
Exploring the Inhibitory Efficacy of Resokaempferol and Tectochrysin on PI3Kα Protein by Combining DFT and Molecular Docking against Wild-Type and H1047R Mutant Forms


Abstract
1. Introduction
2. Materials and Methods
2.1. Evaluation of the Ligands’ Optimized Molecular Structures
2.2. Assessing the Reactivity Profiles of Compounds
2.2.1. Numerical Analysis of Quantum Chemical Attributes
- The HOMO energy of the ligand sample falls below the default thresholds of −8 eV for electrophilic superdelocalizability, −2 eV for nucleophilic superdelocalizability, and −5 eV for radical superdelocalizability.
- The LUMO energy of the ligand sample exceeds the default levels of −8 eV for electrophilic superdelocalizability, −2 eV for nucleophilic superdelocalizability, and −5 eV for radical superdelocalizability.
2.2.2. Bioavailability, Drug-likeness, and Medicinal Chemistry Attributes
2.3. Molecular Docking Analysis
2.3.1. Preparing Molecular Structures for Docking Simulation
2.3.2. Docking Algorithms and Results Analysis
3. Results
3.1. Analysis of the Optimized Molecular Configurations
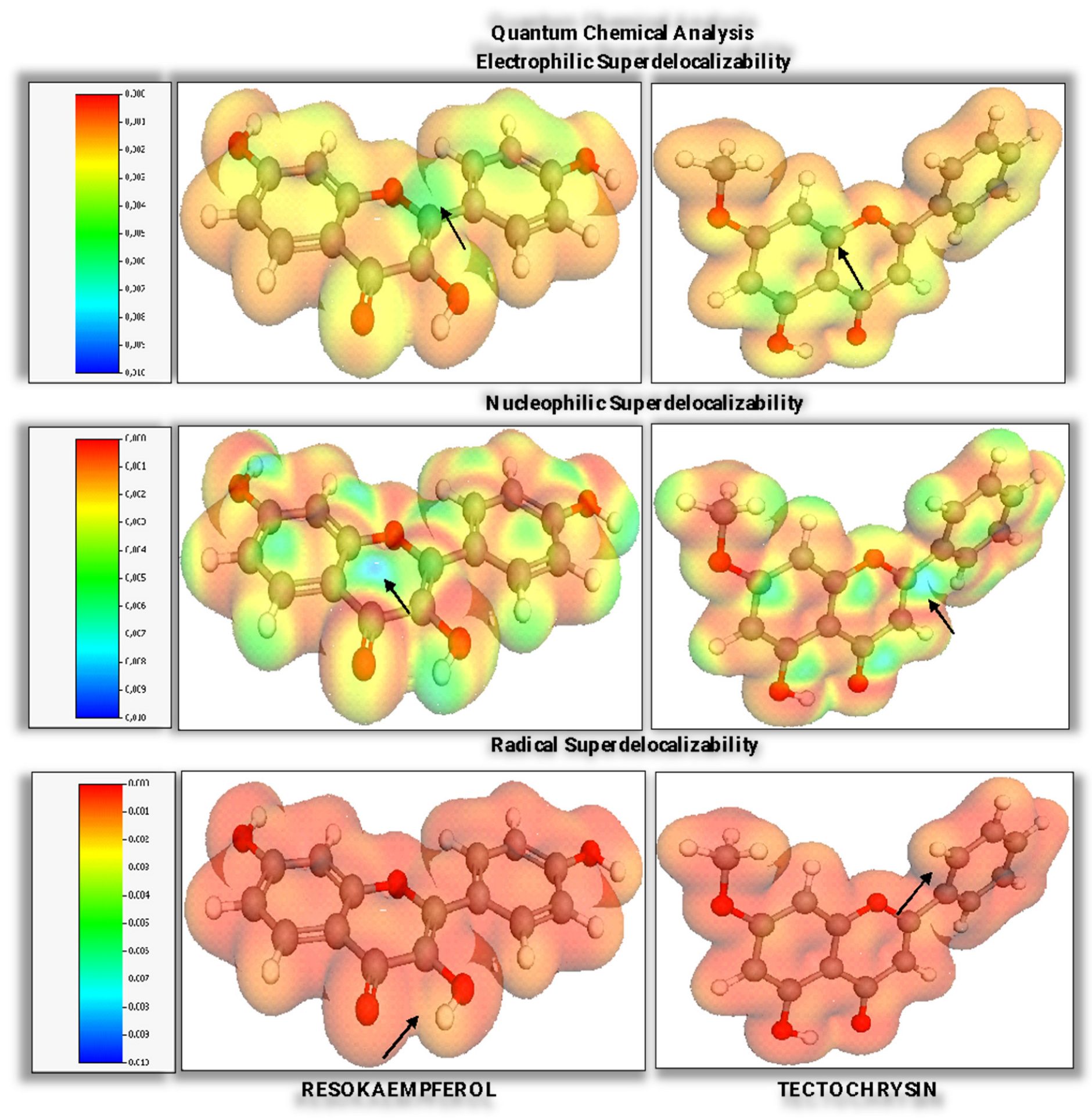
3.2. Quantitative Evaluation of Quantum Chemical Parameters
- The electrophilic, nucleophilic, and radical susceptibilities, as well as their corresponding superdelocalizabilities (see Figure 4), offer deeper insights into the reactivity of these flavonoids and their potential as ligands in biochemical contexts.
- For resokaempferol, with a HOMO energy of −5.96 eV and a LUMO energy of −2.17 eV, the molecule falls within the energy criteria for nucleophilic and radical superdelocalizability (since the HOMO is less than −2 eV and −5 eV, respectively, and the LUMO is greater than −2 eV and −5 eV, respectively). It does not meet the criteria for electrophilic superdelocalizability, as the HOMO energy is not less than the specified default energy of −8 eV.
- For tectochrysin, with a HOMO energy of −6.38 eV and a LUMO energy of −2.26 eV, similar to resokaempferol, also qualifies for nucleophilic and radical superdelocalizability for the same reasons. This compound does not meet the threshold for electrophilic superdelocalizability either since the HOMO energy does not surpass the threshold of −8 eV.
3.3. The Physicochemical and Pharmacokinetic Profiles
3.4. Molecular Docking and Scoring
3.4.1. Self-Docking and Cross-Docking of Native Structures and Ligands of Interest
3.4.2. Structural Alignment Validation and MolProbity Analysis
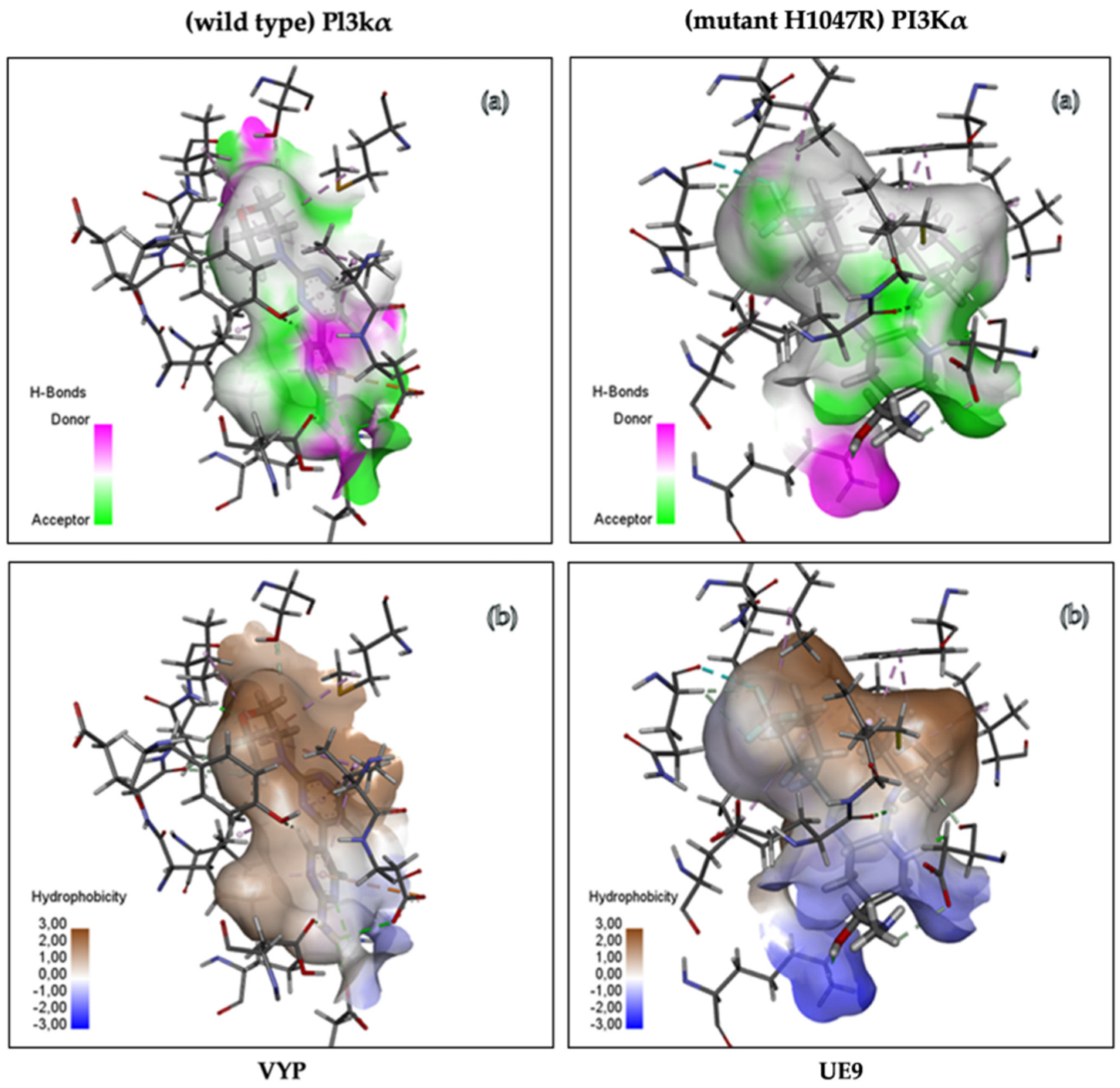
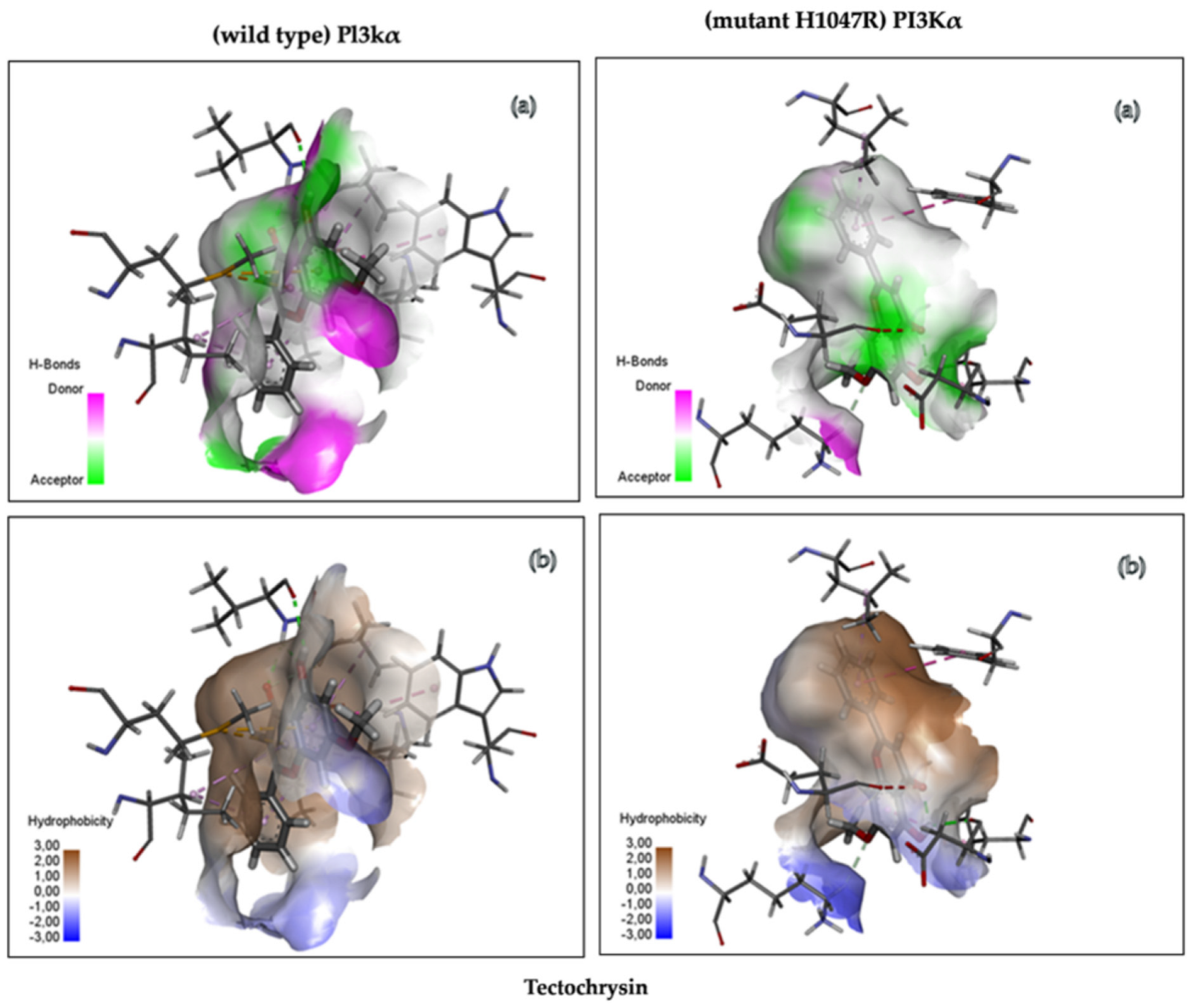
3.4.3. Analysis of Molecular Interactions
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, H.; Tian, Y.; Zhou, Y.; Kan, Y.; Wu, T.; Xiao, W.; Luo, Y. Multi-modular engineering of Saccharomyces cerevisiae for high-titre production of tyrosol and salidroside. Microb. Biotechnol. 2021, 14, 2605–2616. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.E.; Park, E.S.; Yoon, H.S.; Seo, D.W.; Hyun, B.K.; Han, S.B.; Ham, Y.W.; Hwang, B.Y.; Hong, J.T. Anticancer effect of tectochrysin in colon cancer cell via suppression of NF-kappaB activity and enhancement of death receptor expression. Mol. Cancer 2015, 14, 124. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Guo, S.; Loibl, S.; von Minckwitz, G.; Darb-Esfahani, S.; Lederer, B.; Denkert, C. PIK3CA H1047R Mutation Associated with a Lower Pathological Complete Response Rate in Triple-Negative Breast Cancer Patients Treated with Anthracycline-Taxane-Based Neoadjuvant Chemotherapy. Cancer Res. Treat. 2020, 52, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Gosav, S.; Ion, A.; Praisler, M. DFT Characterization of MDMA Methylene Homologue, a Chemical Compound with Psychoactive Properties. In Proceedings of the 10th Jubilee International Conference of the Balkan Physical Union BPU10, Sofia, Bulgaria, 26–30 August 2018; Volume 2075, p. 170027. [Google Scholar]
- Gosav, S.; Paduraru, N.; Maftei, D.; Birsa, M.L.; Praisler, M. Quantum chemical study of a novel derivative of 3-substituted dithiocarbamic flavanone. Spectrochim. Acta-A Mol. Biomol. Spectrosc. 2017, 172, 115–125. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Yang, Y.; Yao, K.; Repasky, M.P.; Leswing, K.; Abel, R.; Shoichet, B.K.; Jerome, S.V. Efficient exploration of chemical space with docking and deep learning. J. Chem. Theory Comput. 2021, 17, 7106–7119. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision Glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Johnston, R.C.; Yao, K.; Kaplan, Z.; Chelliah, M.; Leswing, K.; Seekins, S.; Watts, S.; Calkins, D.; Chief Elk, J.; Jerome, S.V.; et al. Epik: pKa and protonation state prediction through machine learning. J. Chem. Theory Comput. 2023, 19, 2380–2388. [Google Scholar] [CrossRef]
- Schrödinger. Epik; Release 2024-2; Schrödinger LLC: New York, NY, USA, 2024. [Google Scholar]
- Schrödinger. LigPrep; Release 2024-2; Schrödinger LLC: New York, NY, USA, 2024. [Google Scholar]
- Schrödinger. Maestro; Release 2024-2; Schrödinger LLC: New York, NY, USA, 2024. [Google Scholar]
- Schrödinger. Prime; Release 2024-2; Schrödinger LLC: New York, NY, USA, 2024. [Google Scholar]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aid. Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Schrödinger Release 2024-2: Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA; Impact, Schrödinger, LLC: New York, NY, USA; Prime, Schrödinger, LLC: New York, NY, USA, 2024.
- Murphy, R.B.; Repasky, M.P.; Greenwood, J.R.; Tubert-Brohman, I.; Steven Jerome, S.; Annabhimoju, R.; Boyles, N.A.; Schmitz, C.D.; Abel, R.; Farid, R.; et al. WScore: A flexible and accurate treatment of explicit water molecules in ligand–receptor docking. J. Med. Chem. 2016, 59, 4364–4384. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Cheng, H.; Orr, S.T.M.; Bailey, S.; Brooun, A.; Chen, P.; Deal, J.G.; Deng, Y.L.; Edwards, M.P.; Gallego, G.M.; Grodsky, N.; et al. Structure-Based Drug Design and Synthesis of PI3Kα-Selective Inhibitor (PF-06843195). J. Med. Chem. 2021, 64, 644–661. [Google Scholar] [CrossRef]
- Varkaris, A.; Pazolli, E.; Gunaydin, H.; Wang, Q.; Pierce, L.; Boezio, A.A.; Bulku, A.; DiPietro, L.; Fridrich, C.; Frost, A.; et al. Discovery and Clinical Proof-of-Concept of RLY-2608, a First-in-Class Mutant-Selective Allosteric PI3K alpha Inhibitor That Decouples Antitumor Activity from Hyperinsulinemia. Cancer Discov. 2024, 14, 240–257. [Google Scholar] [CrossRef] [PubMed]
- Bliven, S.E.; Bourne, P.E.; Prlić, A. Detection of circular permutations within protein structures using CE-CP. Bioinformatics 2015, 31, 1316–1318. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Jaroszewski, L.; Iyer, M.; Sedova, M.; Godzik, A. FATCAT 2.0: Towards a better understanding of the structural diversity of proteins. Nucleic Acids Res. 2020, 48, W60–W64. [Google Scholar] [CrossRef]
- Ma, J.; Wang, S. Algorithms, Applications, and Challenges of Protein Structure Alignment. Adv. Protein Chem. Struct. Biol. 2014, 94, 121–175. [Google Scholar] [PubMed]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Williams, C.J.; Headd, J.J.; Moriarty, N.W.; Prisant, M.G.; Videau, L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.C.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- PCIDB: PhytoChemical Interactions DB. Available online: https://www.genome.jp/db/pcidb (accessed on 15 May 2024).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView; Version 6.1.1.; Semichem Inc.: Shawnee Mission, KS, USA, 2019. [Google Scholar]
- Mauri, A.; Bertola, M. Alvascience: A New Software Suite for the QSAR Workflow Applied to the Blood–Brain Barrier Permeability. Int. J. Mol. Sci. 2022, 23, 12882. [Google Scholar] [CrossRef]
- Sushko, I.; Novotarskyi1, S.; Körner, R.; Pandey, A.K.; Cherkasov, A.; Li, J.; Gramatica, P.; Hansen, K.; Schroeter, T.; Müller, K.-R.; et al. Applicability domains for classification problems: Benchmarking of distance to models for AMES mutagenicity set. J. Chem. Inf. Model. 2010, 50, 2094–2111. [Google Scholar] [CrossRef]
- Cassano, A.; Manganaro, A.; Martin, T.; Young, D.; Piclin, N.; Pintore, M.; Bigoni, D.; Benfenati, E. The CAESAR models for developmental toxicity. Chem. Cent. J. 2010, 4 (Suppl. S1), S4. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminformatics 2012, 4, 17. [Google Scholar] [CrossRef]
- Meng, E.C.; Goddard, T.D.; Pettersen, E.F.; Couch, G.S.; Pearson, Z.J.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Tools for structure building and analysis. Protein Sci. 2023, 32, e4792. [Google Scholar] [CrossRef]
- BIOVIA, Dassault Systèmes. Discovery Studio, version 24.1.0.23298; Dassault Systèmes: San Diego, CA, USA, 2024. [Google Scholar]
- Fu, L.; Shi, S.; Yi, J.; Wang, N.; He, Y.; Wu, Z.; Peng, J.; Deng, Y.; Wang, W.; Wu, C.; et al. ADMETlab 3.0: An updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res. 2024, 52, W422–W431. [Google Scholar] [CrossRef]
- Pharos Knowledge Management Center (KMC). Available online: https://pharos.nih.gov (accessed on 15 May 2024).
- UniProt (Universal Protein Resource). Available online: https://www.uniprot.org (accessed on 15 May 2024).
- Pubchem. Available online: https://pubchem.ncbi.nlm.nih.gov (accessed on 15 May 2024).
- Paduraru, N.; Gosav, S.; Praisler, M. Chemometric Characterization of Some Flavonoids Active Against HT-29 Human Cancer Cells. In Proceedings of the 2015 E-Health and Bioengineering Conference (EHB 2015), Iasi, Romania, 19–21 November 2015; p. 7391429. [Google Scholar]
- Murray, J.; Politzer, P. Molecular electrostatic potentials and noncovalent interactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1326. [Google Scholar] [CrossRef]
- Martí Gimeno, T.; Marset, X.; Guillem, C.; Ramón, D.; Guillena, G. Electrophilic aromatic substitution in eutectic-type mixtures: From an old concept to new sustainable horizons. RSC Sustain. 2024, 2, 1215–1223. [Google Scholar] [CrossRef]
- Smith, M.B. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 8th ed.; Wiley: Hoboken, NJ, USA, 2020; pp. 3–1605. [Google Scholar]
- Martin, T. User’s Guide for T.E.S.T. (Toxicity Estimation Software Tool) A Java Application to Estimate Toxicities and Physical Properties from Molecular Structure; version 5.1; U.S. Environmental Protection Agency: Washington, DC, USA, 2020.
- Lagorce, D.; Bouslama, L.; Becot, J.; Miteva, M.A.; Villoutreix, B.O. FAF-Drugs4: Free ADME-tox filtering computations for chemical biology and early stages drug discovery. Bioinformatics 2017, 33, 3658–3660. [Google Scholar] [CrossRef]
- Baell, J.B. Broad coverage of commercially available lead-like screening space with fewer than 350,000 compounds. J. Chem. Inf. Model. 2013, 53, 39–55. [Google Scholar] [CrossRef]
- Pihan, E.; Colliandre, L.; Guichou, J.F.; Douguet, D. e-Drug3D: 3D structure collections dedicated to drug repurposing and fragment-based drug design. Bioinformatics 2012, 28, 1540–1541. [Google Scholar] [CrossRef]
- Kralj, S.; Jukič, M.; Bren, U. Comparative Analyses of Medicinal Chemistry and Cheminformatics Filters with Accessible Implementation in Konstanz Information Miner (KNIME). Int. J. Mol. Sci. 2022, 23, 5727. [Google Scholar] [CrossRef]
- Shityakov, S.; Neuhaus, W.; Dandekar, T.; Förster, C. Analysing molecular polar surface descriptors to predict blood-brain barrier permeation. Int. J. Comput. Biol. Drug Des. 2013, 6, 146–156. [Google Scholar] [CrossRef]
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Labuda, J.; Bowater, R.P.; Fojta, M.; Gauglitz, G.; Glatz, Z.; Hapala, I.; Havliš, J.; Kilar, F.; Kilar, A.; Malinovská, L.; et al. Terminology of bioanalytical methods (IUPAC Recommendations 2018). Pure Appl. Chem. 2018, 90, 1121–1198. [Google Scholar] [CrossRef]
- Todeschini, R.; Vighi, M.; Finizio, A.; Gramatica, P. SAR & QSAR. Environ. Res. 1997, 7, 173–193. [Google Scholar]
- Wani, K.A.; Mamta (Eds.) Handbook of Research on the Adverse Effects of Pesticide Pollution in Aquatic Ecosystems, 1st ed.; Engineering Science Reference (IGI Global): Hershey, PA, USA, 2018. [Google Scholar]
- Lea, T. Caco-2 Cell Line. In The Impact of Food Bioactives on Health; Verhoeckx, K., Ed.; Springer: Cham, Switzerland, 2015; pp. 103–111. [Google Scholar]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef]
- Basant, N.; Gupta, S.; Singh, K.P. Predicting human intestinal absorption of diverse chemicals using ensemble learning based QSAR modeling approaches. Comput. Biol. Chem. 2016, 61, 178–196. [Google Scholar] [CrossRef]
- Kim, M.T.; Sedykh, A.; Chakravarti, S.K.; Saiakhov, R.D.; Zhu, H. Critical evaluation of human oral bioavailability for pharmaceutical drugs by using various cheminformatics approaches. Pharm. Res. 2014, 31, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Harder, A.; Fang, Z.; Weinheimer, M.; Laplanche, L.; Mezler, M. Characterization and validation of canine P-glycoprotein-deficient MDCK II cell lines for efflux substrate screening. Pharm. Res. 2020, 37, 194. [Google Scholar] [CrossRef]
- Zhivkova, Z.D. Quantitative structure–pharmacokinetics relationships for plasma protein binding of basic drugs. Int. J. Pharm. Pharm. Sci. 2017, 20, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Mo, F.; Pellerino, A.; Soffietti, R.; Rudà, R. Blood–brain barrier in brain tumors: Biology and clinical relevance. Int. J. Mol. Sci. 2021, 22, 12654. [Google Scholar] [CrossRef]
- Watanabe, R.; Esaki, T.; Kawashima, H.; Natsume-Kitatani, Y.; Nagao, C.; Ohashi, R.; Mizuguchi, K. Predicting Fraction Unbound in Human Plasma from Chemical Structure: Improved Accuracy in the Low Value Ranges. Mol. Pharm. 2018, 15, 5302–5311. [Google Scholar] [CrossRef] [PubMed]
- Kiani, Y.S.; Jabeen, I. Exploring the Chemical Space of Cytochrome P450 Inhibitors Using Integrated Physicochemical Parameters, Drug Efficiency Metrics and Decision Tree Models. Computation 2019, 7, 26. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Barakat, K.H. Binding modes of hERG blockers: An unsolved mystery in the drug design arena. Expert Opin. Drug Discov. 2018, 13, 207–210. [Google Scholar] [CrossRef]
- Ta, G.H.; Weng, C.F.; Leong, M.K. In silico prediction of skin sensitization: Quo vadis? Front. Pharmacol. 2021, 12, 655771. [Google Scholar] [CrossRef]
- Bickerton, G.R.; Paolini, G.V.; Besnard, J.; Muresan, S.; Hopkins, A.L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90–98. [Google Scholar] [CrossRef]
- Skoraczyński, G.; Kitlas, M.; Miasojedow, B.; Gambin, A. Critical assessment of synthetic accessibility scores in computer-assisted synthesis planning. J. Cheminform. 2023, 15, 6. [Google Scholar] [CrossRef]
- Wei, W.; Cherukupalli, S.; Jing, L.; Liu, X.; Zhan, P. Fsp3: A new parameter for drug-likeness. Drug Discov. Today 2020, 25, 1839–1845. [Google Scholar] [CrossRef] [PubMed]
- Ivanenkov, Y.A.; Zagribelnyy, B.A.; Aladinskiy, V.A. Are We Opening the Door to a New Era of Medicinal Chemistry or Being Collapsed to a Chemical Singularity? J. Med. Chem. 2019, 62, 10026–10043. [Google Scholar] [CrossRef] [PubMed]
- Ertl, P.; Roggo, S.; Schuffenhauer, A. Natural product-likeness score and its application for prioritization of compound libraries. J. Chem. Inf. Model. 2007, 48, 68–74. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.; Dress, K.; Edwards, M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorg. Med. Chem. Lett. 2009, 19, 5560–5564. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Tice, R.R.; Austin, C.P.; Kavlock, R.J.; Bucher, J.R. Improving the human hazard characterization of chemicals: A Tox21 update. Environ. Health Perspect. 2013, 121, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zeng, K.; Ma, X.; Song, F.; Jiang, Y.; Tu, P.; Wang, X. Resokaempferol-mediated anti-inflammatory effects on activated macrophages via the inhibition of JAK2/STAT3, NF-κB and JNK/p38 MAPK signaling pathways. Int. Immunopharmacol. 2016, 38, 104–114. [Google Scholar] [CrossRef]
- Saponara, S.; Carosati, E.; Mugnai, P.; Sgaragli, G.; Fusi, F. The flavonoid scaffold as a template for the design of modulators of the vascular Ca(v) 1.2 channels. Br. J. Pharmacol. 2011, 164, 1684–1697. [Google Scholar] [CrossRef]
- Park, M.H.; Hong, J.E.; Hwang, C.J.; Choi, M.; Choi, J.S.; An, Y.J.; Son, D.J.; Hong, J.T. Synergistic inhibitory effect of cetuximab and tectochrysin on human colon cancer cell growth via inhibition of EGFR signal. Arch. Pharm. Res. 2016, 39, 721–729. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | EHOMO (Hartree/eV) | ELUMO (Hartree/eV) | EGAP 1 (Hartree/eV) |
|---|---|---|---|
| Resokaempferol | −0.22/−5.96 | −0.08/−2.17 | 0.14/3.79 |
| Tectochrysin | −0.23/−6.38 | −0.08/−2.26 | 0.15/4.13 |
| Ligand | Dipole Moment (Debye) | Polarizability (ų) |
|---|---|---|
| Resokaempferol | 3.47 | 32.44 |
| Tectochrysin | 6.69 | 32.21 |
| Quantum Chemical Properties * | Resokaempferol (eV) | Tectochrysin (eV) |
|---|---|---|
| IP | 5.96 | 6.38 |
| EA | 2.17 | 2.26 |
| η | 1.89 | 2.06 |
| σ | 0.53 | 0.48 |
| χ | 4.06 | 4.32 |
| ω | 4.35 | 4.52 |
| ADMET Property | Molecular Descriptor * | Resokaempferol | Tectochrysin | ||
|---|---|---|---|---|---|
| Predicted Value/Probability ** | Empirical Decision | Predicted Value/Probability ** | Empirical Decision | ||
| Absorption | Caco-2 | −5.62 | poor | −4.78 | excellent |
| Pgp-inhibitor | 0.14 (−−) | excellent | 0.96 (+++) | poor | |
| Pgp-substrate | 0.27 (−−) | excellent | 0.07 (−−−) | excellent | |
| HIA | 0.03 (−−−) | excellent | 0.01 (−−−) | excellent | |
| F20% | 0.32 (−) | medium | 0.05 (−−−) | excellent | |
| F30% | 0.78 (++) | poor | 0.45 (−) | medium | |
| MDCK | −4.85 | poor | −4.68 | excellent | |
| Distribution | PPB | 96.66 | poor | 98.51 | poor |
| VDss | 0.20 | excellent | 0.77 | excellent | |
| BBB Penetration | 0.01 (−−−) | excellent | 0.2 (−−) | excellent | |
| Fu | 3.33 | poor | 0.81 | poor | |
| Metabolism | CYP1A2 inhibitor | 0.75 (++) | poor | 1.00 (+++) | poor |
| CYP1A2 substrate | 0.12 (−−) | excellent | 0.79 (++) | poor | |
| CYP2C19 inhibitor | 0.25 (−−) | excellent | 0.98 (+++) | poor | |
| CYP2C19 substrate | 0.001 (−−−) | excellent | 0.02 (−−−) | excellent | |
| CYP2C9 inhibitor | 0.89 (++) | poor | 0.038 (−−−) | excellent | |
| CYP2C9 substrate | 0.32 (−) | medium | 0.98 (+++) | poor | |
| CYP2D6 inhibitor | 0.01 (−−−) | excellent | 0.81 (++) | poor | |
| CYP2D6 substrate | 0.75 (++) | poor | 0.98 (+++) | poor | |
| CYP3A4 inhibitor | 0.92 (+++) | poor | 0.98 (+++) | poor | |
| CYP3A4 substrate | 0.01 (−−−) | excellent | 0.01 (−−−) | excellent | |
| Excretion | CLplasma | 7.14 | medium | 5.28 | medium |
| T1/2 | 1.49 | medium | 0.75 | poor | |
| Toxicity | hERG Blockers | 0.11 | excellent | 0.13 | excellent |
| H-HT | 0.40 | medium | 0.46 | medium | |
| DILI | 0.67 | medium | 0.94 | poor | |
| AMES Mutagenicity | 0.55 | medium | 0.64 | medium | |
| FDAMDD | 0.74 | poor | 0.73 | poor | |
| Skin Sensitization | 0.63 | medium | 0.42 | medium | |
| Carcinogenicity | 0.79 | poor | 0.81 | poor | |
| Eye Corrosion | 0.77 | poor | 0.32 | medium | |
| Eye Irritation | 0.99 | poor | 0.99 | poor | |
| Respiratory Toxicity | 0.64 | medium | 0.77 | poor | |
| Medicinal Chemistry 1 | Resokaempferol | Tectochrysin | ||
|---|---|---|---|---|
| Predicted Value | Empirical Decision | Predicted Value | Empirical Decision | |
| Drug-likeness | 0.63 | poor | 0.78 | excellent |
| SAscore | 3.08 | excellent | 3.01 | excellent |
| Fsp3 | 0 | poor | 0.06 | poor |
| MCE-18 | 17 | poor | 16 | poor |
| NPscore | 1.04 | medium | 0.95 | medium |
| Lipinski Rule | 0 | excellent | 0 | excellent |
| Pfizer Rule | 0 | excellent | 2 | poor (2 conditions satisfied) 2 |
| GSK Rule | 0 | excellent | 0 | excellent |
| Golden Triangle | 0 | excellent | 0 | excellent |
| PAINS | 0 | excellent | 0 | excellent |
| BMS | 0 | excellent | 0 | excellent |
| NonBiodegradable | 0 | excellent | 0 | excellent |
| SureChEMBL Rule | 0 | excellent | 0 | excellent |
| Ligand 1 | Best Docking Conformation 2 | Receptor | |
|---|---|---|---|
| Wild-Type | H1047R 3 | ||
| VYP | Free Energy of Binding (kcal/mol) | −8.44 | - |
| Inhibition Constant, Ki (nM) | 653.08 | - | |
| Ligand Efficiency (docking energy) (kcal/mol) | −0.44 | - | |
| Intermolecular energy (kcal/mol) | −9.33 | - | |
| Total Internal Energy (kcal/mol) | −0.77 | - | |
| Electrostatic Energy (kcal/mol) | 0.00 | - | |
| van der Waals + Hydrogen bonds + Desolations. Energy (kcal/mol) | −9.33 | - | |
| Torsional Free Energy (kcal/mol) | 0.89 | - | |
| Unbound System’s Energy (kcal/mol) | −0.77 | - | |
| RMSD from reference structure (Å) | 0.631 | - | |
| UE9 | Free Energy of Binding (kcal/mol) | - | −8.34 |
| Inhibition Constant, Ki (nM) | - | 772.54 | |
| Ligand Efficiency (docking energy) (kcal/mol) | - | −0.26 | |
| Intermolecular energy (kcal/mol) | - | −10.13 | |
| Total Internal Energy (kcal/mol) | - | −1.82 | |
| Electrostatic Energy (kcal/mol) | - | −0.15 | |
| van der Waals + Hydrogen bonds + DE solvation. Energy (kcal/mol) | - | −9.97 | |
| Torsional Free Energy (kcal/mol) | - | 1.79 | |
| Unbound System’s Energy (kcal/mol) | - | −1.82 | |
| RMSD from reference structure (Å) | - | 12.96 | |
| Resokaempferol | Free Energy of Binding (kcal/mol) | −8.73 | −9.22 |
| Inhibition Constant, Ki (nM) | 395.76 | 175.41 | |
| Ligand Efficiency (docking energy) (kcal/mol) | −0.44 | −0.46 | |
| Intermolecular energy (kcal/mol) | −9.93 | −10.41 | |
| Total Internal Energy (kcal/mol) | −1.11 | −1.09 | |
| Electrostatic Energy (kcal/mol) | 0.00 | 0.00 | |
| van der Waals + Hydrogen bonds + desolations. Energy (kcal/mol) | −9.93 | −10.41 | |
| Torsional Free Energy (kcal/mol) | 1.19 | 1.19 | |
| Unbound System’s Energy (kcal/mol) | −1.11 | −1.09 | |
| RMSD from reference structure (Å) | 31.39 | 33.29 | |
| Tectochrysin | Free Energy of Binding (kcal/mol) | −6.45 | −6.16 |
| Inhibition Constant, Ki (nM) | 18.60 | 30.48 | |
| Ligand Efficiency (docking energy) (kcal/mol) | −0.32 | −0.31 | |
| Intermolecular energy (kcal/mol) | −7.35 | −7.06 | |
| Total Internal Energy (kcal/mol) | −1.03 | −1.03 | |
| Electrostatic Energy (kcal/mol) | −0.1 | −0.06 | |
| van der Waals + Hydrogen bonds + DE solvation. Energy (kcal/mol) | −7.25 | −6.99 | |
| Torsional Free Energy (kcal/mol) | 0.89 | 0.89 | |
| Unbound System’s Energy (kcal/mol) | −1.03 | −1.03 | |
| RMSD from reference structure (Å) | 32.38 | 34.39 | |
| Best Docking Conformation | Ligand 1 | Receptor | |
|---|---|---|---|
| Wild-Type | H1047R 2 | ||
| Affinity (kcal/mol) | VYP | −8.0 | - |
| UE9 | - | −11.05 | |
| Resokaempferol | −8.23 | −8.05 | |
| Tectochrysin | −8.25 | −7.84 | |
| Ligand 1 | Best Docking Conformation 2 | Receptor | |
|---|---|---|---|
| Wild-Type | H1047R 3 | ||
| VYP | XP Glide Score | −7.52 | - |
| Glide Ligand Efficiency | −0.39 | - | |
| UE9 | XP Glide Score | - | −10.02 |
| Glide Ligand Efficiency | - | −0.31 | |
| Resokaempferol | XP Glide Score | −9.63 | −7.40 |
| Glide Ligand Efficiency | −0.48 | −0.37 | |
| Tectochrysin | XP Glide Score | −8.54 | −6.47 |
| Glide Ligand Efficiency | −0.43 | −0.32 | |
| Pairwise Structure Alignment 1 | 7K71 RCSB PDB 2 | Glide Re-Docking | AD 4 Re-Docking 3 | Vina Re-Docking |
|---|---|---|---|---|
| Chain | A | A | A | A |
| RMSD | - | 0.17 | 0 | 0 |
| TM-score | - | 1 | 1 | 1 |
| Identity | - | 99% | 100% | 67% |
| Aligned Residues | - | 843 | 843 | 843 |
| Sequence Length | 946 | 843 | 843 | 843 |
| Modeled Residues | 843 | 843 | 843 | 843 |
| Pairwise Structure Alignment 1 | 8TS9 RCSB PDB 2 | Glide Re-Docking | AD 4 Re-Docking 3 | Vina Re-Docking |
|---|---|---|---|---|
| Chain | A | A | A | A |
| RMSD | - | 0.17 | 0 | 0 |
| TM-score | - | 1 | 1 | 1 |
| Identity | - | 99% | 100% | 100% |
| Aligned Residues | - | 1004 | 1004 | 1004 |
| Sequence Length | 1060 | 1004 | 1004 | 1004 |
| Modeled Residues | 1004 | 1004 | 1004 | 1004 |
| Pairwise Structure Alignment 1 | 7K71 RCSB PDB 2 | Glide Molecular Docking | AD 4 Molecular Docking 3 | Vina Molecular Docking |
|---|---|---|---|---|
| Chain | A | A | A | A |
| RMSD | - | 0.29 | 0 | 0 |
| TM-score | - | 1 | 1 | 1 |
| Identity | - | 99% | 100% | 67% |
| Aligned Residues | - | 843 | 843 | 843 |
| Sequence Length | 946 | 843 | 843 | 843 |
| Modeled Residues | 843 | 843 | 843 | 843 |
| Pairwise Structure Alignment 1 | 8TS9 RCSB PDB 2 | Glide Molecular Docking | AD 4 Molecular Docking 3 | Vina Molecular Docking |
|---|---|---|---|---|
| Chain | A | A | A | A |
| RMSD | - | 0.17 | 0 | 0 |
| TM-score | - | 1 | 1 | 1 |
| Identity | - | 99% | 100% | 100% |
| Aligned Residues | - | 1004 | 1004 | 1004 |
| Sequence Length | 1060 | 1004 | 1004 | 1004 |
| Modeled Residues | 1004 | 1004 | 1004 | 1004 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paraschiv, C.; Gosav, S.; Burlacu, C.M.; Praisler, M. Exploring the Inhibitory Efficacy of Resokaempferol and Tectochrysin on PI3Kα Protein by Combining DFT and Molecular Docking against Wild-Type and H1047R Mutant Forms. Inventions 2024, 9, 96. https://doi.org/10.3390/inventions9050096
Paraschiv C, Gosav S, Burlacu CM, Praisler M. Exploring the Inhibitory Efficacy of Resokaempferol and Tectochrysin on PI3Kα Protein by Combining DFT and Molecular Docking against Wild-Type and H1047R Mutant Forms. Inventions. 2024; 9(5):96. https://doi.org/10.3390/inventions9050096
Chicago/Turabian StyleParaschiv, Cristina, Steluța Gosav, Catalina Mercedes Burlacu, and Mirela Praisler. 2024. "Exploring the Inhibitory Efficacy of Resokaempferol and Tectochrysin on PI3Kα Protein by Combining DFT and Molecular Docking against Wild-Type and H1047R Mutant Forms" Inventions 9, no. 5: 96. https://doi.org/10.3390/inventions9050096
APA StyleParaschiv, C., Gosav, S., Burlacu, C. M., & Praisler, M. (2024). Exploring the Inhibitory Efficacy of Resokaempferol and Tectochrysin on PI3Kα Protein by Combining DFT and Molecular Docking against Wild-Type and H1047R Mutant Forms. Inventions, 9(5), 96. https://doi.org/10.3390/inventions9050096