Insights into the Pathogenic Secondary Symptoms Caused by the Primary Loss of Dystrophin



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Animal Models of Muscular Dystrophy
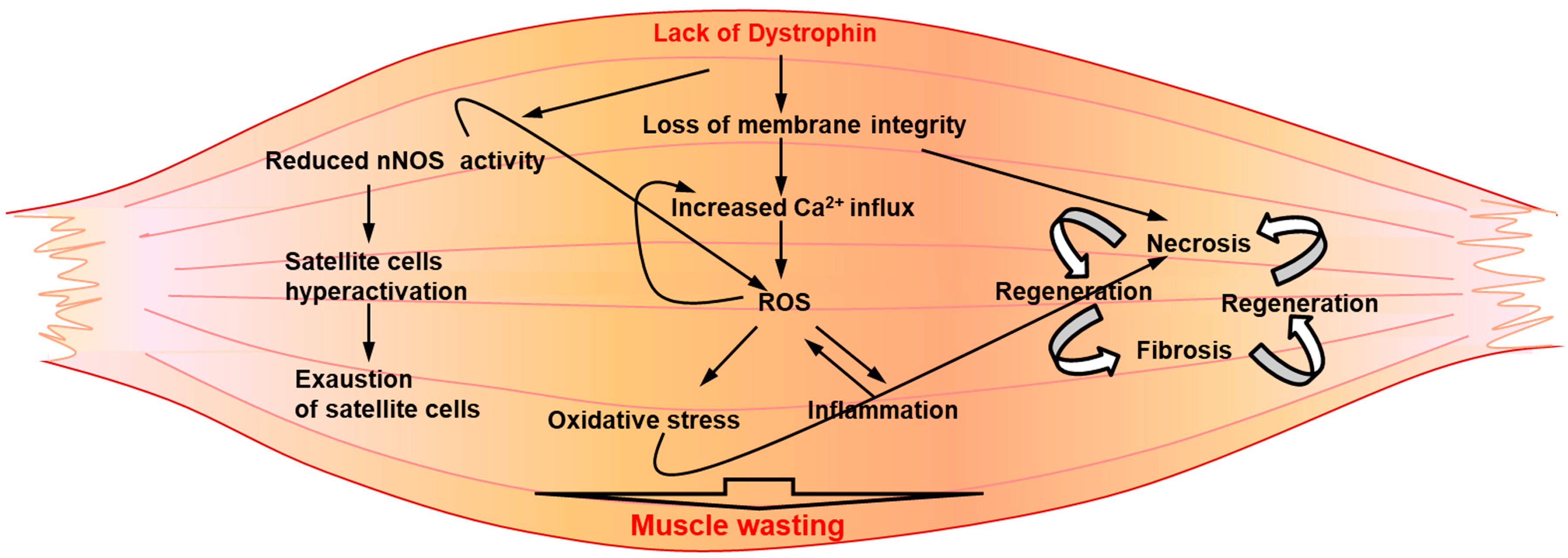
3. Secondary Pathogenic Mechanisms Underlining DMD Pathology
3.1. Continuous Cycles of Myofiber Degeneration and Regeneration
3.2. Inflammation
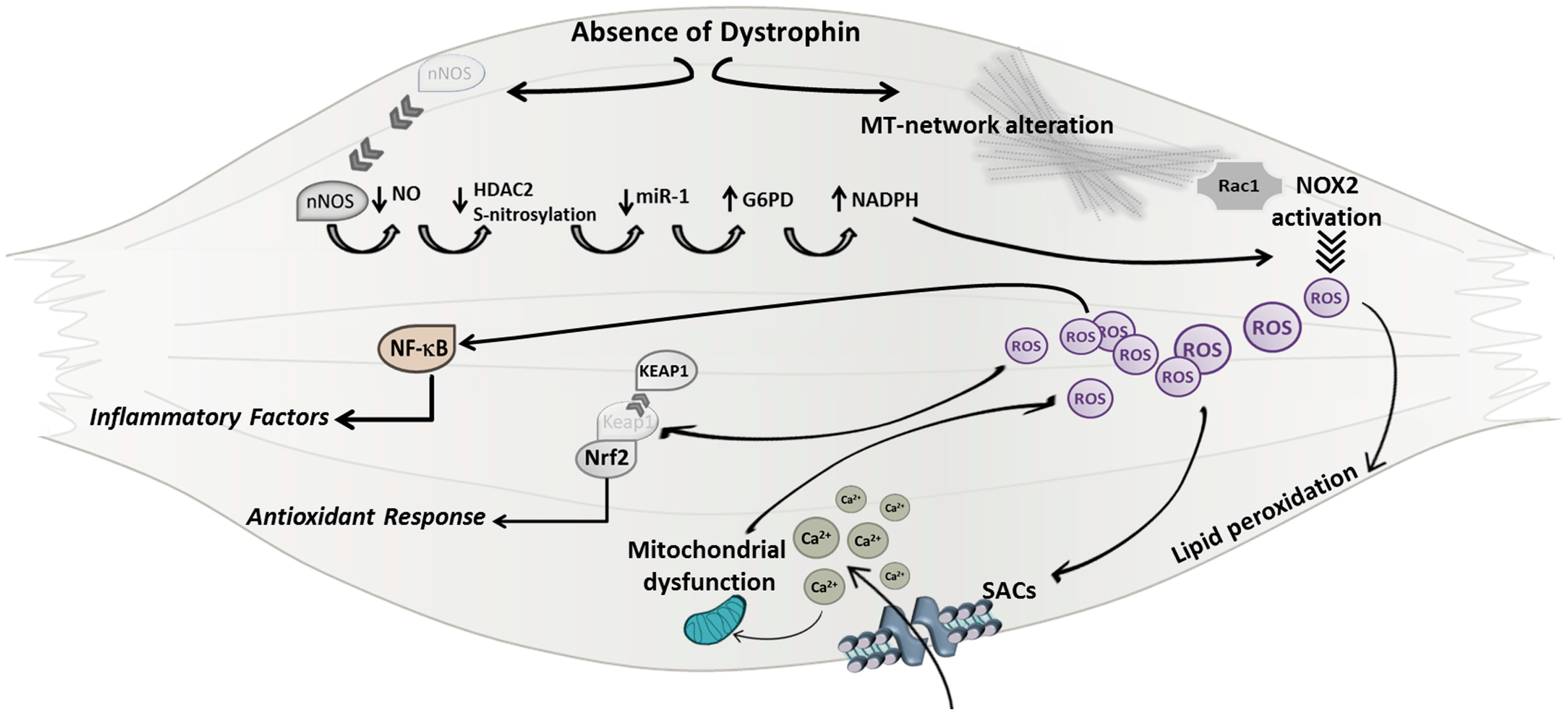
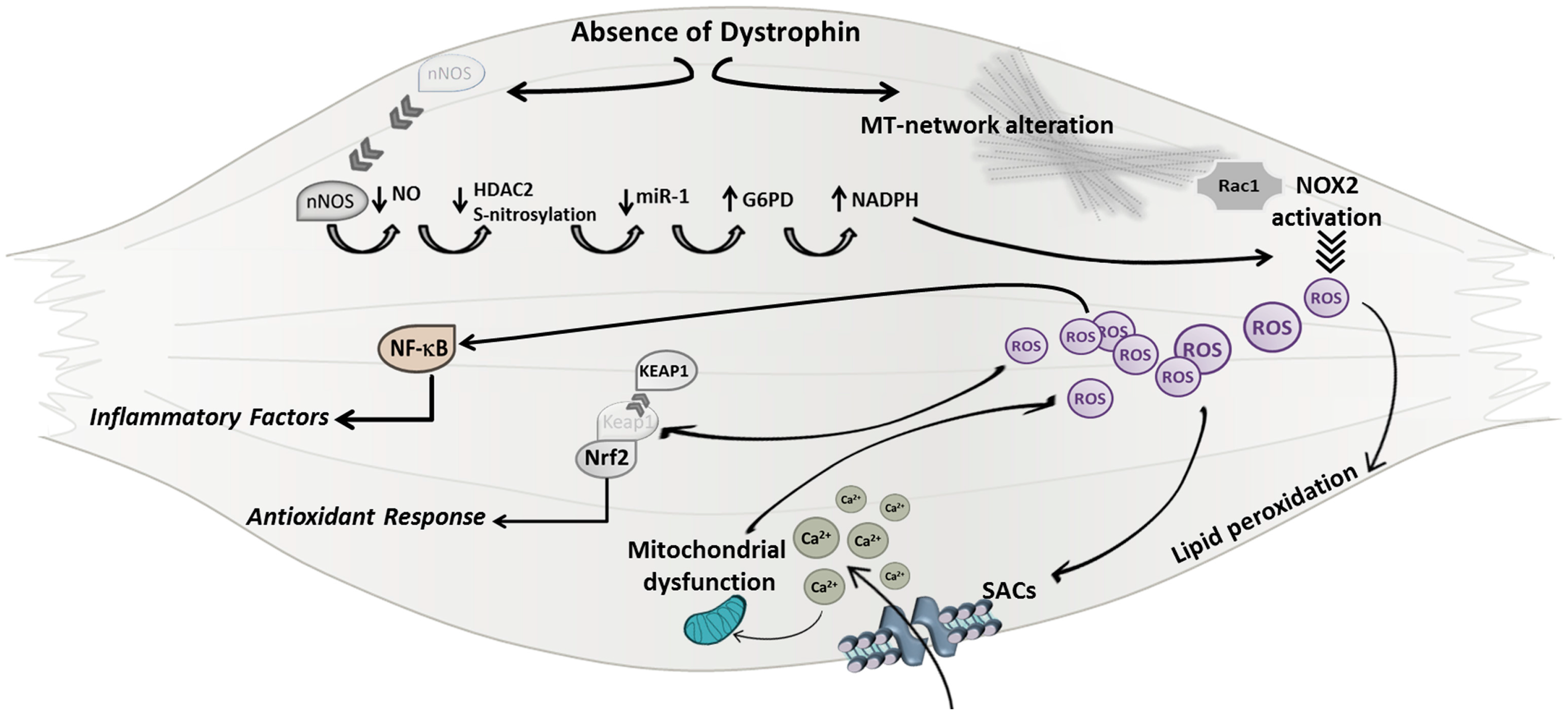
3.3. Oxidative Stress
4. Crosstalk between Oxidative Stress and Inflammatory Response
5. Therapeutic Approaches Direct against Pathogenic Mechanisms Downstream in the Absence of Dystrophin Protein
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Guiraud, S.; Chen, H.; Burns, D.T.; Davies, K.E. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Exp. Physiol. 2015, 100, 1458–1467. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; Edwards, B.; Squire, S.E.; Babbs, A.; Shah, N.; Berg, A.; Chen, H.; Davies, K.E. Identification of serum protein biomarkers for utrophin based DMD therapy. Sci. Rep. 2017, 7, 43697. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; Davies, K.E. Pharmacological advances for treatment in Duchenne muscular dystrophy. Curr. Opin. Pharmacol. 2017, 34, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Neve, R.L.; Colletti-Feener, C.; Bertelson, C.J.; Kurnit, D.M.; Kunkel, L.M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 1986, 323, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Consalvi, S.; Saccone, V.; Giordani, L.; Minetti, G.; Mozzetta, C.; Puri, P.L. Histone deacetylase inhibitors in the treatment of muscular dystrophies: Epigenetic drugs for genetic diseases. Mol. Med. 2011, 17, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.J. Two tales concerning skeletal muscle. J. Clin. Investig. 2007, 117, 2388–2391. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.F.; Kunkel, L.M. Dystrophin and muscular dystrophy: Past, present, and future. Mol. Genet. Metab. 2001, 74, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Radley, H.G.; Lynch, G.S.; Nagaraju, K.; de Luca, A. Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiol. Dis. 2008, 31, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, E.; Abdel-Meguid, I.; Korraa, S.S. Markers of degeneration and regeneration in Duchenne muscular dystrophy. Acta Myol. 2009, 28, 94–100. [Google Scholar] [PubMed]
- Carnwath, J.W.; Shotton, D.M. Muscular dystrophy in the mdx mouse: Histopathology of the soleus and extensor digitorum longus muscles. J. Neurol. Sci. 1987, 80, 39–54. [Google Scholar] [CrossRef]
- Deconinck, N.; Dan, B. Pathophysiology of duchenne muscular dystrophy: Current hypotheses. Pediatr. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.P.; Misyak, S.A.; Robertson, J.L.; Bassaganya-Riera, J.; Grange, R.W. Dysregulated intracellular signaling and inflammatory gene expression during initial disease onset in Duchenne muscular dystrophy. Am. J. Phys. Med. Rehabil. 2009, 88, 502–522. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Long, C.; Yue, Y.; Duan, D. Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum. Mol. Genet. 2009, 18, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Pescatori, M.; Broccolini, A.; Minetti, C.; Bertini, E.; Bruno, C.; D’amico, A.; Bernardini, C.; Mirabella, M.; Silvestri, G.; Giglio, V.; et al. Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007, 21, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.D.; Khanna, S.; Kaminski, H.J.; Rao, J.S.; Merriam, A.P.; Richmonds, C.R.; Leahy, P.; Li, J.; Guo, W.; Andrade, F.H. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2002, 11, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Connolly, A.M.; Keeling, R.M.; Mehta, S.; Pestronk, A.; Sanes, J.R. Three mouse models of muscular dystrophy: The natural history of strength and fatigue in dystrophin-, dystrophin/utrophin-, and laminin α2-deficient mice. Neuromuscul. Disord. 2001, 11, 703–712. [Google Scholar] [CrossRef]
- Deconinck, N.; Tinsley, J.; De Backer, F.; Fisher, R.; Kahn, D.; Phelps, S.; Davies, K.; Gillis, J.M. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat. Med. 1997, 3, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Grady, R.M.; Teng, H.; Nichol, M.C.; Cunningham, J.C.; Wilkinson, R.S.; Sanes, J.R. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: A model for Duchenne muscular dystrophy. Cell 1997, 90, 729–738. [Google Scholar] [CrossRef]
- Megeney, L.A.; Kablar, B.; Garrett, K.; Anderson, J.E.; Rudnicki, M.A. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev. 1996, 10, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Raymackers, J.M.; Debaix, H.; Colson-Van Schoor, M.; De Backer, F.; Tajeddine, N.; Schwaller, B.; Gailly, P.; Gillis, J.M. Consequence of parvalbumin deficiency in the mdx mouse: Histological, biochemical and mechanical phenotype of a new double mutant. Neuromuscul. Disord. 2003, 13, 376–387. [Google Scholar] [CrossRef]
- Guo, C.; Willem, M.; Werner, A.; Raivich, G.; Emerson, M.; Neyses, L.; Mayer, U. Absence of α 7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum. Mol. Genet. 2006, 15, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Mourkioti, F.; Tran, R.; Choi, J.; Llewellyn, M.; Kraft, P.; Shkreli, M.; Delp, S.; Pomerantz, J.H.; Artandi, S.E.; et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 2010, 143, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Hathout, Y.; Marathi, R.L.; Rayavarapu, S.; Zhang, A.; Brown, K.J.; Seol, H.; Gordish-Dressman, H.; Cirak, S.; Bello, L.; Nagaraju, K.; et al. Discovery of serum protein biomarkers in the mdx mouse model and cross-species comparison to Duchenne muscular dystrophy patients. Hum. Mol. Genet. 2014, 23, 6458–6469. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; von Maltzahn, J.; Dumont, N.A.; Stark, D.A.; Wang, Y.X.; Nhan, K.; Frenette, J.; Cornelison, D.D.; Rudnicki, M.A. Wnt7a stimulates myogenic stem cell motility and engraftment resulting in improved muscle strength. J. Cell Biol. 2014, 205, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Consalvi, S.; Mozzetta, C.; Bettica, P.; Germani, M.; Fiorentini, F.; del Bene, F.; Rocchetti, M.; Leoni, F.; Monzani, V.; Mascagni, P.; et al. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol. Med. 2013, 19, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Denti, M.A.; Rosa, A.; D’Antona, G.; Sthandier, O.; de Angelis, F.G.; Nicoletti, C.; Allocca, M.; Pansarasa, O.; Parente, V.; Musaro, A.; et al. Body-wide gene therapy of Duchenne muscular dystrophy in the mdx mouse model. Proc. Natl. Acad. Sci. USA 2006, 103, 3758–3763. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Berardinelli, M.G.; De Pasquale, L.; Nicoletti, C.; D’amico, A.; Carvello, F.; Moneta, G.M.; Catizone, A.; Bertini, E.; de Benedetti, F.; et al. Functional and morphological improvement of dystrophic muscle by interleukin 6 receptor blockade. EBioMedicine 2015, 2, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Quattrocelli, M.; Salamone, I.M.; Page, P.G.; Warner, J.L.; Demonbreun, A.R.; McNally, E.M. Intermittent glucocorticoid dosing improves muscle repair and function in mice with limb-girdle muscular dystrophy. Am. J. Pathol. 2017, 187, 2520–2535. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, A.; Wuebbles, R.D.; Fontelonga, T.M.; Tarchione, A.R.; Mathews Griner, L.A.; Heredia, D.J.; Nunes, A.M.; Duan, S.; Brewer, P.D.; van Ry, T.; et al. SU9516 Increases α7β1 Integrin and ameliorates disease progression in the mdx mouse model of duchenne muscular dystrophy. Mol. Ther. 2017, 25, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, F.S.; Hoshiya, H.; D’Antona, G.; Gerli, M.F.; Messina, G.; Antonini, S.; Tonlorenzi, R.; Benedetti, S.; Berghella, L.; Torrente, Y.; et al. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci. Transl. Med. 2011, 3, 96ra78. [Google Scholar] [CrossRef] [PubMed]
- Vojnits, K.; Pan, H.; Dai, X.; Sun, H.; Tong, Q.; Darabi, R.; Huard, J.; Li, Y. Functional neuronal differentiation of injury-induced muscle-derived stem cell-like cells with therapeutic implications. Sci. Rep. 2017, 7, 1177. [Google Scholar] [CrossRef] [PubMed]
- Blau, H.M.; Webster, C.; Chiu, C.P.; Guttman, S.; Chandler, F. Differentiation properties of pure populations of human dystrophic muscle cells. Exp. Cell Res. 1983, 144, 495–503. [Google Scholar] [CrossRef]
- Dadgar, S.; Wang, Z.; Johnston, H.; Kesari, A.; Nagaraju, K.; Chen, Y.W.; Hill, D.A.; Partridge, T.A.; Giri, M.; Freishtat, R.J.; et al. Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J. Cell Biol. 2014, 207, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Chevalier, F.P.; Rudnicki, M.A. Satellite cells in muscular dystrophy—Lost in polarity. Trends Mol. Med. 2016, 22, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Musaro, A. The basis of muscle regeneration. Adv. Biol. 2014, 1–16. [Google Scholar] [CrossRef]
- Almada, A.E.; Wagers, A.J. Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat. Rev. Mol. Cell Biol. 2016, 17, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Sica, G.; Musaro, A. Stem cells and tissue niche: Two faces of the same coin of muscle regeneration. Eur. J. Transl. Myol. 2016, 26, 6125. [Google Scholar] [CrossRef] [PubMed]
- Wada, E.; Tanihata, J.; Iwamura, A.; Takeda, S.; Hayashi, Y.K.; Matsuda, R. Treatment with the anti-IL-6 receptor antibody attenuates muscular dystrophy via promoting skeletal muscle regeneration in dystrophin-/utrophin-deficient mice. Skelet. Muscle 2017, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Kottlors, M.; Kirschner, J. Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res. 2010, 340, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 is required for the specification of myogenic satellite cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Martone, J.; Girardi, E.; Cesana, M.; Incitti, T.; Morlando, M.; Nicoletti, C.; Santini, T.; Sthandier, O.; Barberi, L.; et al. MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab. 2010, 12, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Nagaraju, K.; Bakay, M.; McIntyre, O.; Rawat, R.; Shi, R.; Hoffman, E.P. Early onset of inflammation and later involvement of TGF β in Duchenne muscular dystrophy. Neurology 2005, 65, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Radley, H.G.; Gebski, B.L.; Bogoyevitch, M.A.; Shavlakadze, T. Implications of cross-talk between tumour necrosis factor and insulin-like growth factor-1 signalling in skeletal muscle. Clin. Exp. Pharmacol. Physiol. 2008, 35, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Echigoya, Y.; Fukada, S.I.; Yokota, T. Current Translational research and murine models for Duchenne muscular dystrophy. J. Neuromuscul. Dis. 2016, 3, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.R.; Morrison, K.E.; Salmon, M.; Buckley, C.D. Why does inflammation persist: A dominant role for the stromal microenvironment? Expert Rev. Mol. Med. 2002, 4, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Kurek, J.B.; Nouri, S.; Kannourakis, G.; Murphy, M.; Austin, L. Leukemia inhibitory factor and interleukin-6 are produced by diseased and regenerating skeletal muscle. Muscle Nerve 1996, 19, 1291–1309. [Google Scholar] [CrossRef]
- Serrano, A.L.; Baeza-Raja, B.; Perdiguero, E.; Jardi, M.; Munoz-Canoves, P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008, 7, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.N. Interleukin-6 induces skeletal muscle protein breakdown in rats. Proc. Soc. Exp. Biol. Med. 1994, 205, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Zaldivar, F.; Cooper, D.M.; Adams, G.R. IL-6-induced skeletal muscle atrophy. J. Appl. Physiol. 2005, 98, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Tsujinaka, T.; Ebisui, C.; Fujita, J.; Kishibuchi, M.; Morimoto, T.; Ogawa, A.; Katsume, A.; Ohsugi, Y.; Kominami, E.; Monden, M. Muscle undergoes atrophy in association with increase of lysosomal cathepsin activity in interleukin-6 transgenic mouse. Biochem. Biophys. Res. Commun. 1995, 207, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Vita, G.L.; Aguennouz, M.; Sframeli, M.; Romeo, S.; Rodolico, C.; Vita, G. Activation of NF-kappaB pathway in Duchenne muscular dystrophy: Relation to age. Acta Myol. 2011, 30, 16–23. [Google Scholar] [PubMed]
- Pelosi, L.; Berardinelli, M.G.; Forcina, L.; Spelta, E.; Rizzuto, E.; Nicoletti, C.; Camilli, C.; Testa, E.; Catizone, A.; De Benedetti, F.; et al. Increased levels of interleukin-6 exacerbate the dystrophic phenotype in mdx mice. Hum. Mol. Genet. 2015, 24, 6041–6053. [Google Scholar] [CrossRef] [PubMed]
- Kostek, M.C.; Nagaraju, K.; Pistilli, E.; Sali, A.; Lai, S.H.; Gordon, B.; Chen, Y.W. IL-6 signaling blockade increases inflammation but does not affect muscle function in the mdx mouse. BMC Musculoskelet. Disord. 2012, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Price, F.D.; von Maltzahn, J.; Bentzinger, C.F.; Dumont, N.A.; Yin, H.; Chang, N.C.; Wilson, D.H.; Frenette, J.; Rudnicki, M.A. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 2014, 20, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Tierney, M.T.; Aydogdu, T.; Sala, D.; Malecova, B.; Gatto, S.; Puri, P.L.; Latella, L.; Sacco, A. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med. 2014, 20, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.R.; McMillan, D.C.; Crilly, A.; McArdle, C.S.; Milroy, R. The relationship between weight loss and interleukin 6 in non-small-cell lung cancer. Br. J. Cancer 1996, 73, 1560–1562. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.J.; Marino, M.W.; Winckler, W.M. Altered pathological progression of diaphragm and quadriceps muscle in TNF-deficient, dystrophin-deficient mice. Neuromuscul. Disord. 2000, 10, 612–619. [Google Scholar] [CrossRef]
- Arthur, P.G.; Grounds, M.D.; Shavlakadze, T. Oxidative stress as a therapeutic target during muscle wasting: Considering the complex interactions. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Moylan, J.S.; Reid, M.B. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 2007, 35, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kwak, H.B.; Thompson, L.V.; Lawler, J.M. Contribution of oxidative stress to pathology in diaphragm and limb muscles with Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rando, T.A. Oxidative stress and the pathogenesis of muscular dystrophies. Am. J. Phys. Med. Rehabil. 2002, 81, S175–S186. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Mendell, J.R. Emerging drugs for Duchenne muscular dystrophy. Expert Opin. Emerg. Drugs 2012, 17, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Musaro, A.; Fulle, S.; Fano, G. Oxidative stress and muscle homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Pelosi, L.; Sica, G.; Musaro, A. The physiopathologic role of oxidative stress in skeletal muscle. Mech. Ageing Dev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.J.; Herting, D.C.; Hurst, V.; Finch, S.C.; Spiro, H.M. Tocopherol deficiency in man. N. Engl. J. Med. 1965, 273, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yang, C.; Xue, R.; Li, S.; Zhang, T.; Pan, L.; Ma, X.; Wang, L.; Li, D. Sulforaphane alleviates muscular dystrophy in mdx mice by activation of Nrf2. J. Appl. Physiol. 2015, 118, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, S.; Li, D. Sulforaphane mitigates muscle fibrosis in mdx mice via Nrf2-mediated inhibition of TGF-β/Smad signaling. J. Appl. Physiol. 2016, 120, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.C.; Li, S.J.; Yang, C.L.; Xue, R.L.; Xi, Y.Y.; Wang, L.; Zhao, Q.L.; Li, D.J. Sulforaphane Attenuates Muscle Inflammation in Dystrophin-deficient mdx Mice via NF-E2-related Factor 2 (Nrf2)-mediated Inhibition of NF-κB Signaling Pathway. J. Biol. Chem. 2015, 290, 17784–17795. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Pelosi, L.; Piemonte, F.; Travaglini, L.; Forcina, L.; Catteruccia, M.; Petrini, S.; Verardo, M.; D’amico, A.; Musaro, A.; et al. Oxidative stress in Duchenne muscular dystrophy: Focus on the NRF2 redox pathway. Hum. Mol. Genet. 2017, 26, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, R.J.; Shi, G.; Sbrana, F.; Prosser, B.L.; Borroto, C.; Mazaitis, M.J.; Hoffman, E.P.; Mahurkar, A.; Sachs, F.; Sun, Y.; et al. Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci. Signal. 2012, 5, ra56. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Forcina, L.; Nicoletti, C.; Scicchitano, B.M.; Musaro, A. Increased circulating levels of Interleukin-6 Induce Perturbation in redox-regulated signaling cascades in muscle of dystrophic mice. Oxid. Med. Cell Longev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Canton, M.; Menazza, S.; Di Lisa, F. Oxidative stress in muscular dystrophy: From generic evidence to specific sources and targets. J. Muscle Res. Cell Motil. 2014, 35, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Prosser, B.L.; Khairallah, R.J.; Ziman, A.P.; Ward, C.W.; Lederer, W.J. X-ROS signaling in the heart and skeletal muscle: Stretch-dependent local ROS regulates [Ca2+]i. J. Mol. Cell Cardiol. 2013, 58, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Sanchez, G.; Barrientos, G.; Aracena-Parks, P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S-glutathionylation. J. Biol. Chem. 2006, 281, 26473–26482. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.Y.; Turner, P.R.; Denetclaw, W.F.; Steinhardt, R.A. Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science 1990, 250, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Franco, A., Jr.; Lansman, J.B. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature 1990, 344, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Gumerson, J.D.; Michele, D.E. The dystrophin-glycoprotein complex in the prevention of muscle damage. J. Biomed. Biotechnol. 2011, 2011, 210797. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; Westwood, T.; Regen, C.M.; Steinhardt, R.A. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature 1988, 335, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Yeung, E.W.; Whitehead, N.P.; Suchyna, T.M.; Gottlieb, P.A.; Sachs, F.; Allen, D.G. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 2005, 562, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Isaeva, E.V.; Shirokova, N. Metabolic regulation of Ca2+ release in permeabilized mammalian skeletal muscle fibres. J. Physiol. 2003, 547, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Isaeva, E.V.; Shkryl, V.M.; Shirokova, N. Mitochondrial redox state and Ca2+ sparks in permeabilized mammalian skeletal muscle. J. Physiol. 2005, 565, 855–872. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Shkryl, V.M.; Martins, A.S.; Ullrich, N.D.; Nowycky, M.C.; Niggli, E.; Shirokova, N. Reciprocal amplification of ROS and Ca2+ signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch. 2009, 458, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs. preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.W.; Humston, J.L.; Mehta, A.; Tate, V.; Ralston, E.; Ervasti, J.M. Dystrophin is a microtubule-associated protein. J. Cell Biol. 2009, 186, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Yoshida, M.; Yamamoto, H.; Ozawa, E. Glycoprotein-binding site of dystrophin is confined to the cysteine-rich domain and the first half of the carboxy-terminal domain. FEBS Lett. 1992, 308, 154–160. [Google Scholar] [CrossRef]
- Tidball, J.G.; Wehling-Henricks, M. The role of free radicals in the pathophysiology of muscular dystrophy. J. Appl. Physiol. 2007, 102, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Bray-French, K.; Drewe, J. Pharmacokinetic evaluation of idebenone. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, M.; Vankan, P.; Dreier, M.; Kutz, K.W.; Drewe, J. Pharmacokinetics and metabolism of idebenone in healthy male subjects. Eur. J. Clin. Pharmacol. 2009, 65, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; Van der, M.G.; Erb, M.; D’hooge, J.; Herijgers, P.; Verbeken, E.; Jara, A.; Van Den, B.A.; Mertens, L.; Courdier-Fruh, I.; et al. Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: Cardiac protection and improved exercise performance. Eur. Heart J. 2009, 30, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Gillis, J.C.; Benefield, P.; McTavish, D. Idebenone, A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in age-related cognitive disorders. Drugs Aging 1994, 5, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. How NF-kappaB is activated: The role of the I κB kinase (IKK) complex. Oncogene 1999, 18, 6867–6874. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Delhase, M. The I kappa B kinase (IKK) and NF-kappa B: Key elements of proinflammatory signalling. Semin. Immunol. 2000, 12, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Boriek, A.M. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: A possible role in Duchenne muscular dystrophy. FASEB J. 2003, 17, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Altavilla, D.; Aguennouz, M.; Seminara, P.; Minutoli, L.; Monici, M.C.; Bitto, A.; Mazzeo, A.; Marini, H.; Squadrito, F.; et al. Lipid peroxidation inhibition blunts nuclear factor-kappaB activation, reduces skeletal muscle degeneration, and enhances muscle function in mdx mice. Am. J. Pathol. 2006, 168, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Karin, M. The IKK/NF-kappa B pathway. Crit. Care Med. 2002, 30, S18–S26. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Bitto, A.; Aguennouz, M.; Minutoli, L.; Monici, M.C.; Altavilla, D.; Squadrito, F.; Vita, G. Nuclear factor kappa-B blockade reduces skeletal muscle degeneration and enhances muscle function in Mdx mice. Exp. Neurol. 2006, 198, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Traynor, K. Deflazacort approved for Duchenne muscular dystrophy. Am. J. Health Syst. Pharm. 2017, 74, 368. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Shimizu-Motohashi, Y.; Takeda, S.; Aoki, Y. Anti-inflammatory drugs for Duchenne muscular dystrophy: Focus on skeletal muscle-releasing factors. Drug Des. Dev. Ther. 2016, 10, 2745–2758. [Google Scholar] [CrossRef]
- Buyse, G.M.; Voit, T.; Schara, U.; Straathof, C.S.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.M.; Finkel, R.S.; Goemans, N.; McDonald, C.M.; et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): A double-blind randomised placebo-controlled phase 3 trial. Lancet 2015, 385, 1748–1757. [Google Scholar] [CrossRef]
- Buyse, G.M.; Voit, T.; Schara, U.; Straathof, C.S.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.M.; Finkel, R.S.; Goemans, N.; Rummey, C.; et al. Treatment effect of idebenone on inspiratory function in patients with Duchenne muscular dystrophy. Pediatr. Pulmonol. 2017, 52, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Sadowska, A.M.; Manuel, Y.K.; De Backer, W.A. Antioxidant and anti-inflammatory efficacy of NAC in the treatment of COPD: Discordant in vitro and in vivo dose-effects: A review. Pulm. Pharmacol. Ther. 2007, 20, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Capogrosso, R.F.; Cozzoli, A.; Mantuano, P.; Camerino, G.M.; Massari, A.M.; Sblendorio, V.T.; De Bellis, M.; Tamma, R.; Giustino, A.; Nico, B.; et al. Assessment of resveratrol, apocynin and taurine on mechanical-metabolic uncoupling and oxidative stress in a mouse model of duchenne muscular dystrophy: A comparison with the gold standard, α-methyl prednisolone. Pharmacol. Res. 2016, 106, 101–113. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forcina, L.; Pelosi, L.; Miano, C.; Musarò, A. Insights into the Pathogenic Secondary Symptoms Caused by the Primary Loss of Dystrophin. J. Funct. Morphol. Kinesiol. 2017, 2, 44. https://doi.org/10.3390/jfmk2040044
Forcina L, Pelosi L, Miano C, Musarò A. Insights into the Pathogenic Secondary Symptoms Caused by the Primary Loss of Dystrophin. Journal of Functional Morphology and Kinesiology. 2017; 2(4):44. https://doi.org/10.3390/jfmk2040044
Chicago/Turabian StyleForcina, Laura, Laura Pelosi, Carmen Miano, and Antonio Musarò. 2017. "Insights into the Pathogenic Secondary Symptoms Caused by the Primary Loss of Dystrophin" Journal of Functional Morphology and Kinesiology 2, no. 4: 44. https://doi.org/10.3390/jfmk2040044
APA StyleForcina, L., Pelosi, L., Miano, C., & Musarò, A. (2017). Insights into the Pathogenic Secondary Symptoms Caused by the Primary Loss of Dystrophin. Journal of Functional Morphology and Kinesiology, 2(4), 44. https://doi.org/10.3390/jfmk2040044