Employment of Microencapsulated Sertoli Cells as a New Tool to Treat Duchenne Muscular Dystrophy


 ,
, 

Abstract
1. Duchenne Muscular Dystrophy (DMD)
1.1. Therapeutic Approaches to DMD
1.1.1. Corticosteroids
1.1.2. Cell Therapy
1.1.3. Gene Therapy
1.1.4. Exon Skipping
1.1.5. Induction of Utrophin Expression
1.1.6. Alternative Approaches
2. Sertoli Cells
2.1. Sertoli Cells: Multiple Roles for a Single Cell Type
2.2. The Immunomodulatory Properties of SeC
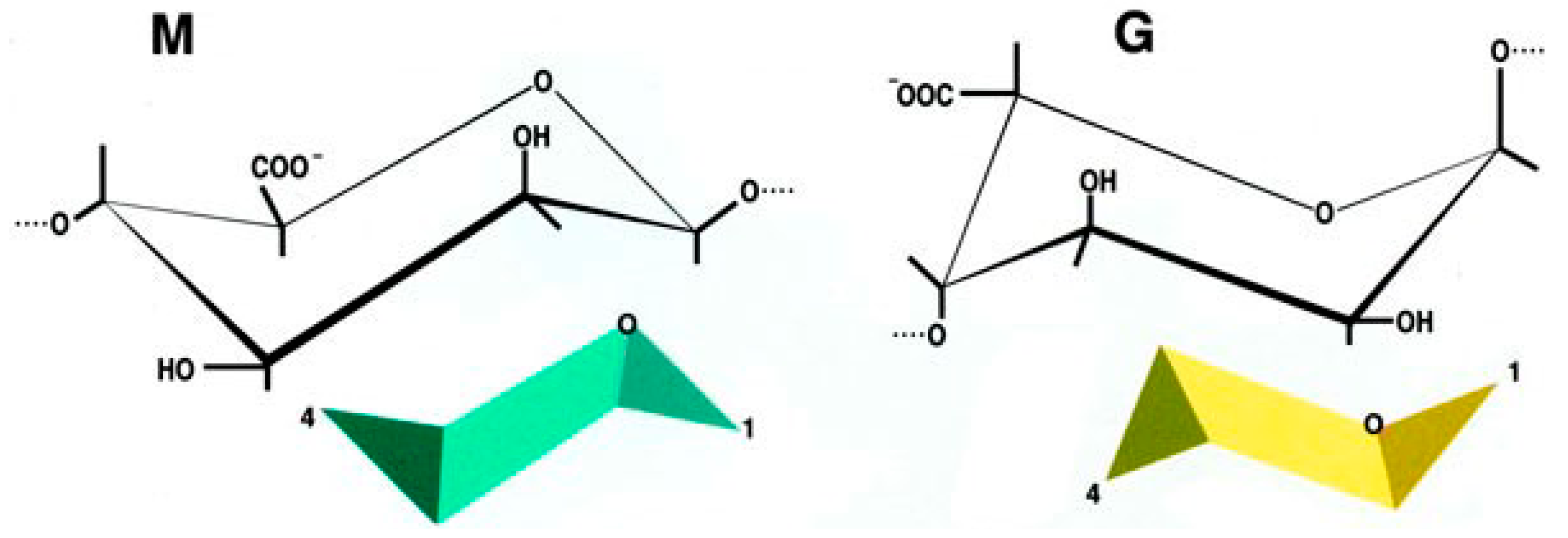
2.3. The Encapsulation Chance
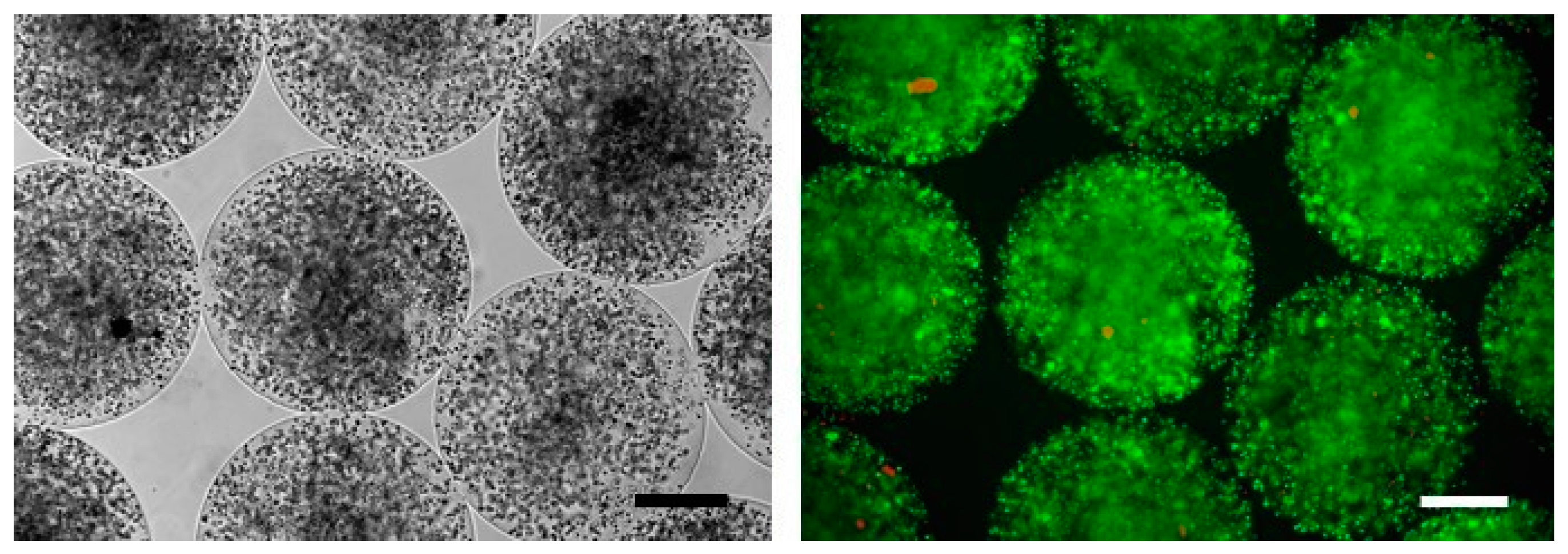
2.4. Pre-Clinical Studies Using Microencapsulated SeC
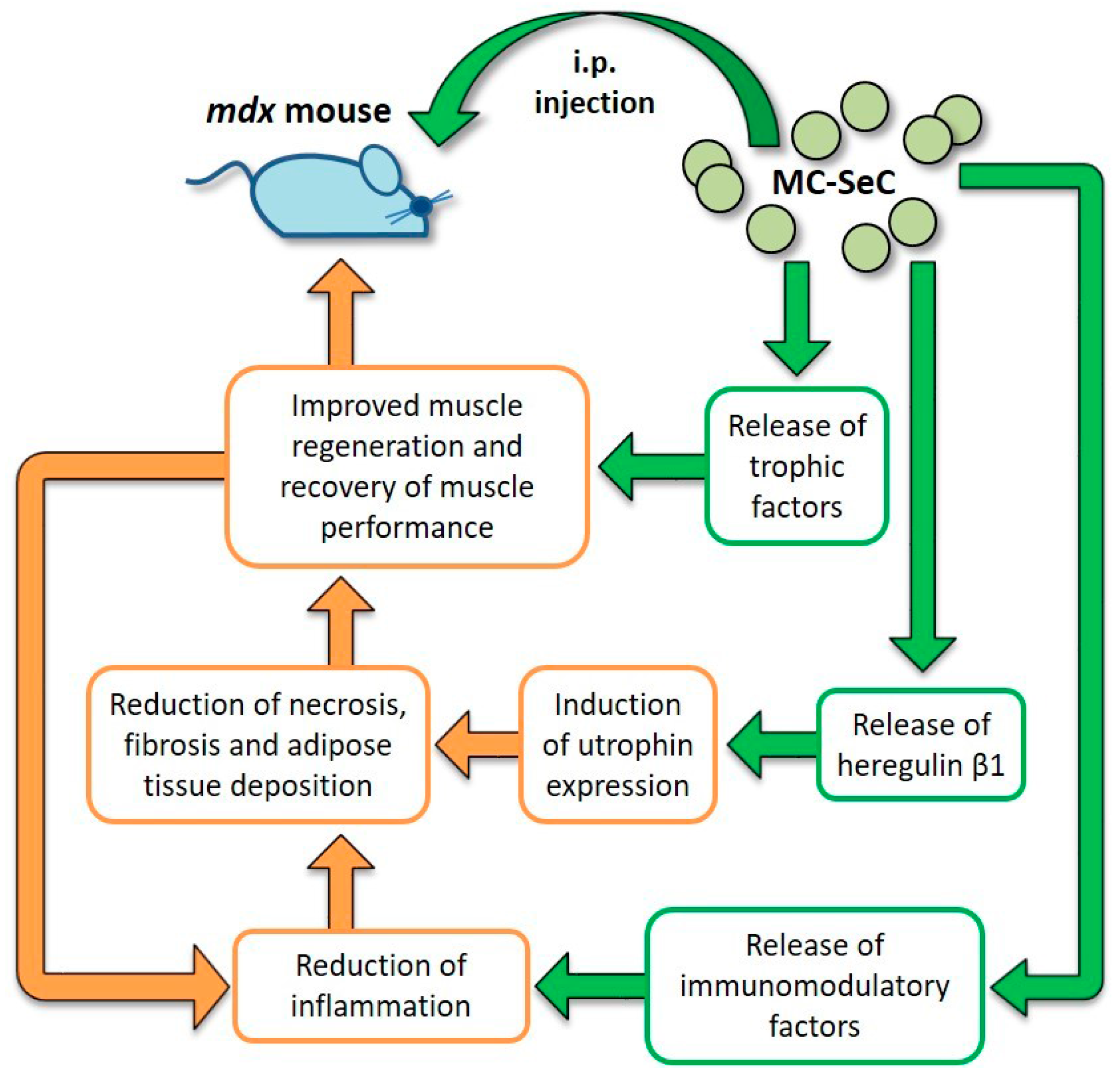
2.5. Use of MC-SeC in DMD
3. Conclusions and Remarks
Acknowledgments
Conflicts of Interest
Abbreviations
| AAV | Adeno-associated virus |
| AON | Antisense oligonucleotide |
| BTB | Blood-testis barrier |
| DAPC | Dystrophin-associated protein complex |
| DMD | Duchenne muscular dystrophy |
| E-MC | Empty microcapsules |
| GRMD | Golden retriever muscular dystrophy |
| HDAC | Histone deacetylase |
| HRGβ1 | Heregulin β 1 |
| IDO | Indoleamine 2,3-dioxygenase |
| IGF-1 | Insulin-like growth factor 1 |
| iPSC | Induced pluripotent stem cells |
| MC-SeC | Microencapsulated Sertoli cells |
| NMJ | Neuromuscular junction |
| NOD | Non-obese diabetic |
| NRG1 | Neuregulin 1 |
| PDE5 | Phosphodiesterase 5 |
| SeC | Sertoli cells |
| SPF | Specific-pathogens free |
| TGF-β | Transforming growth factor β |
References
- Dalkilic, I.; Kunkel, L.M. Muscular dystrophies: Genes to pathogenesis. Curr. Opin. Genet. Dev. 2003, 13, 231–238. [Google Scholar] [CrossRef]
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell Biol. 2006, 10, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.P.; Misyak, S.A.; Robertson, J.L.; Bassaganya−Riera, J.; Grange, R.W. Immune mediated mechanisms potentially regulate the disease time course of Duchenne muscular dystrophy and provide targets for therapeutic intervention. Am. J. Phys. Med. Rehabil. 2009, 1, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Zhao, P.; Borup, R.; Hoffman, E.P. Expression profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J. Cell Biol. 2000, 151, 1321–1336. [Google Scholar] [CrossRef] [PubMed]
- Narici, M.V.; Maffulli, N. Sarcopenia: Characteristics, mechanisms and functional significance. Br. Med. Bull. 2010, 95, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Meng, S.-J.; Yu, L.-J. Oxidative stress, molecular inflammation and sarcopenia. Int. J. Mol. Sci. 2010, 11, 1509–1526. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G. Sarcopenia and exercise “The state of the art”. J. Funct. Morphol. Kinesiol. 2017, 2, 40. [Google Scholar] [CrossRef]
- Berardi, E. Muscular dystrophies and cancer cachexia: Similarities in chronic skeletal muscle degeneration. J. Funct. Morphol. Kinesiol. 2017, 2, 39. [Google Scholar] [CrossRef]
- Muntoni, F.; Fisher, I.; Morgan, J.E.; Abraham, D. Steroids in Duchenne muscular dystrophy: From clinical trials to genomic research. Neuromuscul. Disord. 2002, 12, S162–S165. [Google Scholar] [CrossRef]
- Falzarano, M.S.; Scotton, C.; Passarelli, C.; Ferlini, A. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules 2015, 20, 18168–18184. [Google Scholar] [CrossRef] [PubMed]
- Fenichel, G.M.; Florence, J.M.; Pestronk, A.; Mendell, J.R.; Moxley, R.T., 3rd; Griggs, R.C.; Brooke, M.H.; Miller, J.P.; Robison, J.; King, W.; et al. Long-term benefit from prednisone therapy in Duchenne muscular dystrophy. Neurology 1991, 41, 1874–1877. [Google Scholar] [CrossRef] [PubMed]
- Mozzetta, C.; Minetti, G.; Puri, P.L. Regenerative pharmacology in the treatment of genetic diseases: The paradigm of muscular dystrophy. Int. J. Biochem. Cell Biol. 2008, 41, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Ichim, T.E.; Alexandrescu, D.T.; Solano, F.; Lara, F.; De Necochea Campion, R.; Paris, E.; Woods, E.J.; Murphy, M.P.; Dasanu, C.A.; Patel, A.N.; et al. Mesenchymal stem cells as anti−inflammatories: Implications for treatment of Duchenne muscular dystrophy. Cell. Immunol. 2010, 260, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Peterle, E. Old and new therapeutic developments in steroid treatment in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 9–15. [Google Scholar] [PubMed]
- Manzur, A.Y.; Kuntzer, T.; Pike, M.; Swan, A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2008, 1, CD003725. [Google Scholar] [CrossRef]
- Bruscoli, S.; Donato, V.; Velardi, E.; Di Sante, M.; Migliorati, G.; Donato, R.; Riccardi, C. Glucocorticoid-induced leucine zipper (GILZ) and long GILZ inhibit myogenic differentiation and mediate anti-myogenic effects of glucocorticoids. J. Biol. Chem. 2010, 285, 10385–10396. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.R.; Damsker, J.M.; Yu, Q.; Dillingham, B.C.; Huynh, T.; van der Meulen, J.H.; Sali, A.; Miller, B.K.; Phadke, A.; Scheffer, L.; et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol. Med. 2013, 5, 1569–1585. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Motohashi, Y.; Miyatake, S.; Komaki, H.; Takeda, S.; Aoki, Y. Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: From discovery to clinical trials. Am. J. Transl. Res. 2016, 8, 2471–2489. [Google Scholar] [PubMed]
- Partridge, T.A.; Morgan, J.E.; Coulton, G.R.; Hoffman, E.P.; Kunkel, L.M. Conversion of mdx myofibres from dystrophin-negative to positive by injection of normal myoblasts. Nature 1989, 337, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Skuk, D.; Goulet, M.; Roy, B.; Chapdelaine, P.; Bouchard, J.P.; Roy, R.; Dugre, F.J.; Sylvain, M.; Lachance, J.G.; Deschenes, L.; et al. Dystrophin expression in muscles of duchenne muscular dystrophy patients after high-density injections of normal myogenic cells. J. Neuropathol. Exp. Neurol. 2006, 65, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Sampaolesi, M.; Blot, S.; D’Antona, G.; Granger, N.; Tonlorenzi, R.; Innocenzi, A.; Mognol, P.; Thibaud, J.L.; Galvez, B.G.; Barthelemy, I.; et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 2006, 444, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Cossu, G.; Previtali, S.C.; Napolitano, S.; Cicalese, M.P.; Tedesco, F.S.; Nicastro, F.; Noviello, M.; Roostalu, U.; Natali Sora, M.G.; Scarlato, M.; et al. Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol. Med. 2015, 7, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Darabi, R.; Arpke, R.W.; Irion, S.; Dimos, J.T.; Grskovic, M.; Kyba, M.; Perlingeiro, R.C. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 2012, 10, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Giacomazzi, G.; Holvoet, B.; Trenson, S.; Caluwé, E.; Kravic, B.; Grosemans, H.; Cortés-Calabuig, Á.; Deroose, C.M.; Huylebroeck, D.; Hashemolhosseini, S.; et al. MicroRNAs promote skeletal muscle differentiation of mesodermal iPSC-derived progenitors. Nat. Commun. 2017, 8, 1249. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.Q.; Hauser, M.A.; DelloRusso, C.; Duan, D.; Crawford, R.W.; Phelps, S.F.; Harper, H.A.; Robinson, A.S.; Engelhardt, J.F.; Brooks, S.V.; et al. Modular flexibility of dystrophin: Implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002, 8, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, J.; Xiao, X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl. Acad. Sci. USA 2000, 97, 13714–13719. [Google Scholar] [CrossRef] [PubMed]
- Gregorevic, P.; Allen, J.M.; Minami, E.; Blankinship, M.J.; Haraguchi, M.; Meuse, L.; Finn, E.; Adams, M.E.; Froehner, S.C.; Murry, C.E.; et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006, 12, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Pan, X.; Hakim, C.H.; Kodippili, K.; Zhang, K.; Shin, J.H.; Yang, H.T.; McDonald, T.; Duan, D. Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum. Mol. Genet. 2015, 24, 5880–5890. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.E.; McPhee, S.W.; Li, C.; Gray, S.J.; Samulski, J.J.; Camp, A.S.; Li, J.; Wang, B.; Monahan, P.E.; Rabinowitz, J.E.; et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. 2012, 20, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Cirak, S.; Partridge, T. What Can We Learn From Clinical Trials of Exon Skipping for DMD? Mol. Ther. Nucleic Acids 2014, 3, e152. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; De Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Relizani, K.; Griffith, G.; Echevarría, L.; Zarrouki, F.; Facchinetti, P.; Vaillend, C.; Leumann, C.; Garcia, L.; Goyenvalle, A. Efficacy and Safety Profile of Tricyclo-DNA Antisense Oligonucleotides in Duchenne Muscular Dystrophy Mouse Model. Mol. Ther. Nucleic Acids 2017, 8, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.D.; Campbell, K.P. Molecular basis of muscular dystrophies. Muscle Nerve 2000, 23, 1456–1471. [Google Scholar] [CrossRef]
- Khurana, T.S.; Watkins, S.C.; Chafey, P.; Chelly, J.; Tomé, F.M.; Fardeau, M.; Kaplan, J.C.; Kunkel, L.M. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul. Disord. 1991, 1, 185–194. [Google Scholar] [CrossRef]
- Nguyen, T.M.; Ellis, J.M.; Love, D.R.; Davies, K.E.; Gatter, K.C.; Dickson, G.; Morris, G.E. Localization of the DMDL gene-encoded dystrophin-related protein using a panel of nineteen monoclonal antibodies: Presence at neuromuscular junctions, in the sarcolemma of dystrophic skeletal muscle, in vascular and other smooth muscles, and in proliferating brain cell lines. J. Cell Biol. 1991, 115, 1695–1700. [Google Scholar] [PubMed]
- Clerk, A.; Morris, G.E.; Dubowitz, V.; Davies, K.E.; Sewry, C.A. Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem. J. 1993, 25, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.M.; Fairclough, R.J.; Storer, R.; Wilkes, F.J.; Potter, A.C.; Squire, S.E.; Powell, D.S.; Cozzoli, A.; Capogrosso, R.F.; Lambert, A.; et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS ONE 2011, 6, e19189. [Google Scholar] [CrossRef] [PubMed]
- Ricotti, V.; Spinty, S.; Roper, H.; Hughes, I.; Tejura, B.; Robinson, N.; Layton, G.; Davies, K.; Muntoni, F.; Tinsley, J. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to pediatric patients with duchenne muscular dystrophy. PLoS ONE 2016, 11, e0152840. [Google Scholar] [CrossRef] [PubMed]
- Basu, U.; Gyrd-Hansen, M.; Baby, S.M.; Lozynska, O.; Krag, T.O.; Jensen, C.J.; Frödin, M.; Khurana, T.S. Heregulin-induced epigenetic regulation of the utrophin-A promoter. FEBS Lett. 2007, 581, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Krag, T.O.; Bogdanovich, S.; Jensen, C.J.; Fischer, M.D.; Hansen-Schwartz, J.; Javazon, E.H.; Flake, A.W.; Edvinsson, L.; Khurana, T.S. Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc. Natl. Acad. Sci. USA 2004, 101, 13856–13860. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 2009, 105, 19183–19187. [Google Scholar] [CrossRef] [PubMed]
- Consalvi, S.; Mozzetta, C.; Bettica, P.; Germani, M.; Fiorentini, F.; del Bene, F.; Rocchetti, M.; Leoni, F.; Monzani, V.; Mascagni, P.; et al. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol. Med. 2013, 19, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Percival, J.M.; Whitehead, N.P.; Adams, M.E.; Adamo, C.M.; Beavo, J.A.; Froehner, S.C. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J. Pathol. 2012, 228, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.D.; Rader, F.; Tang, X.; Tavyev, J.; Nelson, S.F.; Miceli, M.C.; Elashoff, R.M.; Sweeney, H.L.; Victor, R.G. PDE5 inhibition alleviates functional muscle ischemia in boys with Duchenne muscular dystrophy. Neurology 2014, 82, 2085–2091. [Google Scholar] [CrossRef] [PubMed]
- Bodanovsky, A.; Guttman, N.; Barzilai-Tutsch, H.; Genin, O.; Levy, O.; Pines, M.; Halevy, O. Halofuginone improves muscle-cell survival in muscular dystrophies. Biochim. Biophys. Acta 2014, 1843, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; Goemans, N.; van den Hauwe, M.; Meier, T. Effects of glucocorticoids and idebenone on respiratory function in patients with duchenne muscular dystrophy. Pediatr. Pulmonol. 2013, 48, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; Voit, T.; Schara, U.; Straathof, C.S.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.M.; Finkel, R.S.; Goemans, N.; McDonald, C.M.; et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): A double-blind randomised placebo-controlled phase 3 trial. Lancet 2015, 385, 1748–1757. [Google Scholar] [CrossRef]
- McDonald, C.M.; Meier, T.; Voit, T.; Schara, U.; Straathof, C.S.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.M.; Finkel, R.S.; Goemans, N.; et al. DELOS Study Group. Idebenone reduces respiratory complications in patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2016, 26, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Ito, K.; Miyagi, T.; Hiranuma, T.; Yoshioka, K.; Ozasa, S.; Matsukura, M.; Ikezawa, M.; Matsuo, M.; Takeshima, Y.; et al. A novel approach to identify Duchenne muscular dystrophy patients for aminoglycoside antibiotics therapy. Brain Dev. 2005, 27, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Kayali, R.; Ku, J.M.; Khitrov, G.; Jung, M.E.; Prikhodko, O.; Bertoni, C. Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum. Mol. Genet. 2012, 21, 4007–4020. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bonnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS ONE 2013, 8, e81302. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014, 50, 477–487. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Clinical Evaluator Training Group; ACT DMD Study Group. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACTDMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; McPherron, A.C.; Winik, N.; Lee, S.J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann. Neurol. 2002, 52, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Bogdanovich, S.; Krag, T.O.; Barton, E.R.; Morris, L.D.; Whittemore, L.A.; Ahima, R.S.; Khurana, T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002, 420, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Rong, H.; Gordi, T.; Bosley, J.; Bhattacharya, I. Translational Pharmacokinetic/Pharmacodynamic Analysis of MYO-029 Antibody for Muscular Dystrophy. Clin. Transl. Sci. 2016, 9, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, I.; Manukyan, Z.; Chan, P.; Heatherington, A.; Harnisch, L. Application of Quantitative Pharmacology Approaches in Bridging Pharmacokinetics and Pharmacodynamics of Domagrozumab From Adult Healthy Subjects to Pediatric Patients With Duchenne Muscular Disease. J. Clin. Pharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Van Deutekom, J.C.; van Ommen, G.J. Advances in Duchenne muscular dystrophy gene therapy. Nat. Rev. Genet. 2003, 4, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Bettica, P.; Petrini, S.; D’Oria, V.; D’Amico, A.; Catteruccia, M.; Pane, M.; Sivo, S.; Magri, F.; Brajkovic, S.; Messina, S.; et al. Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul. Disord. 2016, 26, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Victor, R.G.; Sweeney, H.L.; Finkel, R.; McDonald, C.M.; Byrne, B.; Eagle, M.; Goemans, N.; Vandenborne, K.; Dubrovsky, A.L.; Topaloglu, H.; et al. Tadalafil DMD Study Group. A phase 3 randomized placebo-controlled trial of tadalafil for Duchenne muscular dystrophy. Neurology 2017, 89, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Akashi Administration. Dosing and Enrollment in HT-100 Trial Suspended. Available online: http://akashirx.com/news/dosing-and-enrollment-in-ht-100-trial-suspended (accessed on 14 December 2017).
- Bhattacharya, I.; Pawlak, S.; Marraffino, S.; Christensen, J.; Sherlock, S.P.; Alvey, C.; Morris, C.; Arkin, S.; Binks, M. Safety, tolerability, pharmacokinetics, and pharmacodynamics of domagrozumab (PF-06252616), an antimyostatin monoclonal antibody, in healthy subjects. Clin. Pharmacol. Drug Dev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.D.; Griswold, M.D. The Sertoli Cell; Cache River Press: Clearwater, FL, USA, 1993. [Google Scholar]
- Mital, P.; Kaur, G.; Dufour, J.M. Immunoprotective Sertoli cells: Making allogeneic and xenogeneic transplantation feasible. Reproduction 2010, 139, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Qvist, G. John Hunter 1728–1793; William Heinemann Medical Books Ltd.: London, UK, 1981; ISBN 13:9780433270959. [Google Scholar]
- Meinhardt, A.; Hedger, M.P. Immunological, paracrine and endocrine aspects of testicular immune privilege. Mol. Cell. Endocrinol. 2011, 335, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Thompson, L.A.; Dufour, J.M. Sertoli cells—Immunological sentinels of spermatogenesis. Semin. Cell Dev. Biol. 2014, 30, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Selawry, H.P.; Cameron, D.F. Sertoli cell-enriched fractions in successful islet cell transplantation. Cell Transplant. 1993, 2, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.M.; Rajotte, R.V.; Kin, T.; Korbutt, G.S. Immunoprotection of rat islet xenografts by cotransplantation with Sertoli cells and a single injection of antilymphocyte serum. Transplantation 2003, 75, 1594–1596. [Google Scholar] [CrossRef] [PubMed]
- Shamekh, R.; El-Badri, N.S.; Saporta, S.; Pascual, C.; Sanberg, P.R.; Cameron, D.F. Sertoli cells induce systemic donor-specific tolerance in xenogenic transplantation model. Cell Transplant. 2006, 15, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Sanberg, P.R.; Borlongan, C.V.; Saporta, S.; Cameron, D.F. Testis-derived Sertoli cells survive and provide localized immunoprotection for xenografts in rat brain. Nat. Biotechnol. 1996, 14, 1692–1695. [Google Scholar] [CrossRef] [PubMed]
- Willing, A.E.; Sudberry, J.J.; Othberg, A.I.; Saporta, S.; Poulos, S.G.; Poulos, S.G.; Cameron, D.F.; Freeman, T.B.; Sanberg, P.R. Sertoli cells decrease microglial response and increase engraftment of human hNT neurons in the hemiparkinsonian rat striatum. Brain Res. Bull. 1999, 48, 441–444. [Google Scholar] [CrossRef]
- Lee, H.M.; Lim, H.G.; Oh, B.C.; Park, C.S.; Lee, D.S.; Lee, J.R. Systemic immune modulation using chemokine receptor 7 expressing porcine Sertoli cells. Xenotransplantation 2007, 14, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.G.; Lee, H.M.; Oh, B.C.; Lee, J.R. Cell-mediated immunomodulation of chemokine receptor 7-expressing porcine sertoli cells in murine heterotopic heart transplantation. J. Heart Lung Transplant. 2009, 28, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Valdés-González, R.A.; White, D.J.; Dorantes, L.M.; Terán, L.; Garibay-Nieto, G.N.; Bracho-Blanchet, E.; Dávila-Pérez, R.; Evia-Viscarra, L.; Ormsby, C.E.; Ayala-Sumuano, J.T.; et al. Three-yr follow-up of a type 1 diabetes mellitus patient with an islet xenotransplant. Clin. Transplant. 2007, 21, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Esquivel-Pérez, R.; Rodriguez-Ventura, A.L.; Dorantes, L.M.; Ramírez-González, B.; López-Santos, M.G.; Valdés-Gonzalez, R. Correlation between insulin requirements and anti-galactose antibodies in patients with type 1 diabetes transplanted with neonatal pig islets. Clin. Exp. Immunol. 2011, 165, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.I.; Willing, A.E.; Saporta, S.; Cameron, D.F.; Sanberg, P.R. Effects of Sertoli cells transplants in a 3 nitroproprionic acid model of early Huntington’s disease: A preliminary study. Neurotox. Res. 2003, 5, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Hemendinger, R.; Wang, J.; Malik, S.; Persinski, R.; Copeland, J.; Emerich, D.; Gores, P.; Halberstadt, C.; Rosenfeld, J. Sertoli cells improve survival of motor neurons in SOD1 transgenic mice, a model of amyotrophic lateral sclerosis. Exp. Neurol. 2005, 196, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Selawry, H.P.; Kotb, M.; Herrod, H.G.; Lu, Z.N. Production of a factor, or factors, suppressing IL-2 production and T cell proliferation by Sertoli cell-enriched preparations. A potential role for islet transplantation in an immunologically privileged site. Transplantation 1991, 52, 846–850. [Google Scholar] [CrossRef] [PubMed]
- De Cesaris, P.; Filippini, A.; Cervelli, C.; Riccioli, A.; Muci, S.; Starace, G.; Stefanini, M.; Ziparo, E. Immunosuppressive molecules produced by Sertoli cells cultured in vitro: Biological effects on lymphocytes. Biochem. Biophys. Res. Commun. 1992, 186, 1639–1646. [Google Scholar] [CrossRef]
- Takeda, Y.; Gotoh, M.; Dono, K.; Nishihara, M.; Grochowiecki, T.; Kimura, F.; Yoshida, T.; Ohta, Y.; Ota, H.; Ohzato, H.; et al. Protection of islet allografts transplanted together with Fas ligand expressing testicular allografts. Diabetologia 1998, 41, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Pinzon, W.; Korbutt, G.S.; Power, R.; Hooton, J.; Rajotte, R.V.; Rabinovitch, A. Testicular Sertoli cells protect islet β-cells from autoimmune destruction in NOD mice by a transforming growth factor beta1- dependent mechanism. Diabetes 2000, 49, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Luca, G.; Calvitti, M.; Mancuso, F.; Nastruzzi, C.; Fioretti, M.C.; Grohmann, U.; Becchetti, E.; Burgevin, A.; Kratzer, R.; et al. Therapy of experimental type 1 diabetes by isolated Sertoli cell xenografts alone. J. Exp. Med. 2009, 206, 2511–2526. [Google Scholar] [CrossRef] [PubMed]
- Winnall, W.R.; Muir, J.A.; Hedger, M.P. Rat resident testicular macrophages have an alternatively activated phenotype and constitutively produce interleukin-10 in vitro. J. Leukoc. Biol. 2011, 90, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Feng, X.; Han, D. Mechanisms of testicular immune privilege. Front. Biol. 2011, 6, 19–30. [Google Scholar] [CrossRef]
- Campese, A.F.; Grazioli, P.; de Cesaris, P.; Riccioli, A.; Bellavia, D.; Pelullo, M.; Padula, F.; Noce, C.; Verkhovskaia, S.; Filippini, A.; et al. Mouse Sertoli cells sustain de novo generation of regulatory T cells by triggering the notch pathway through soluble JAGGED1. Biol. Reprod. 2014, 90, 53. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.M.; Hamilton, M.; Rajotte, R.V.; Korbutt, G.S. Neonatal porcine Sertoli cells inhibit human natural antibody-mediated lysis. Biol. Reprod. 2005, 72, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Sipione, S.; Simmen, K.C.; Lord, S.J.; Motyka, B.; Ewen, C.; Shostak, I.; Rayat, G.R.; Dufour, J.M.; Korbutt, G.S.; Rajotte, R.V.; et al. Identification of a novel human granzyme B inhibitor secreted by cultured sertoli cells. J. Immunol. 2006, 177, 5051–5058. [Google Scholar] [CrossRef] [PubMed]
- Riccioli, A.; Starace, D.; Galli, R.; Fuso, A.; Scarpa, S.; Palombi, F.; de Cesaris, P.; Ziparo, E.; Filippini, A. Sertoli cells initiate testicular innate immune responses through TLR activation. J. Immunol. 2006, 177, 7122–7130. [Google Scholar] [CrossRef] [PubMed]
- Paredes Juárez, G.A.; Spasojevic, M.; Faas, M.M.; de Vos, P. Immunological and technical considerations in application of alginate-based microencapsulation systems. Front. Bioeng. Biotechnol. 2014, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Lim, F.; Sun, A.M. Microencapsulated islets as bioartificial endocrine pancreas. Science 1980, 210, 908–910. [Google Scholar] [CrossRef] [PubMed]
- Orive, G.; Hernández, R.M.; Rodríguez Gascón, A.; Calafiore, R.; Chang, T.M.; de Vos, P.; Hortelano, G.; Hunkeler, D.; Lacík, I.; Pedraz, J.L. History, challenges and perspectives of cell microencapsulation. Trends Biotechnol. 2004, 22, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Calafiore, R.; Basta, G.; Luca, G.; Lemmi, A.; Montanucci, M.P.; Calabrese, G.; Racanicchi, L.; Mancuso, F.; Brunetti, P. Microencapsulated pancreatic islet allografts into nonimmunosuppressed patients with type 1 diabetes: First two cases. Diabetes Care 2006, 29, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Calafiore, R.; Basta, G.; Luca, G.; Lemmi, A.; Racanicchi, L.; Mancuso, F.; Montanucci, M.P.; Brunetti, P. Standard technical procedures for microencapsulation of human islets for graft into nonimmunosuppressed patients with type 1 diabetes mellitus. Transplant. Proc. 2006, 38, 1156–1157. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed]
- Remminghorst, U.; Rehm, B.H. Bacterial alginates: From biosynthesis to applications. Biotechnol. Lett. 2006, 28, 1701–1712. [Google Scholar] [CrossRef] [PubMed]
- Orive, G.; Ponce, S.; Hernández, R.M.; Gascón, A.R.; Igartua, M.; Pedraz, J.L. Biocompatibility of microcapsules for cell immobilization elaborated with different type of alginates. Biomaterials 2002, 23, 3825–3831. [Google Scholar] [CrossRef]
- Zimmermann, H.; Shirley, S.G.; Zimmermann, U. Alginate-based encapsulation of cells: Past, present, and future. Curr. Diabetes Rep. 2007, 7, 314–320. [Google Scholar] [CrossRef]
- Luca, G.; Calvitti, M.; Nastruzzi, C.; Bilancetti, L.; Becchetti, E.; Angeletti, G.; Mancuso, F.; Calafiore, R. Encapsulation, in vitro characterization, and in vivo biocompatibility of Sertoli cells in alginate-based microcapsules. Tissue Eng. 2007, 13, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Aomatsu, Y.; Iwata, H.; Kin, T.; Kanehiro, H.; Hisanga, M.; Ko, S.; Nagao, M.; Harb, G.; Nakajima, Y. Survival of microencapsulated islets at 400 days posttransplantation in the omental pouch of NOD mice. Cell Transplant. 2006, 15, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Montanucci, P.; Luca, G.; Boselli, C.; Noya, G.; Barbaro, B.; Qi, M.; Kinzer, K.P.; Oberholzer, J.; Calafiore, R. Long-term metabolic and immunological follow-up of nonimmunosuppressed patients with type 1 diabetes treated with microencapsulated islet allografts: Four cases. Diabetes Care 2011, 34, 2406–2409. [Google Scholar] [CrossRef] [PubMed]
- Luca, G.; Calvitti, M.; Mancuso, F.; Falabella, G.; Arato, I.; Bellucci, C.; List, E.O.; Bellezza, E.; Angeli, G.; Lilli, C.; et al. Reversal of experimental Laron Syndrome by xenotransplantation of microencapsulated porcine Sertoli cells. J. Controll. Release 2013, 165, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Chiappalupi, S.; Luca, G.; Mancuso, F.; Madaro, L.; Fallarino, F.; Nicoletti, C.; Calvitti, M.; Arato, I.; Falabella, G.; Salvadori, L.; et al. Intraperitoneal injection of microencapsulated Sertoli cells restores muscle morphology and performance in dystrophic mice. Biomaterials 2016, 75, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Soon-Shiong, P.; Heintz, R.E.; Merideth, N.; Yao, Q.X.; Yao, Z.; Zheng, T.; Murphy, M.; Moloney, M.K.; Schmehl, M.; Harris, M.; et al. Insulin independence in a type 1 diabetic patient after encapsulated islet transplantation. Lancet 1994, 343, 950–951. [Google Scholar] [CrossRef]
- Luca, G.; Calafiore, R.; Basta, G.; Ricci, M.; Calvitti, M.; Neri, L.; Nastruzzi, C.; Becchetti, E.; Capitani, S.; Brunetti, P.; et al. Improved function of rat islets upon co-microencapsulation with Sertoli’s cells in alginate/poly-Lornithine. AAPS PharmSciTech 2001, 2, E15. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T.M.; Diakanov, I.; Selden, C.; Hodgson, H. Cotransplantation of encapsulated HepG2 and rat Sertoli cells improves outcome in a thioacetamide induced rat model of acute hepatic failure. Transplant. Int. 2005, 18, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Luca, G.; Fallarino, F.; Calvitti, M.; Mancuso, F.; Nastruzzi, C.; Arato, I.; Falabella, G.; Grohmann, U.; Becchetti, E.; Puccetti, P.; et al. Xenograft of microencapsulated sertoli cells reverses T1DM in NOD mice by inducing neogenesis of β-cells. Transplantation 2010, 90, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Bistoni, G.; Calvitti, M.; Mancuso, F.; Arato, I.; Falabella, G.; Cucchia, R.; Fallarino, F.; Becchetti, A.; Baroni, T.; Mazzitelli, S.; et al. Prolongation of skin allograft survival in rats by the transplantation of microencapsulated xenogeneic neonatal porcine Sertoli cells. Biomaterials 2012, 33, 5333–5340. [Google Scholar] [CrossRef] [PubMed]
- Luca, G.; Bellezza, I.; Arato, I.; di Pardo, A.; Mancuso, F.; Calvitti, M.; Falabella, G.; Bartoli, S.; Maglione, V.; Amico, E.; et al. Terapeutic Potential of Microencapsulated Sertoli Cells in Huntington Disease. CNS Neurosci. Ther. 2016, 22, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Luca, G.; Cameron, D.F.; Arato, I.; Mancuso, F.; Linden, E.H.; Calvitti, M.; Falabella, G.; Szekeres, K.; Bodo, M.; Ricci, G.; et al. Xenograft of microencapsulated Sertoli cells for the cell therapy of type 2 diabetes mellitus in spontaneously diabetic nonhuman primates: Preliminary data. Transplant. Proc. 2014, 46, 1999–2001. [Google Scholar] [CrossRef] [PubMed]
- Chiappalupi, S.; Luca, G.; Mancuso, F.; Madaro, L.; Fallarino, F.; Nicoletti, C.; Calvitti, M.; Arato, I.; Falabella, G.; Salvadori, L.; et al. Effects of intraperitoneal injection of microencapsulated Sertoli cells on chronic and presymptomatic dystrophic mice. Data Brief 2015, 15, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E.H. Duchenne Muscular Dystrophy, 2nd ed.; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Musarò, A.; McCullagh, K.; Paul, A.; Houghton, L.; Dobrowolny, G.; Molinaro, M.; Barton, E.R.; Sweeney, H.L.; Rosenthal, N. Localized IGF-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat. Genet. 2001, 27, 195–200. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Approach | Mechanism of Action | Positive Effects | Side Effects/Limitations | Clinical Trial | Refs. |
|---|---|---|---|---|---|
| Givinostat (HDAC inhibitor) | Not completely clarified in DMD.Downregulation of myostatin due to increased expression of follistatin | Improved muscle morphology and increase in myofiber size in mdx mice and DMD patients. | Platelet reduction, gastrointestinal adverse effects. | Phase II ongoing | [47,48,69] |
| Tadalafil and Sildenafil (PDE5 inhibitors) | Increased levels of cGMP inducing vasodilatation. | Prevention of exercise-induced ischemia, injury and fatigue in mdx mice. | Headache, fall, upper respiratory tract infection, gastrointestinal adverse effects. | Phase III failed in protecting versus ambulatory decline | [19,49,50,70] |
| Halofuginone (HT-100) | Anti-fibrotic agent | Reduction of fibrotic tissue deposition | No serious adverse effects reported for low doses | Phase II suspended for death of a patient receiving high doses | [51,71] |
| Idebenone (Catena/Raxone) | Antioxidant inducing mitochondrial electron reflux and cellular energy production; inhibition of lipid peroxidation | Improved cardiac and muscle functionality in mdx mice; improved respiratory function in DMD patients | Nasopharyngitis, headache and mild diarrhoea | Phase III completed | [52,53,54] |
| Ataluren (Translarna, PTC124) | Premature stop codons readthrough | Restoration of dystrophin expression in mdx mice | No severe adverse effects reported. Specific for nonsense mutations | Phase III completed | [55,56,57,58,59] |
| Domagrozumab (Human anti-myostatin mAb) | Blockade of myostatin activity | Increment in muscle mass and strength in mdx mice and healthy subject | Headache, fatigue, upper respiratory tract infections, and muscle spasms. | Phase II recruiting | [60,61,62,63,64,72] |
| CRISPR/Cas9 | Cleavage of specific DNA sequences to remove the mutated exon | Improved skeletal and cardiac muscle morphology and functionality in mdx mice | Mutation-dependent. No adverse effects reported | Preclinical | [65,66,67,73] |
| Growth Factors/Cytokines | Immunomodulatory Factors | Maturative Factors/Hormones |
|---|---|---|
| BDNF (Brain Derived Neurotrophic Factor) and GDNF (Glial cell-Derived Neurotrophic Factor) | Activin A | Activins/Inhibins |
| bFGF (basic Fibroblast Growth Factor) | Clusterin | Dhh (Desert hedgehog) |
| EGF (Epidermal Growth Factor) | Complement cascade inhibitors | Estrogens |
| HRG (Heregulin)-β1 | FasL (Fas Ligand) | KL/SCF (Kit Ligand/Stem Cell Factor) |
| hSCSGF (human Sertoli Cell Secreted Growth Factor) | IDO (Indolamine 2,3-Dioxygenase) | MIS/AMH (Müllerian Inhibiting Substance/Anti Müllerian Hormone) |
| IFN (Interferon)-γ | IL-2 suppressor factors | |
| IGF (Insulin-like Growth Factor)-I and -II | JAG1 (soluble JAGGED1) | Antiapoptotic factors |
| IL (Interleukin)-1 and -6 | MIF (Macrophages Inhibitor Factor) | BCL-w |
| NT (Neurotrophin)-3 | Serpins (serine protease inhibitors) | |
| PDGF (Platelet Derived Growth Factor) | TGF-β | |
| SGP (Sulfated Glycoprotein-1/Prosaposin) | Transferrin | |
| TGF (Transforming Growth Factor)-α and -β | ||
| VEGF (Vascular Endothelial Growth Factor) |
| Approach | Mechanism of Action | Positive Effects | Side Effects | Clinical Trial | Refs. |
|---|---|---|---|---|---|
| MC-SeC | A cocktail of antiinflammatory and trophic factors secreted by SeC reaches muscle tissue from the peritoneal cavity. Induction of utrophin expression via SeC-released HRGβ1 | Reduction of muscle inflammation, necrosis and fibrosis. Increased expression of utrophin. Improved muscle morphology and functionality; incr eased resistance to exercise-induced muscle damage | No side effects observed | Preclinical | [114,122] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiappalupi, S.; Salvadori, L.; Luca, G.; Riuzzi, F.; Calafiore, R.; Donato, R.; Sorci, G. Employment of Microencapsulated Sertoli Cells as a New Tool to Treat Duchenne Muscular Dystrophy. J. Funct. Morphol. Kinesiol. 2017, 2, 47. https://doi.org/10.3390/jfmk2040047
Chiappalupi S, Salvadori L, Luca G, Riuzzi F, Calafiore R, Donato R, Sorci G. Employment of Microencapsulated Sertoli Cells as a New Tool to Treat Duchenne Muscular Dystrophy. Journal of Functional Morphology and Kinesiology. 2017; 2(4):47. https://doi.org/10.3390/jfmk2040047
Chicago/Turabian StyleChiappalupi, Sara, Laura Salvadori, Giovanni Luca, Francesca Riuzzi, Riccardo Calafiore, Rosario Donato, and Guglielmo Sorci. 2017. "Employment of Microencapsulated Sertoli Cells as a New Tool to Treat Duchenne Muscular Dystrophy" Journal of Functional Morphology and Kinesiology 2, no. 4: 47. https://doi.org/10.3390/jfmk2040047
APA StyleChiappalupi, S., Salvadori, L., Luca, G., Riuzzi, F., Calafiore, R., Donato, R., & Sorci, G. (2017). Employment of Microencapsulated Sertoli Cells as a New Tool to Treat Duchenne Muscular Dystrophy. Journal of Functional Morphology and Kinesiology, 2(4), 47. https://doi.org/10.3390/jfmk2040047