Inherited Retinal Disease Therapies Targeting Precursor Messenger Ribonucleic Acid


Abstract
1. Introduction
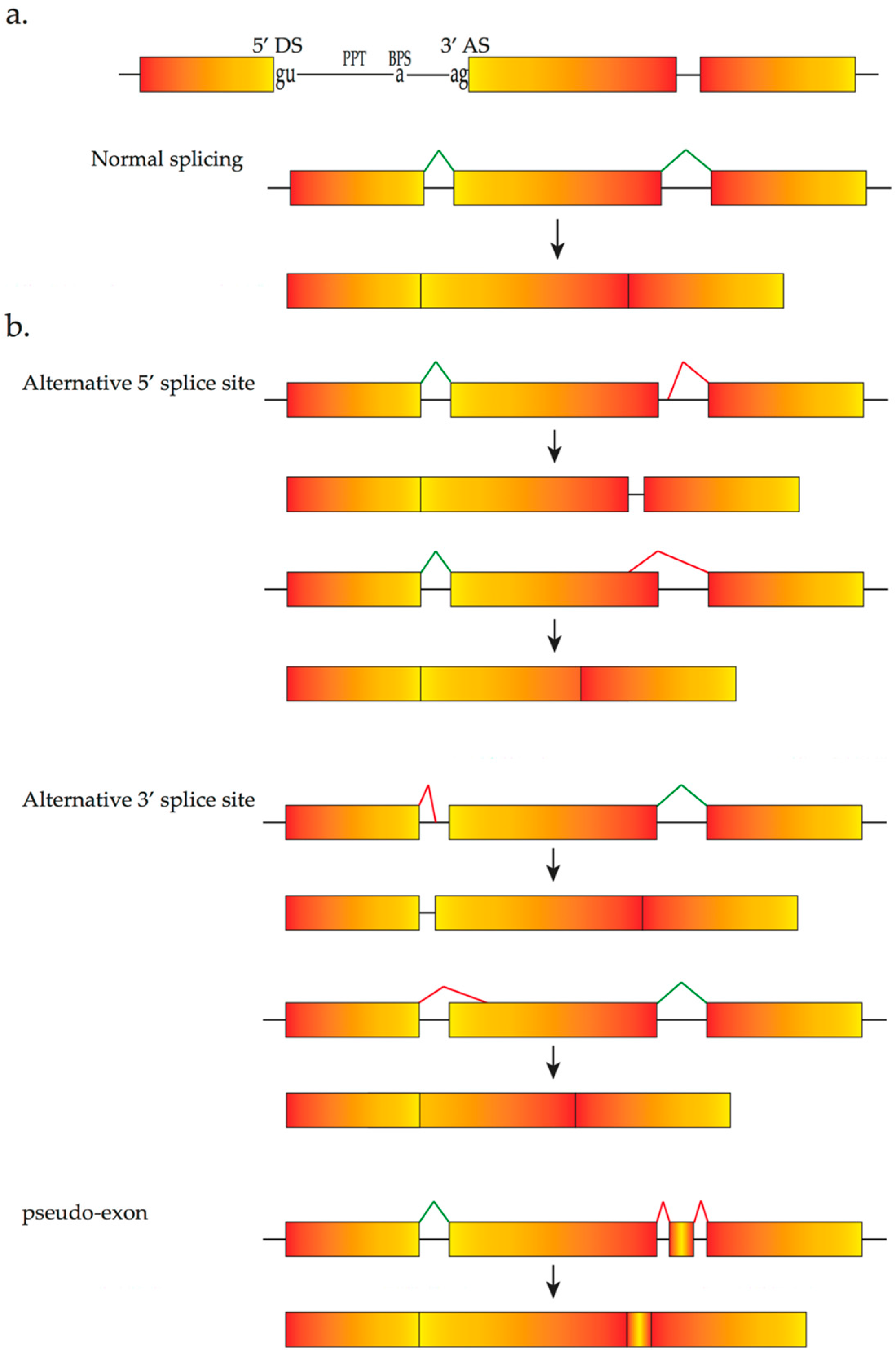
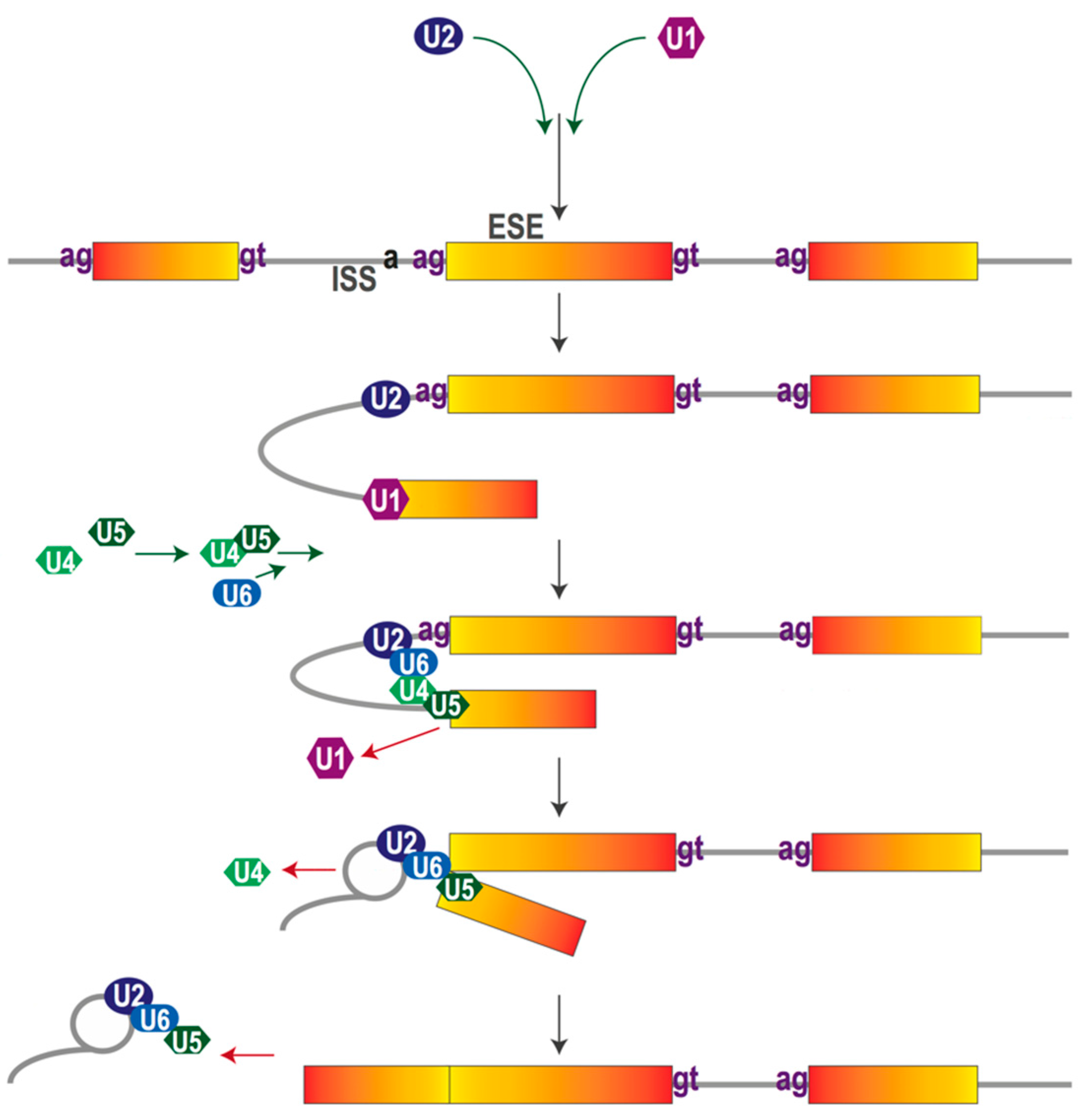
2. The Pre-mRNA Splicing Process
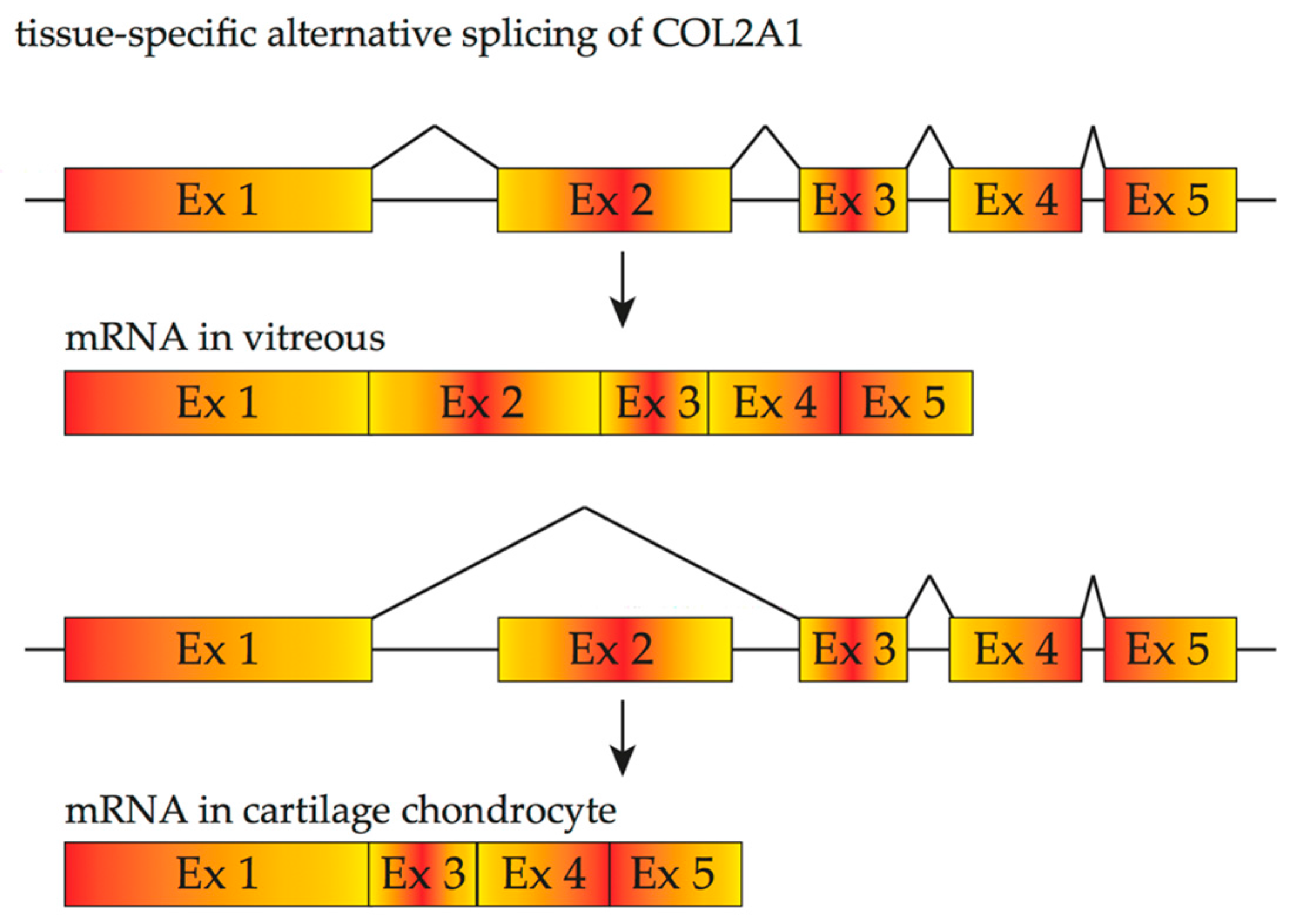
3. Inherited Retinal Diseases Due to Mutations That Affect Splicing and Spliceosome
4. Therapeutic Induced Alternative Splicing
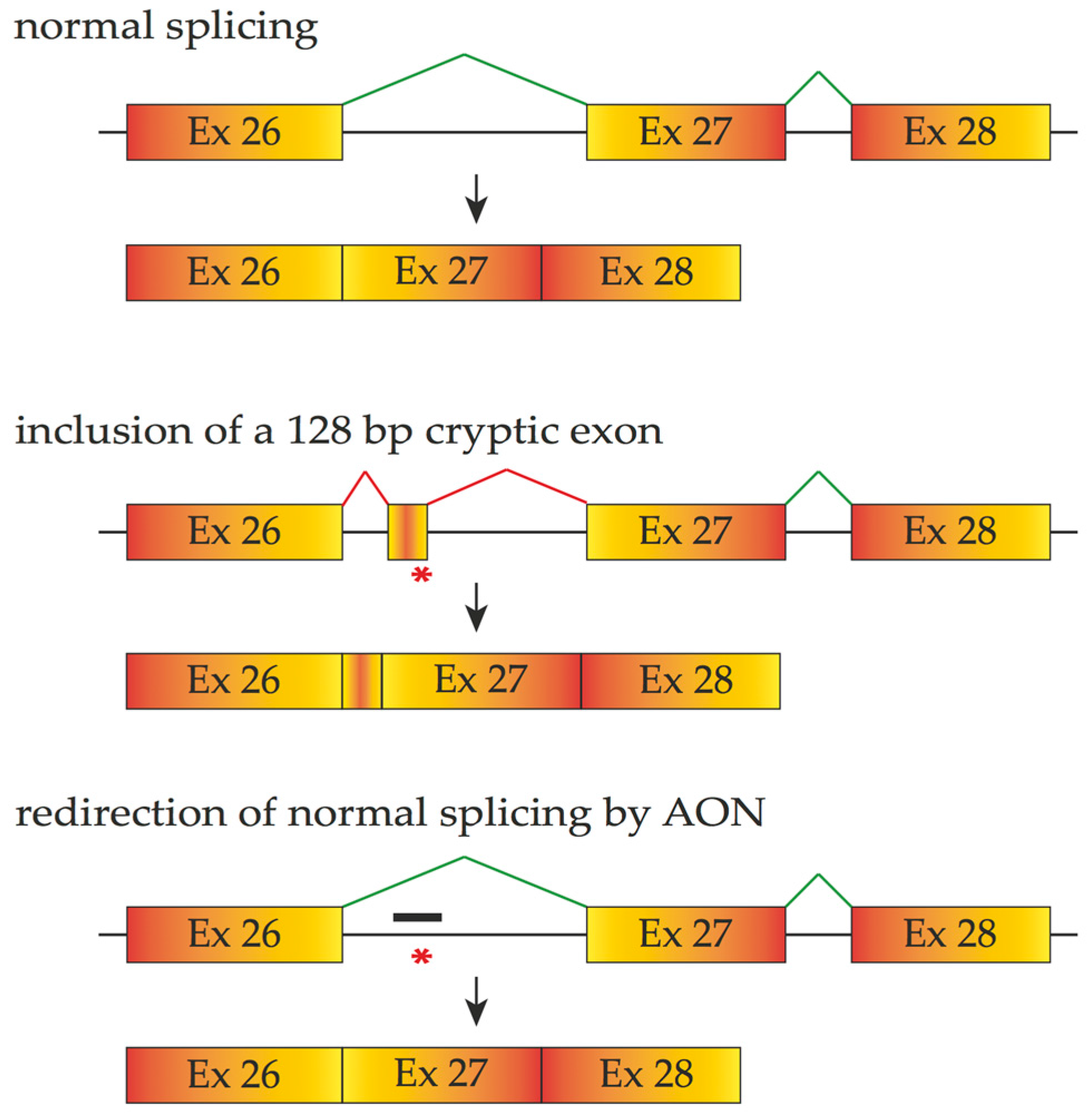
4.1. Antisense Oligonucleotides
4.2. Engineered Small Nuclear Ribonucleic Acid (snRNA)
4.3. Splicesome Protein Modulators
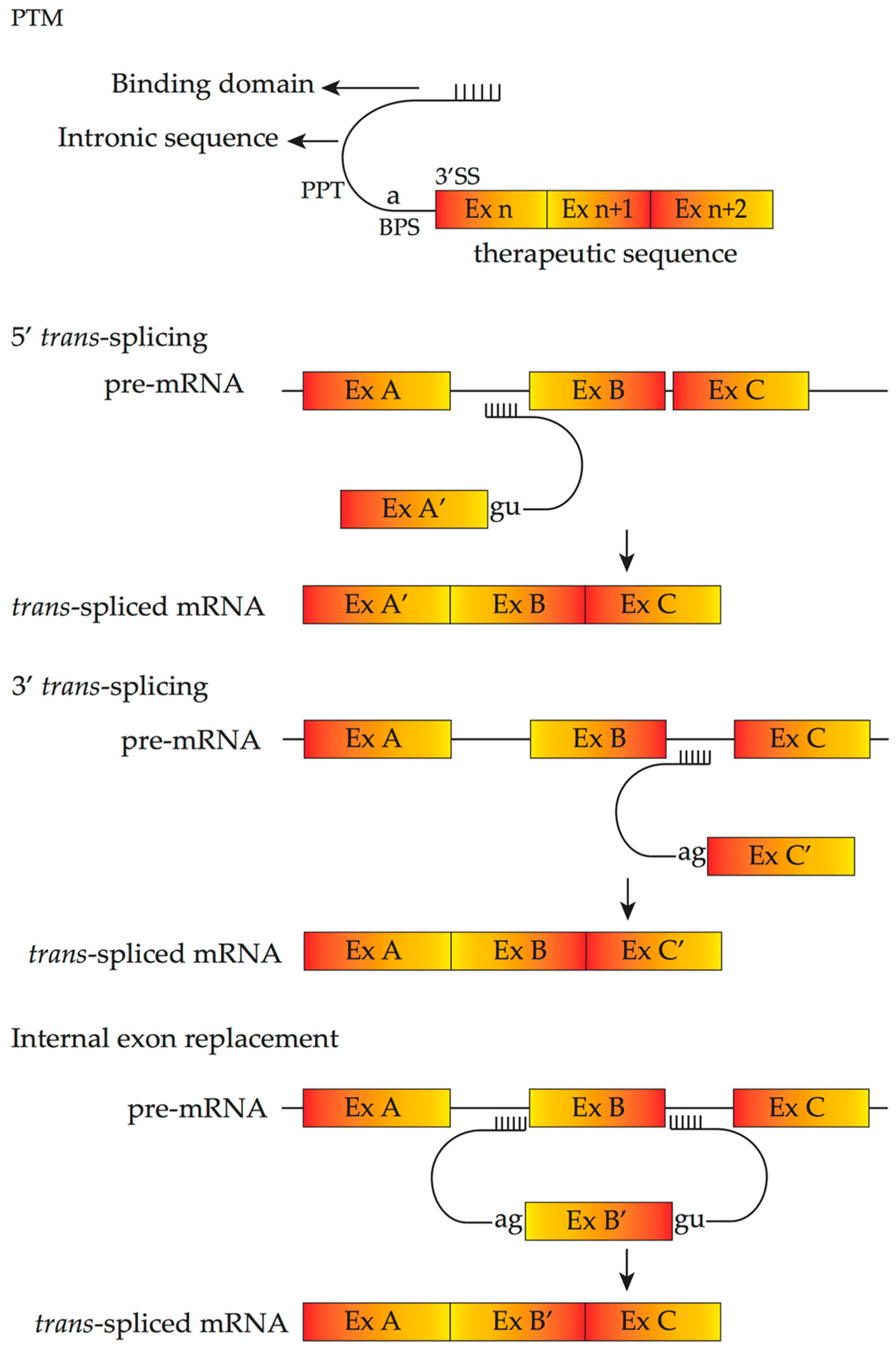
4.4. Pre-Trans-Splicing Molecules
5. Delivery of Therapeutics Inducing Alternative Splicing
6. Future Directions
Acknowledgments
Conflicts of Interest
Disclosure
References
- Hamel, C. Retinitis pigmentosa. Orphanet J. Rare Dis. 2006, 1, 40. [Google Scholar] [CrossRef] [PubMed]
- Hamel, C.P. Cone rod dystrophies. Orphanet J. Rare Dis. 2007, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Bainbridge, J.W.; Ali, R.R. Gene supplementation therapy for recessive forms of inherited retinal dystrophies. Gene Ther. 2012, 19, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Sahel, J.A.; Marazova, K.; Audo, I. Clinical characteristics and current therapies for inherited retinal degenerations. Cold Spring Harbor Perspect. Med. 2015, 5, a017111. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.; Yao, F.; Liang, X.; Xu, F.; Li, H.; Sui, R.; Dong, F. De novo mutations in the cone-rod homeobox gene associated with leber congenital amaurosis in Chinese patients. Ophthalmic Genet. 2015, 36, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Liew, G.; Michaelides, M.; Bunce, C. A comparison of the causes of blindness certifications in England and Wales in working age adults (16–64 years), 1999–2000 with 2009–2010. BMJ Open. 2014, 4, e004015. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A.; Jacobson, S.G.; Alexander, K.R.; Cideciyan, A.V.; Birch, D.G.; Weleber, R.G.; Hood, D.C. Outcome measures and their application in clinical trials for retinal degenerative diseases: Outline, review, and perspective. Retina 2005, 25, 772–777. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Khanna, H.; Punzo, C. Advances in Gene Therapy for Diseases of the Eye. Hum. Gene Ther. 2016, 27, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Scholl, H.P.; Strauss, R.W.; Singh, M.S.; Dalkara, D.; Roska, B.; Picaud, S.; Sahel, J.A. Emerging therapies for inherited retinal degeneration. Sci. Transl. Med. 2016, 8, 368rv6. [Google Scholar] [CrossRef] [PubMed]
- Campochiaro, P.A.; Strauss, R.W.; Lu, L.; Hafiz, G.; Wolfson, Y.; Shah, S.M.; Sophie, R.; Mir, T.A.; Scholl, H.P. Is There Excess Oxidative Stress and Damage in Eyes of Patients with Retinitis Pigmentosa? Antioxid. Redox Signal. 2015, 23, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Mutter, M.; Swietek, N.; Munch, T.A. Salvaging ruins: Reverting blind retinas into functional visual sensors. Methods Mol. Biol. 2014, 1148, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.C.; Humayun, M.S.; Dorn, J.D.; da Cruz, L.; Dagnelie, G.; Handa, J.; Barale, P.O.; Sahel, J.A.; Stanga, P.E.; Hafezi, F.; et al. Long-Term Results from an Epiretinal Prosthesis to Restore Sight to the Blind. Ophthalmology 2015, 122, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Da Cruz, L.; Coley, B.F.; Dorn, J.; Merlini, F.; Filley, E.; Christopher, P.; Chen, F.K.; Wuyyuru, V.; Sahel, J.; Stanga, P.; et al. The Argus II epiretinal prosthesis system allows letter and word reading and long-term function in patients with profound vision loss. Br. J. Ophthalmol. 2013, 97, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Wiley, L.A.; Burnight, E.R.; Songstad, A.E.; Drack, A.V.; Mullins, R.F.; Stone, E.M.; Tucker, B.A. Patient-specific induced pluripotent stem cells (iPSCs) for the study and treatment of retinal degenerative diseases. Prog. Retin. Eye Res. 2015, 44, 15–35. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.L.; Greenberg, P.B.; Borton, D.A. Advances in Retinal Prosthetic Research: A Systematic Review of Engineering and Clinical Characteristics of Current Prosthetic Initiatives. Curr. Eye Res. 2017, 42, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Sterne-Weiler, T.; Howard, J.; Mort, M.; Cooper, D.N.; Sanford, J.R. Loss of exon identity is a common mechanism of human inherited disease. Genome Res. 2011, 21, 1563–1571. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, P.K.; Symons, R.C.; Shah, S.M.; Quinlan, E.J.; Tabandeh, H.; Do, D.V.; Reisen, G.; Lockridge, J.A.; Short, B.; Guerciolini, R.; et al. RNAi-based treatment for neovascular age-related macular degeneration by Sirna-027. Am. J. Ophthalmol. 2010, 150, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Guerin, K.; Gregory-Evans, C.Y.; Hodges, M.D.; Moosajee, M.; Mackay, D.S.; Gregory-Evans, K.; Flannery, J.G. Systemic aminoglycoside treatment in rodent models of retinitis pigmentosa. Exp. Eye Res. 2008, 87, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Nagel-Wolfrum, K.; Moller, F.; Penner, I.; Wolfrum, U. Translational read-through as an alternative approach for ocular gene therapy of retinal dystrophies caused by in-frame nonsense mutations. Vis. Neurosci. 2014, 31, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Bacchi, N.; Casarosa, S.; Denti, M.A. Splicing-correcting therapeutic approaches for retinal dystrophies: Where endogenous gene regulation and specificity matter. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3285–3294. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S.; Moore, M.J. Pre-mRNA splicing: Awash in a sea of proteins. Mol. Cell. 2003, 12, 5–14. [Google Scholar] [CrossRef]
- Zhou, Z.; Fu, X.D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Jean-Philippe, J.; Paz, S.; Caputi, M. hnRNP A1: The Swiss army knife of gene expression. Int. J. Mol. Sci. 2013, 14, 18999–19024. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Hertel, K.J. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip. Rev. RNA 2012, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Matera, A.G.; Wang, Z. A day in the life of the spliceosome. Nat. Rev. Mol. Cell. Biol. 2014, 15, 108–121. [Google Scholar] [CrossRef] [PubMed]
- De Conti, L.; Baralle, M.; Buratti, E. Exon and intron definition in pre-mRNA splicing. Wiley Interdiscip. Rev. RNA. 2013, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.D.; Berglund, J.A. Alternative pre-mRNA splicing. Methods Mol. Biol. 2014, 1126, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Sahebi, M.; Hanafi, M.M.; van Wijnen, A.J.; Azizi, P.; Abiri, R.; Ashkani, S.; Taheri, S. Towards understanding pre-mRNA splicing mechanisms and the role of SR proteins. Gene 2016, 587, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Passacantilli, I.; Sette, C. Alternative splicing and cell survival: From tissue homeostasis to disease. Cell. Death Differ. 2016. [CrossRef] [PubMed]
- De Klerk, E.; AC’t Hoen, P. Alternative mRNA transcription, processing, and translation: Insights from RNA sequencing. Trends Genet. 2015, 31, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.M.; Zack, D.J. Alternative splicing and retinal degeneration. Clin. Genet. 2013, 84, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Utz, V.M.; Beight, C.D.; Marino, M.J.; Hagstrom, S.A.; Traboulsi, E.I. Autosomal dominant retinitis pigmentosa secondary to pre-mRNA splicing-factor gene PRPF31 (RP11): Review of disease mechanism and report of a family with a novel 3-base pair insertion. Ophthalmic Genet. 2013, 34, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lee, J.A.; Black, D.L. Neuronal regulation of alternative pre-mRNA splicing. Nat. Rev. Neurosci. 2007, 8, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Norris, A.D.; Calarco, J.A. Emerging Roles of Alternative Pre-mRNA Splicing Regulation in Neuronal Development and Function. Front. Neurosci. 2012, 6, 122. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Black, D.L. Alternative pre-mRNA splicing in neurons: Growing up and extending its reach. Trends Genet. 2013, 29, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Raj, B.; Blencowe, B.J. Alternative Splicing in the Mammalian Nervous System: Recent Insights into Mechanisms and Functional Roles. Neuron 2015, 87, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Tanackovic, G.; Ransijn, A.; Thibault, P.; Abou Elela, S.; Klinck, R.; Berson, E.L.; Chabot, B.; Rivolta, C. PRPF mutations are associated with generalized defects in spliceosome formation and pre-mRNA splicing in patients with retinitis pigmentosa. Hum. Mol. Genet. 2011, 20, 2116–2130. [Google Scholar] [CrossRef] [PubMed]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.; Cieply, B.; Carstens, R.; Ramamurthy, V.; Stoilov, P. The Musashi 1 Controls the Splicing of Photoreceptor-Specific Exons in the Vertebrate Retina. PLoS Genet. 2016, 12, e1006256. [Google Scholar] [CrossRef] [PubMed]
- Susaki, K.; Kaneko, J.; Yamano, Y.; Nakamura, K.; Inami, W.; Yoshikawa, T.; Ozawa, Y.; Shibata, S.; Matsuzaki, O.; Okano, H.; et al. Musashi-1, an RNA-binding protein, is indispensable for survival of photoreceptors. Exp. Eye Res. 2009, 88, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Kousal, B.; Skalicka, P.; Valesova, L.; Fletcher, T.; Hart-Holden, N.; O’Grady, A.; Chakarova, C.F.; Michaelides, M.; Hardcastle, A.J.; Liskova, P. Severe retinal degeneration in women with a c.2543del mutation in ORF15 of the RPGR gene. Mol. Vis. 2014, 20, 1307–1317. [Google Scholar] [PubMed]
- Glaus, E.; Schmid, F.; Da Costa, R.; Berger, W.; Neidhardt, J. Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR splice defect in patient-derived cells. Mol. Ther. 2011, 19, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Neidhardt, J.; Glaus, E.; Barthelmes, D.; Zeitz, C.; Fleischhauer, J.; Berger, W. Identification and characterization of a novel RPGR isoform in human retina. Hum. Mutat. 2007, 28, 797–807. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Black, G.C.; Rice, J.M.; Hart-Holden, N.; Jones, A.; O’Grady, A.; Ramsden, S.; Wright, A.F. RPGR mutation analysis and disease: An update. Hum. Mutat. 2007, 28, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Donoso, L.A.; Edwards, A.O.; Frost, A.T.; Ritter, R., 3rd; Ahmad, N.; Vrabec, T.; Rogers, J.; Meyer, D.; Parma, S. Clinical variability of Stickler syndrome: Role of exon 2 of the collagen COL2A1 gene. Survey Ophthalmol. 2003, 48, 191–203. [Google Scholar] [CrossRef]
- Parma, E.S.; Korkko, J.; Hagler, W.S.; Ala-Kokko, L. Radial perivascular retinal degeneration: A key to the clinical diagnosis of an ocular variant of Stickler syndrome with minimal or no systemic manifestations. Am. J. Ophthalmol. 2002, 134, 728–734. [Google Scholar] [CrossRef]
- Tran-Viet, K.N.; Soler, V.; Quiette, V.; Powell, C.; Yanovitch, T.; Metlapally, R.; Luo, X.; Katsanis, N.; Nading, E.; Young, T.L. Mutation in collagen II alpha 1 isoforms delineates Stickler and Wagner syndrome phenotypes. Mol. Vis. 2013, 19, 759–766. [Google Scholar] [PubMed]
- Gupta, S.K.; Leonard, B.C.; Damji, K.F.; Bulman, D.E. A frame shift mutation in a tissue-specific alternatively spliced exon of collagen 2A1 in Wagner’s vitreoretinal degeneration. Am. J. Ophthalmol. 2002, 133, 203–210. [Google Scholar] [CrossRef]
- Riazuddin, S.A.; Iqbal, M.; Wang, Y.; Masuda, T.; Chen, Y.; Bowne, S.; Sullivan, L.S.; Waseem, N.H.; Bhattacharya, S.; Daiger, S.P.; et al. A splice-site mutation in a retina-specific exon of BBS8 causes nonsyndromic retinitis pigmentosa. Am. J. Hum. Genet. 2010, 86, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.; Singh, R.; Kolandaivelu, S.; Ramamurthy, V.; Stoilov, P. Alternative Splicing Shapes the Phenotype of a Mutation in BBS8 To Cause Nonsyndromic Retinitis Pigmentosa. Mol. Cell. Biol. 2015, 35, 1860–1870. [Google Scholar] [CrossRef] [PubMed]
- Rebeh, I.B.; Moriniere, M.; Ayadi, L.; Benzina, Z.; Charfedine, I.; Feki, J.; Ayadi, H.; Ghorbel, A.; Baklouti, F.; Masmoudi, S. Reinforcement of a minor alternative splicing event in MYO7A due to a missense mutation results in a mild form of retinopathy and deafness. Mol. Vis. 2010, 16, 1898–1906. [Google Scholar] [PubMed]
- Wang, Y.; Liu, Y.; Nie, H.; Ma, X.; Xu, Z. Alternative splicing of inner-ear-expressed genes. Front. Med. 2016, 10, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Khateb, S.; Zelinger, L.; Ben-Yosef, T.; Merin, S.; Crystal-Shalit, O.; Gross, M.; Banin, E.; Sharon, D. Exome sequencing identifies a founder frameshift mutation in an alternative exon of USH1C as the cause of autosomal recessive retinitis pigmentosa with late-onset hearing loss. PLoS ONE 2012, 7, e51566. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.X.; Cartegni, L.; Zhang, M.Q.; Krainer, A.R. A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat. Genet. 2001, 27, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Pagani, F.; Baralle, F.E. Genomic variants in exons and introns: Identifying the splicing spoilers. Nat. Rev. Genet. 2004, 5, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Puisac, B.; Teresa-Rodrigo, M.E.; Arnedo, M.; Gil-Rodriguez, M.C.; Perez-Cerda, C.; Ribes, A.; Pie, A.; Bueno, G.; Gomez-Puertas, P.; Pie, J. Analysis of aberrant splicing and nonsense-mediated decay of the stop codon mutations c.109G>T and c.504_505delCT in 7 patients with HMG-CoA lyase deficiency. Mol. Genet. MeTable 2013, 108, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Den Hollander, A.I.; Koenekoop, R.K.; Yzer, S.; Lopez, I.; Arends, M.L.; Voesenek, K.E.; Zonneveld, M.N.; Strom, T.M.; Meitinger, T.; Brunner, H.G.; et al. Mutations in the CEP290 (NPHP6) gene are a frequent cause of Leber congenital amaurosis. Am. J. Hum. Genet. 2006, 79, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Ruzickova, S.; Stanek, D. Mutations in spliceosomal proteins and retina degeneration. RNA Biol. 2017, 14, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Jarver, P.; O’Donovan, L.; Gait, M.J. A chemical view of oligonucleotides for exon skipping and related drug applications. Nucleic Acid Ther. 2014, 24, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T. Molecular Mechanisms of Antisense Oligonucleotides. Nucleic Acid Ther. 2017, 27, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Mulamba, G.B.; Hu, A.; Azad, R.F.; Anderson, K.P.; Coen, D.M. Human cytomegalovirus mutant with sequence-dependent resistance to the phosphorothioate oligonucleotide fomivirsen (ISIS 2922). Antimicrob. Agents Chemother. 1998, 42, 971–973. [Google Scholar] [PubMed]
- Anderson, K.P.; Fox, M.C.; Brown-Driver, V.; Martin, M.J.; Azad, R.F. Inhibition of human cytomegalovirus immediate-early gene expression by an antisense oligonucleotide complementary to immediate-early RNA. Antimicrob. Agents Chemother. 1996, 40, 2004–2011. [Google Scholar] [PubMed]
- Iversen, P.L.; Zhu, S.; Meyer, A.; Zon, G. Cellular uptake and subcellular distribution of phosphorothioate oligonucleotides into cultured cells. Antisense Res. Dev. 1992, 2, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Brown, H.E.; Okano, H.J.; Pfaff, D.W. Cellular uptake of intracerebrally administered oligodeoxynucleotides in mouse brain. Regul. Pept. 1995, 59, 143–149. [Google Scholar] [CrossRef]
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 2015, 87, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Temsamani, J.; Iadarola, P.L.; Jiang, Z.; Agrawal, S. Effect of different chemically modified oligodeoxynucleotides on immune stimulation. Biochem. Pharmacol. 1996, 51, 173–182. [Google Scholar] [CrossRef]
- Saleh, A.F.; Arzumanov, A.A.; Gait, M.J. Overview of alternative oligonucleotide chemistries for exon skipping. Methods Mol. Biol. 2012, 867, 365–378. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Bellgard, M.I.; Price, L.; Akkari, A.P.; Wilton, S.D. Translational development of splice-modifying antisense oligomers. Expert Opin. Biol. Ther. 2017, 17, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L.; Ming, X.; Nakagawa, O. The chemistry and biology of oligonucleotide conjugates. Acc. Chem. Res. 2012, 45, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Goemans, N.; Lowes, L.P.; Alfano, L.N.; Berry, K.; Shao, J.; Kaye, E.M.; Mercuri, E. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann. Neurol. 2016, 79, 257–271. [Google Scholar] [CrossRef] [PubMed]
- McClorey, G.; Wood, M.J. An overview of the clinical application of antisense oligonucleotides for RNA-targeting therapies. Curr. Opin. Pharmacol. 2015, 24, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Rigo, F.; Hua, Y.; Krainer, A.R.; Bennett, C.F. Antisense-based therapy for the treatment of spinal muscular atrophy. J. Cell. Biol. 2012, 199, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Hoy, S.M. Nusinersen: First Global Approval. Drugs 2017, 77, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Bao, T.L.; Veedu, R.N.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide development for the treatment of muscular dystrophies. Expert Opin. Orphan Drugs 2015, 4, 139–152. [Google Scholar] [CrossRef]
- Wilton, S.D.; Veedu, R.N.; Fletcher, S. The emperor’s new dystrophin: Finding sense in the noise. Trends Mol. Med. 2015, 21, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Veedu, R.N.; Wengel, J. Locked Nucleic Acids-Promising Nucleic Acid Analogs for Therapeutic Applications. Chem. Biodivers. 2010, 7, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Veedu, R.N.; Wengel, J. Locked nucleic acid as a novel class of therapeutic agents. RNA Biol. 2014, 6, 321–323. [Google Scholar] [CrossRef]
- Gerard, X.; Perrault, I.; Hanein, S.; Silva, E.; Bigot, K.; Defoort-Delhemmes, S.; Rio, M.; Munnich, A.; Scherman, D.; Kaplan, J.; et al. AON-mediated Exon Skipping Restores Ciliation in Fibroblasts Harboring the Common Leber Congenital Amaurosis CEP290 Mutation. Mol. Ther. Nucleic Acids 2012, 1, e29. [Google Scholar] [CrossRef] [PubMed]
- Collin, R.W.; den Hollander, A.I.; van der Velde-Visser, S.D.; Bennicelli, J.; Bennett, J.; Cremers, F.P. Antisense Oligonucleotide (AON)-based Therapy for Leber Congenital Amaurosis Caused by a Frequent Mutation in CEP290. Mol. Ther. Nucleic Acids 2012, 1, e14. [Google Scholar] [CrossRef] [PubMed]
- Garanto, A.; Chung, D.C.; Duijkers, L.; Corral-Serrano, J.C.; Messchaert, M.; Xiao, R.; Bennett, J.; Vandenberghe, L.H.; Collin, R.W. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum. Mol. Genet. 2016, 25, 2552–2563. [Google Scholar] [CrossRef] [PubMed]
- Hnik, P.; Boyer, D.S.; Grillone, L.R.; Clement, J.G.; Henry, S.P.; Green, E.A. Antisense oligonucleotide therapy in diabetic retinopathy. J. Diabetes Sci. Technol. 2009, 3, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Tanner, G.; Glaus, E.; Barthelmes, D.; Ader, M.; Fleischhauer, J.; Pagani, F.; Berger, W.; Neidhardt, J. Therapeutic strategy to rescue mutation-induced exon skipping in rhodopsin by adaptation of U1 snRNA. Hum. Mutat. 2009, 30, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.; Glaus, E.; Barthelmes, D.; Fliegauf, M.; Gaspar, H.; Nurnberg, G.; Nurnberg, P.; Omran, H.; Berger, W.; Neidhardt, J. U1 snRNA-mediated gene therapeutic correction of splice defects caused by an exceptionally mild BBS mutation. Hum. Mutat. 2011, 32, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Schmid, F.; Hiller, T.; Korner, G.; Glaus, E.; Berger, W.; Neidhardt, J. A gene therapeutic approach to correct splice defects with modified U1 and U6 snRNPs. Hum. Gene Ther. 2013, 24, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Wright, J.; Babbs, A.; Wilkins, V.; Garcia, L.; Davies, K.E. Engineering multiple U7snRNA constructs to induce single and multiexon-skipping for Duchenne muscular dystrophy. Mol. Ther. 2012, 20, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Hastings, M.L.; Berniac, J.; Liu, Y.H.; Abato, P.; Jodelka, F.M.; Barthel, L.; Kumar, S.; Dudley, C.; Nelson, M.; Larson, K.; et al. Tetracyclines that promote SMN2 exon 7 splicing as therapeutics for spinal muscular atrophy. Sci. Transl. Med. 2009, 1, 5ra12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Feng, Z.; Ling, K.K.; Mollin, A.; Sheedy, J.; Yeh, S.; Petruska, J.; Narasimhan, J.; Dakka, A.; Welch, E.M.; et al. Pharmacokinetics, pharmacodynamics, and efficacy of a small-molecule SMN2 splicing modifier in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2016, 25, 1885–1899. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, R.; Kaida, D.; Furuno, M.; Burroughs, A.M.; Noma, S.; Suzuki, H.; Kawamura, Y.; Hayashizaki, Y.; Mayeda, A.; Yoshida, M. Global analysis of pre-mRNA subcellular localization following splicing inhibition by spliceostatin A. RNA 2017, 23, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Voukkalis, N.; Koutroumani, M.; Zarkadas, C.; Nikolakaki, E.; Vlassi, M.; Giannakouros, T. SRPK1 and Akt Protein Kinases Phosphorylate the RS Domain of Lamin B Receptor with Distinct Specificity: A Combined Biochemical and In Silico Approach. PLoS ONE 2016, 11, e0154198. [Google Scholar] [CrossRef] [PubMed]
- Erkelenz, S.; Mueller, W.F.; Evans, M.S.; Busch, A.; Schoneweis, K.; Hertel, K.J.; Schaal, H. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA 2013, 19, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Oltean, S.; Gammons, M.; Hulse, R.; Hamdollah-Zadeh, M.; Mavrou, A.; Donaldson, L.; Salmon, A.H.; Harper, S.J.; Ladomery, M.R.; Bates, D.O. SRPK1 inhibition in vivo: Modulation of VEGF splicing and potential treatment for multiple diseases. Biochem. Soc. Trans. 2012, 40, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Lorain, S.; Josephine, C.; Desrosiers, M.; Peccate, C.; Voit, T.; Garcia, L.; Sahel, J.A.; Bemelmans, A.P. Repair of rhodopsin mRNA by spliceosome-mediated RNA trans-splicing: A new approach for autosomal dominant retinitis pigmentosa. Mol. Ther. 2015, 23, 918–930. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Maire, S.; Gaillard, M.C.; Sahel, J.A.; Hantraye, P.; Bemelmans, A.P. mRNA trans-splicing in gene therapy for genetic diseases. Wiley Interdiscip. Rev. RNA 2016, 7, 487–498. [Google Scholar] [CrossRef] [PubMed]
- Wally, V.; Murauer, E.M.; Bauer, J.W. Spliceosome-mediated trans-splicing: The therapeutic cut and paste. J. Investig. Dermatol. 2012, 132, 1959–1966. [Google Scholar] [CrossRef] [PubMed]
- Monjaret, F.; Bourg, N.; Suel, L.; Roudaut, C.; Le Roy, F.; Richard, I.; Charton, K. Cis-splicing and translation of the pre-trans-splicing molecule combine with efficiency in spliceosome-mediated RNA trans-splicing. Mol. Ther. 2014, 22, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Gerard, X.; Perrault, I.; Munnich, A.; Kaplan, J.; Rozet, J.M. Intravitreal Injection of Splice-switching Oligonucleotides to Manipulate Splicing in Retinal Cells. Mol. Ther. Nucleic Acids 2015, 4, e250. [Google Scholar] [CrossRef] [PubMed]
- Murray, S.F.; Jazayeri, A.; Matthes, M.T.; Yasumura, D.; Yang, H.; Peralta, R.; Watt, A.; Freier, S.; Hung, G.; Adamson, P.S.; et al. Allele-Specific Inhibition of Rhodopsin with an Antisense Oligonucleotide Slows Photoreceptor Cell Degeneration. Investig. Ophthalmol. Vis. Sci. 2015, 56, 6362–6375. [Google Scholar] [CrossRef] [PubMed]
- Gomes Dos Santos, A.L.; Bochot, A.; Fattal, E. Intraocular delivery of oligonucleotides. Curr. Pharm. Biotechnol. 2005, 6, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Fattal, E.; Bochot, A. Ocular delivery of nucleic acids: Antisense oligonucleotides, aptamers and siRNA. Adv. Drug Deliv. Rev. 2006, 58, 1203–1223. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, D.; Fletcher, S.; Wilton, S.D.; Palmer, N.; McLenachan, S.; Mackey, D.A.; Chen, F.K. Inherited Retinal Disease Therapies Targeting Precursor Messenger Ribonucleic Acid. Vision 2017, 1, 22. https://doi.org/10.3390/vision1030022
Huang D, Fletcher S, Wilton SD, Palmer N, McLenachan S, Mackey DA, Chen FK. Inherited Retinal Disease Therapies Targeting Precursor Messenger Ribonucleic Acid. Vision. 2017; 1(3):22. https://doi.org/10.3390/vision1030022
Chicago/Turabian StyleHuang, Di, Sue Fletcher, Steve D. Wilton, Norman Palmer, Samuel McLenachan, David A. Mackey, and Fred K. Chen. 2017. "Inherited Retinal Disease Therapies Targeting Precursor Messenger Ribonucleic Acid" Vision 1, no. 3: 22. https://doi.org/10.3390/vision1030022
APA StyleHuang, D., Fletcher, S., Wilton, S. D., Palmer, N., McLenachan, S., Mackey, D. A., & Chen, F. K. (2017). Inherited Retinal Disease Therapies Targeting Precursor Messenger Ribonucleic Acid. Vision, 1(3), 22. https://doi.org/10.3390/vision1030022