.9 Å, the DR hydroxyl group (O*H) always, respectively, never, dissociates to form H
3. A similar influence of hydration on NH bond dissociation was found in a uracil molecule embedded in water, when the core hole was localized on the nitrogen atoms [
16].
2.1.3. Indirect Effect
We have investigated the ultrafast dissociation of the water molecules which belong to the deoxyribose primary hydration shell, after a core ionization was placed on the oxygen atom (O*), but before Auger decay. One of the O*H bonds always exhibits a longer elongation than the other. It reaches on average 1.66 ± 0.23 Å at t = 9.7 fs, compared to 1.12 ± 0.19 Å for the second bond. These values do not seem to depend on whether the water molecule accepts or donates an HB to the sugar. The species formed at the end of the 9.7 fs dynamics are listed in
Table 3.
Let us consider the water molecules which donate an HB to the sugar. In most cases (24/37), the core-ionized water molecule emits only one proton, either towards the sugar (DR−H+ + O*H) or towards another water molecule (H3O+ + O*H). In a few additional cases (4/37), the emitted hydrogen is less than 1.5 Å away from both the core-ionized and HB acceptor molecules ((DR−H−O*H)+ or (H2O−H−O*H)+). More rarely, both O*H bonds undergo significant elongation (in seven cases out of 37).
Let us now consider the water molecules which accept an HB from the sugar hydroxyl groups. In 11 cases, the water molecule has one broken bond leading to the formation of an H3O+, the second O*H bond is significantly elongated in 3 out of these 11 MD. One dynamics results in the complete fragmentation of the core-ionized molecule.
As a conclusion, two species are formed (O*H and H
3O
+) in the vicinity of the sugar, and/or protons may be transferred towards the sugar during the lifetime of the core vacancy. The species thus formed could in turn damage the sugar by an indirect effect. It should however be mentioned that, in most cases, Auger decay will take place well before the molecules dissociation since the lifetime of a K-shell vacancy is ≃4.1 fs in water [
21]. We have in fact estimated that, in liquid water, only ~16% of the core-ionized water molecules will have one or both
H bonds broken before Auger decay [
16].
2.2. Dissociation of Doubly Ionized DR after Auger Effect
How to localize the double hole after Auger effect? In Born Oppenheimer MD (BOMD), one cannot localize the initial charges on a single molecule in the condensed phase. The Ehrenfest time-dependent density functional theory (TDDFT) method must thus be applied to propagate a dicationic electronic state prepared by emptying one of the localized valence molecular orbitals of the sample [
18]. It has to be noticed that Auger effect can lead to many different channels since the Auger electron can be ejected from different orbitals. The calculation of the branching ratios of the Auger deexcitation channels would be prohibitive in view of the number of MO of the system. Here, we chose to remove the two electrons from the deepest valence MO localized on the oxygen atom which had the K-shell vacancy before Auger decay (O*). Although the selected deexcitation process might not be the most probable, it is the most energetic leading to the fastest fragmentation process, and it may thus be modeled in a reasonable computational time.
As this method is very computationally consuming, we restrict our study to a single initial configuration, and to the dissociation induced by the removal of a K-shell electron from the DR oxygen atoms (direct effects). The initial configuration (t = 0) is the one shown in
Figure 1, and the four core–hole dynamics, one for each oxygen atom, are those described in details in [
17]. The geometry of the sugar evolves while the oxygen atom has a K-shell vacancy. In fact, the O*H bond is broken at the end of two out of the four core–hole dynamics. It should however be stressed that the lifetime of the oxygen K vacancy is only 4.7 fs [
22]. As a consequence, approximately 87% of the core-ionized molecules undergo Auger decay before 9.7 fs. Furthermore, Auger decay will most probably happen almost immediately after core ionization, so that the DR
2+ geometry will be very similar to that of the initial configuration (shown in
Figure 1). We have therefore modeled two extreme scenarios. In the first scenario, the DR undergoes Auger decay at t = 0, when no structural change has been induced by the K-shell vacancy. We have thus performed four DR
2+ Ehrenfest TDDFT MD, all starting with the same geometry, that is the initial configuration shown in
Figure 1, but removing the two electrons from each of the four deepest valence MO localized on the DR oxygen atoms. In the second scenario, the DR undergoes Auger decay at the end of the 9.7 fs core hole dynamics, when the structural change induced by the K vacancy is maximal. We have thus performed four DR
2+ Ehrenfest TDDFT MD, starting from four different geometries, those generated at the end of the four core hole dynamics, and removing the two electrons from the deepest valence MO localized on the core-ionized atom (O*).
Figure 2 shows the dissociation dynamics induced by a double hole localized on the DR ring oxygen (O4) after Auger decay. In the first scenario (
Figure 2, top), the O4 core hole decays at t = 0. The C1-O4 and C4-O4 bonds then break very rapidly, at 8.7 and 9.0 fs, respectively. It should be noted that these bonds only elongated by 0.14 and 0.18 Å, respectively, when a core hole was localized on O4 during 9.7 fs. In the second scenario (
Figure 2, bottom), the O4 core hole decays at 9.7 fs. The C1-O4 and C4-O4 bonds then break at 15.4 and 18.9 fs, respectively. The presence of a core vacancy on O4 thus seems only to delay the dissociation of the C-O4 bonds. In both scenarios, the O5-H5 bond length increases slightly during the 9.6 fs DR
2+ dynamics, whereas the O1-H1 elongates more in the first scenario than in the second.
Figure 3 shows the dissociation dynamics induced by a double hole localized on O1 after Auger decay. In the first scenario (
Figure 3, top), the O1 core hole decays at t = 0. The O1-H1 and C1-O1 bonds then break within a few femtoseconds (see
Table 4). H1 first collides on a neighboring water oxygen (O
w1) at ~5 fs, after which it moves further away from O1. The kinetic energy of H1 exhibits large variations. It first increases up to a maximum value of 5.3 eV as H1 departs from the DR. It then decreases to a minimum value of 2.6 eV when H1 is closest to the water molecule. After the collision, it reaches a second maximum of 4.3 eV, and then gradually decreases again. Meanwhile, the kinetic energy of O1 increases up to 0.1 eV during about 5 fs, and then remains fairly constant. In the second scenario (
Figure 3, bottom), the O1 core hole decays at 9.7 fs. It should be noted that the O1-H1 bond breaks during the O1 core–hole dynamics, at t = 8.7 fs (see
Table 4). During the subsequent DR
2+ dynamics, H1 also collides with O
w1, but it is retrodiffused back towards O1 so that the O1-H1 bond forms again at t = 15.0 fs. In the meantime, the C1-O1 bond elongates up to 1.86 Å at t = 19.3 fs. The maximum kinetic energy reached by both H1 and O1 is lower in the second than in the first scenario (see
Table 4).
Figure 4 shows the dissociation dynamics induced by a double hole localized on O3 after Auger decay. In the first scenario (
Figure 4, top), the O3 core hole decays at t = 0. The O3-H3 and C3-O3 bonds then break within a few femtoseconds (see
Table 5). H3 first collides with a neighboring water oxygen (O
w3) at ~6.3 fs, but the deviation angle is small and it moves further away from O3. In the second scenario (
Figure 4, bottom), the O3 core hole decays at 9.7 fs. The O3-H3 and C3-O3 bonds only elongate slightly during the O3 core hole dynamics, but then break very rapidly during the subsequent DR
2+ dynamics (see
Table 5). H3 collides with O
w3 at ~14.8 fs. The deviation angle is larger than in the first scenario, but H3 also moves away from O3 after the collision. The maximum kinetic energy reached by both H3 and O3 is lower in the second than in the first scenario (see
Table 5). Interestingly, a bond forms between C3 and O4 in both scenarios.
Figure 5 shows the dissociation dynamics induced by a double hole localized on O5 after Auger decay. In the first scenario (
Figure 5, top), the O5 core hole decays at t = 0. The O5-H5 and C5-O5 bonds then break within a few femtoseconds (see
Table 6). H5 first collides on a neighboring water oxygen (O
w5). It is then scattered back towards O5 so that the O5-H5 distance diminishes towards the end of the dynamics. In the second scenario (
Figure 5, bottom), the O5 core hole decays at 9.7 fs. It should be noted that the O5-H5 bond breaks during the O5 core–hole dynamics, at t = 6.1 fs. H5 then binds to O
w5. This bond undergoes large vibrations, between 0.81 and 1.26 Å, but H5 remains bound to O
w5 during the subsequent DR
2+. The C5-O5 bond breaks at 16.6 fs. The maximum kinetic energy reached by both H5 and O5 is lower in the second than in the first scenario (see
Table 5).
To summarize, when a double hole is localized on the DR oxygen atoms, the bonds involving this oxygen atom are very rapidly broken, typically after 3 fs for the OH bonds, and 7 to 9 fs for the CO bonds. The elongation of these bonds is much faster than that induced by a core vacancy on the same oxygen atom. Moreover, in most cases, the CO bond breakage during the DR
2+ dynamics does not seem to depend much on the time at which Auger decay occurs. This is not the case for the protons dynamics. For one part, their kinetic energy is lower when the OH bond breaks during the core hole dynamics, than when it breaks during the DR
2+ dynamics. However, the protons trajectories also differ when the OH bond breaks after Auger decay in the two scenarios. In fact, we have recently shown that the trajectories of the protons ejected from an H
2O
2+ molecule are very sensitive to slight perturbations of the system [
19].

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}