Nanostructured TiC Layer is Highly Suitable Surface for Adhesion, Proliferation and Spreading of Cells

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
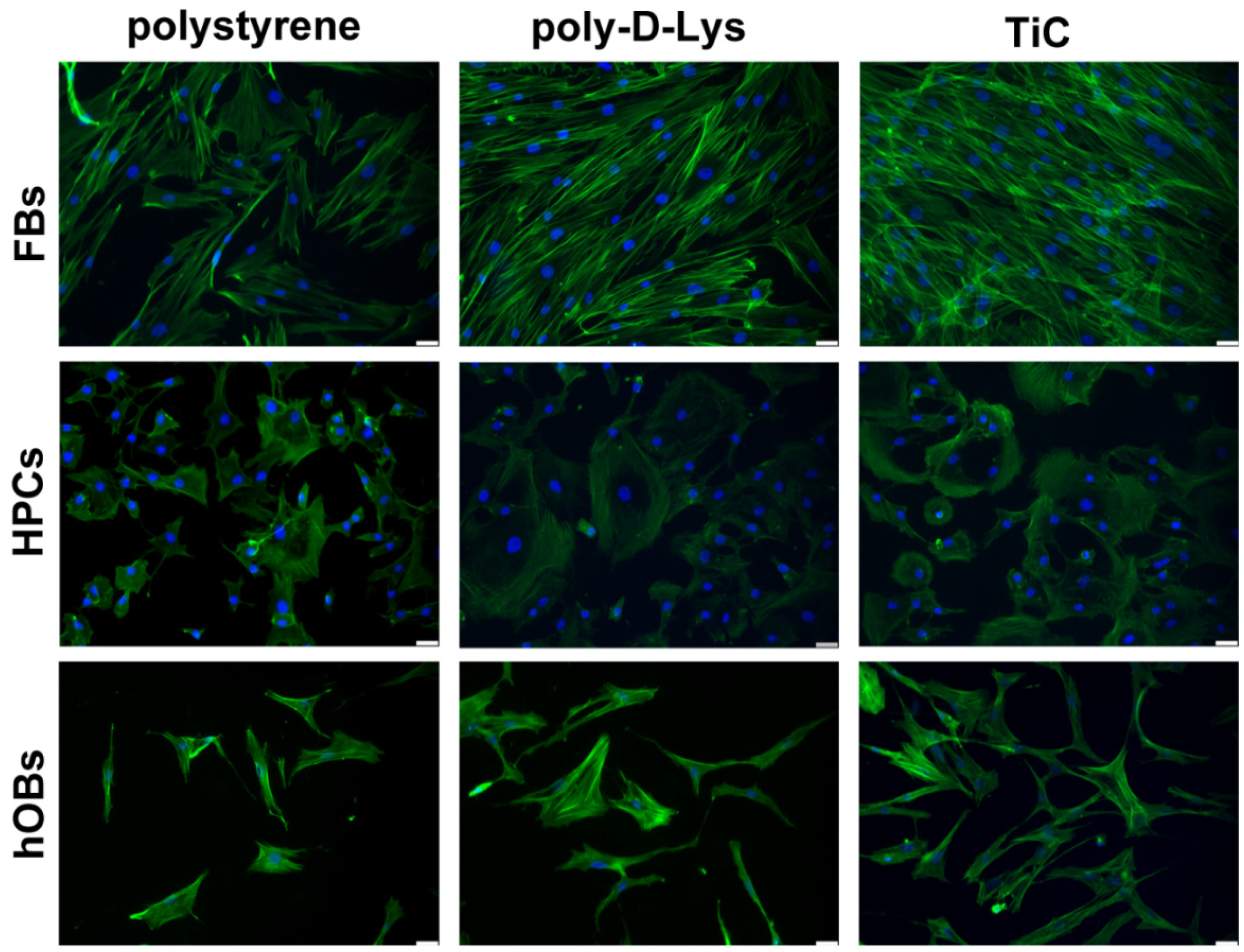
2.1. Cell Adhesion on Polystyrene, Poly-d-Lys and TiC
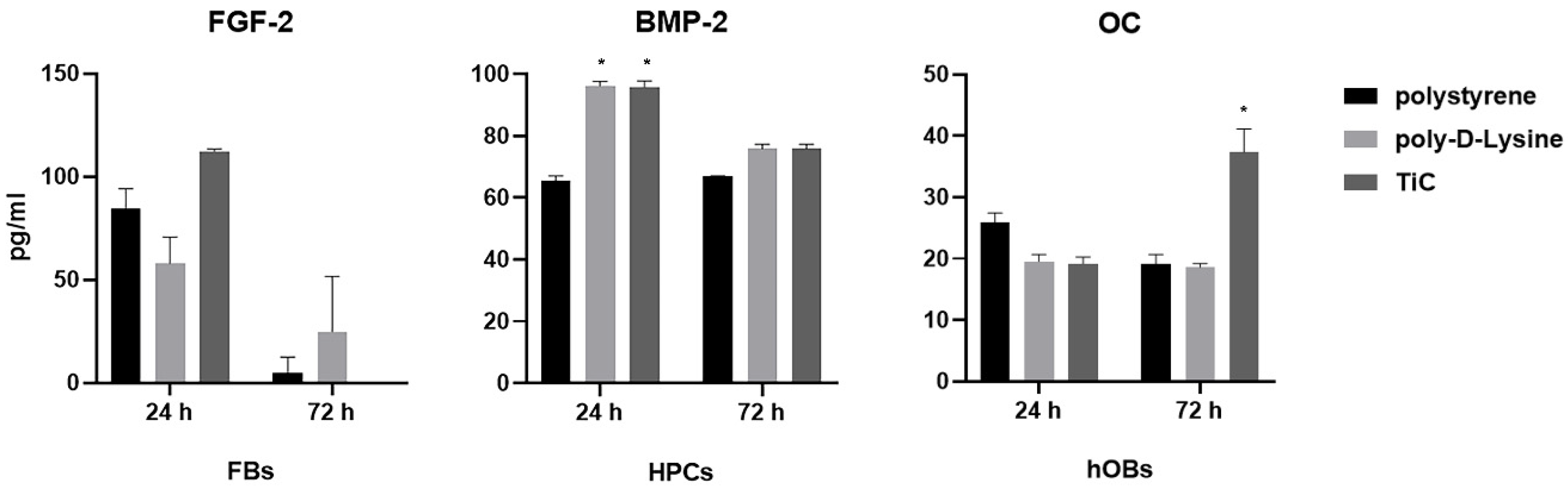
2.2. Production of Osteocalcin, Fibroblast Growth Factor and Bone Morphogenetic Protein 2
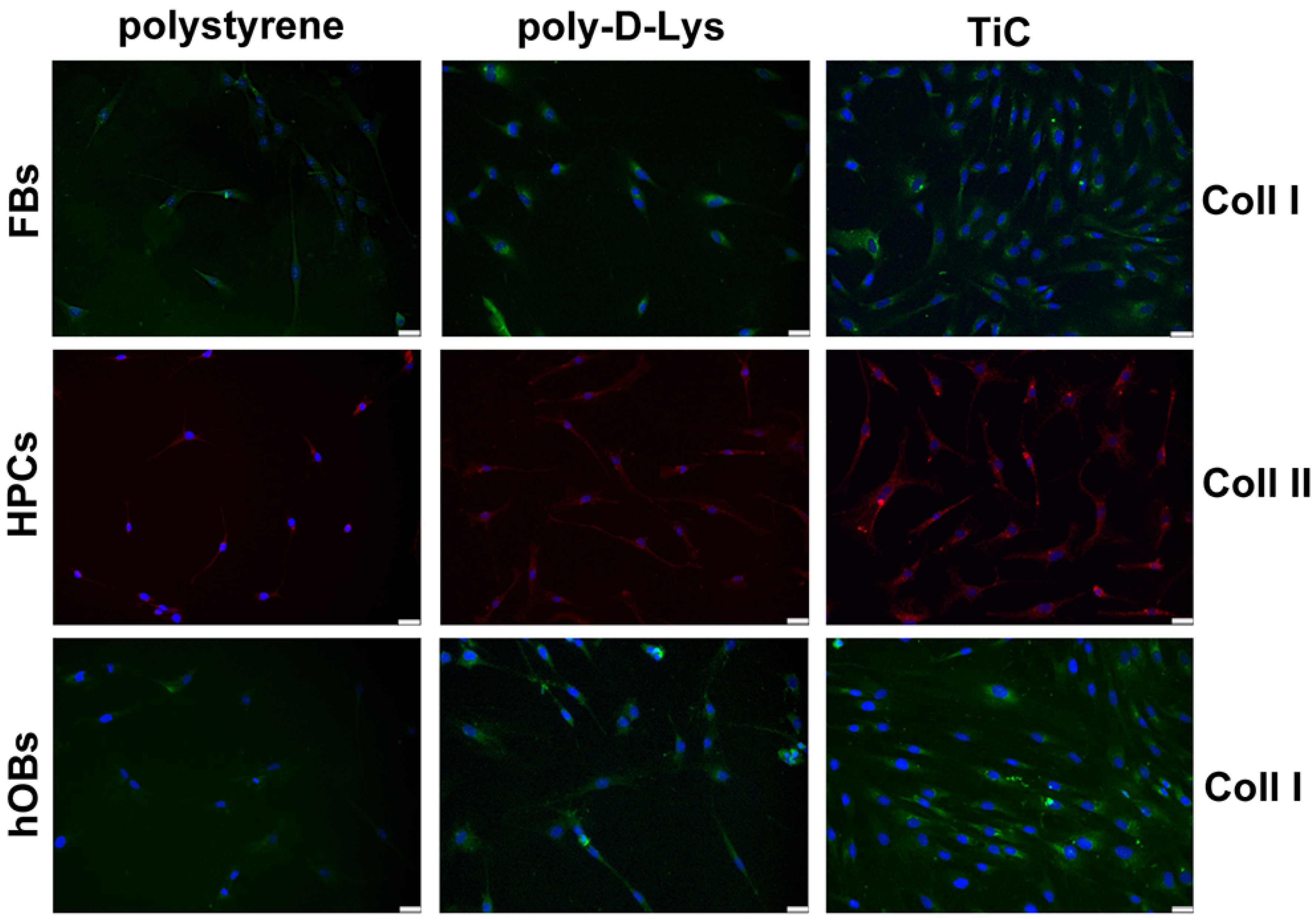
2.3. Collagens Production
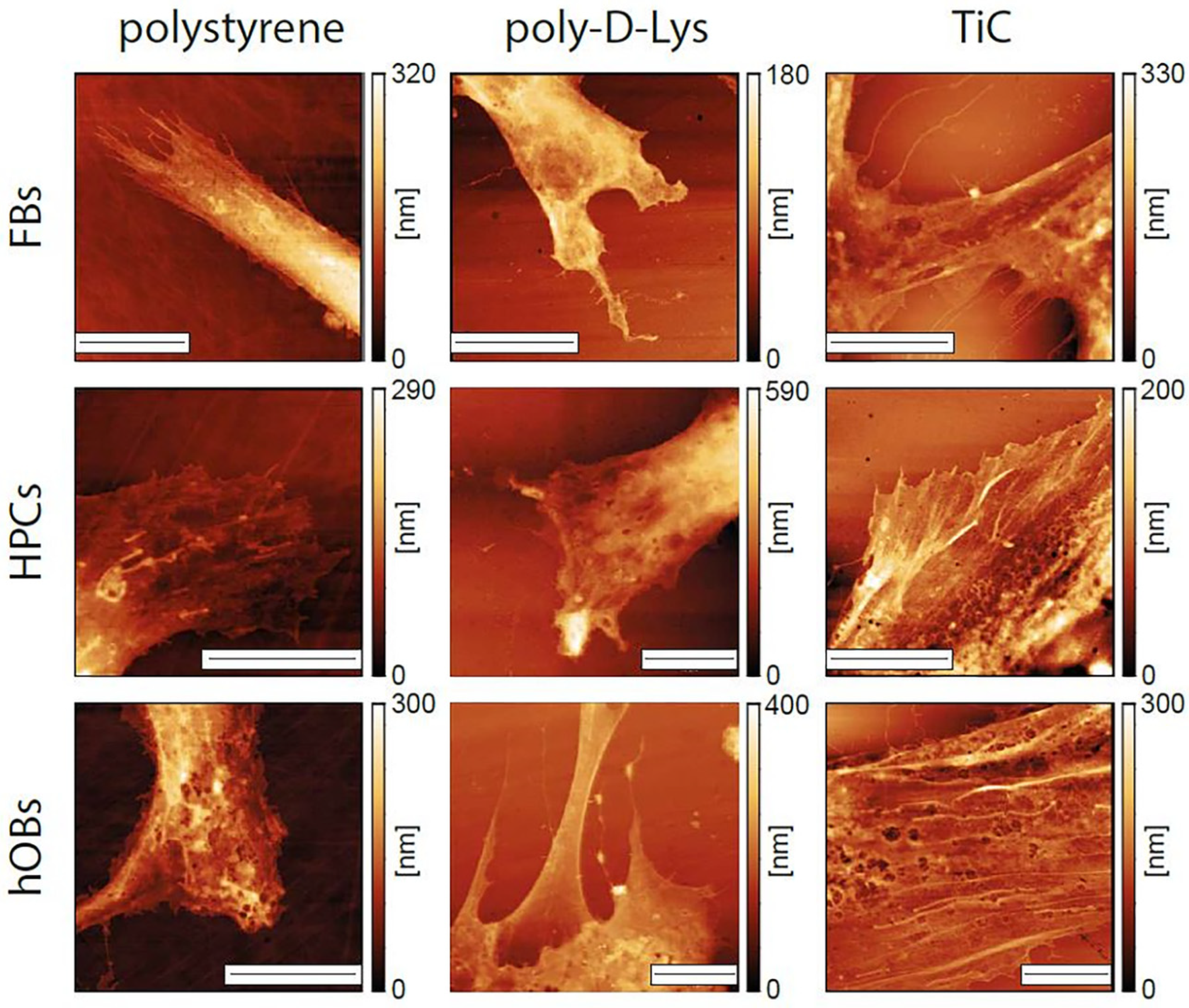
2.4. AFM Analysis
3. Discussion
4. Materials and Methods
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pamies, D.; Bal-Price, A.; Simeonov, A.; Tagle, D.; Allen, D.; Gerhold, D.; Yin, D.; Pistollato, F.; Inutsuka, T.; Sullivan, K.; et al. Good cell culture practice for stem cells & stem-cell-derived models. ALTEX 2017, 34, 95–132. [Google Scholar]
- Lerman, M.J.; Lembong, J.; Muramoto, S.; Gillen, G.; Fisher, J.P. The Evolution of Polystyrene as a Cell Culture Material. Tissue Eng. Part B Rev. 2018, 24, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.M.; Engler, A.J.; Slone, R.D.; Galante, L.L.; Schwarzbauer, J.E. Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation. Cancer Res. 2008, 69, 3185–3192. [Google Scholar] [CrossRef] [PubMed]
- Brafman, D.A.; Shah, K.D.; Fellner, T.; Chien, S.; Willert, K. Defining long-term maintenance conditions of human embryonic stem cells with arrayed cellular microenvironment technology. Stem Cells Dev. 2009, 18, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.; Yu, D.; Davidsion, M.C.; Silva, G.A. Comparison of standard surface chemistries for culturing mesenchymal stem cells prior to neural differentiation. Biomaterials 2006, 27, 4333–4339. [Google Scholar] [CrossRef] [PubMed]
- DeQuach, J.A.; Mezzano, V.; Miglani, A.; Lange, S.; Keller, G.M.; Sheikh, F.; Christman, K.L. Simple and high yielding method for preparing tissue specific extracellular matrix coatings for cell culture. PLoS ONE 2010, 5, e13039. [Google Scholar] [CrossRef] [PubMed]
- Pollard, T.D. Actin and actin-binding proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef]
- Fletcher, D.A.; Mullins, R.D. Cell mechanics and the cytoskeleton. Nature 2010, 463, 485–492. [Google Scholar] [CrossRef]
- Lotz, E.M.; Cohen, D.J.; Schwartz, Z.; Boyan, B.D. Titanium implant surface properties enhance osseointegration in ovariectomy induced osteoporotic rats without pharmacologic intervention. Clin. Oral Implant Res. 2020, in press. [Google Scholar] [CrossRef]
- Sumbal, J.; Koledova, Z. FGF signaling in mammary gland fibroblasts regulates multiple fibroblast functions and mammary epithelial morphogenesis. Development 2019, 146, 185306. [Google Scholar] [CrossRef]
- Wang, Z.; Hutton, W.C.; Tim Yoon, S. ISSLS prize winner: Effect of link protein peptide on human intervertebral disc cells. Spine 2013, 38, 1501–1507. [Google Scholar] [CrossRef] [PubMed]
- Florencio-Silva, R.; Sasso, G.R.D.S.; Sasso-Cerri, E.; Simões, M.J.; Cerri, P.S. Biology of Bone Tissue: Structure, Function, and Factors That Influence Bone Cells. Biomed Res. Int. 2015, 2015, 421746. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B. Update on the biology of the chondrocyte and new approaches to treating cartilage diseases. Best Pract. Res. Clin. Rheumatol. 2006, 20, 1003–1025. [Google Scholar] [CrossRef] [PubMed]
- Thulabandu, V.; Chen, D.; Atit, R.P. Dermal fibroblast in cutaneous development and healing. Wiley Interdiscip. Rev. Dev. Biol. 2018, 7, e307. [Google Scholar] [CrossRef]
- Dinarelli, S.; Girasole, M.; Longo, G. Methods for atomic force microscopy of biological and living specimens. Methods Mol. Biol. 2018, 1814, 529–539. [Google Scholar]
- Kasas, S.; Longo, G.; Dietler, G. Mechanical properties of biological specimens explored by atomic force microscopy. J. Phys. D Appl. Phys. 2013, 46, 133001. [Google Scholar] [CrossRef]
- Longo, G.; Kasas, S. Effects of antibacterial agents and drugs monitored by atomic force microscopy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2014, 6, 230–244. [Google Scholar] [CrossRef]
- Japaridze, A.; Muskhelishvili, G.; Benedetti, F.; Gavriilidou, A.F.M.; Zenobi, R.; De Los Rios, P.; Longo, G.; Dietler, G. Hyperplectonemes: A Higher Order Compact and Dynamic DNA Self-Organization. Nano Lett. 2017, 17, 1938–1948. [Google Scholar] [CrossRef]
- Jiangtao, Z.; Francesco, S.R.; Manuela, R.Z.; Georg, M.; Giovanni, L.; Sergey, K.S. Effects of sedimentation, microgravity hydrodynamic mixing and air-water interface on α-synuclein amyloid formation. Chem. Sci. 2020, 11, 3687–3693. [Google Scholar]
- Ruggeri, F.S.; Longo, G.; Faggiano, S.; Lipiec, E.; Pastore, A.; Dietler, G. Infrared nanospectroscopy characterization of oligomeric and fibrillar aggregates during amyloid formation. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Longo, G.; Rio, L.M.; Roduit, C.; Trampuz, A.; Bizzini, A.; Dietler, G.; Kasas, S. Force volume and stiffness tomography investigation on the dynamics of stiff material under bacterial membranes. J. Mol. Recognit. 2012, 25, 278–284. [Google Scholar] [CrossRef]
- Longo, G.; Rio, L.M.; Trampuz, A.; Dietler, G.; Bizzini, A.; Kasas, S. Antibiotic-induced modifications of the stiffness of bacterial membranes. J. Microbiol. Methods 2013, 93, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Girasole, M.; Pompeo, G.; Cricenti, A.; Longo, G.; Boumis, G.; Bellelli, A.; Amiconi, S. The how, when, and why of the aging signals appearing on the human erythrocyte membrane: An atomic force microscopy study of surface roughness. Nanomed. Nanotechnol. Biol. Med. 2010, 6, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Longo, G.; Ioannidu, C.A.; Scotto d’Abusco, A.; Superti, F.; Misiano, C.; Zanoni, R.; Politi, L.; Mazzola, L.; Iosi, F.; Mura, F.; et al. Improving osteoblast response in vitro by a nanostructured thin film with titanium carbide and titanium oxides clustered around graphitic carbon. PLoS ONE 2016, 11, e0152566. [Google Scholar] [CrossRef]
- Veronesi, F.; Giavaresi, G.; Fini, M.; Longo, G.; Ioannidu, C.A.; Scotto d’Abusco, A.; Superti, F.; Panzini, G.; Misiano, C.; Palattella, A.; et al. Osseointegration is improved by coating titanium implants with a nanostructured thin film with titanium carbide and titanium oxides clustered around graphitic carbon. Mater. Sci. Eng. C 2017, 70, 264–271. [Google Scholar] [CrossRef]
- Anselme, K. Biomaterials and interface with bone. Osteoporos. Int. 2011, 22, 2037–2042. [Google Scholar] [CrossRef]
- Brama, M.; Rhodes, N.; Hunt, J.; Ricci, A.; Teghil, R.; Migliaccio, S.; Rocca, C.D.; Leccisotti, S.; Lioi, A.; Scandurra, M.; et al. Effect of titanium carbide coating on the osseointegration response in vitro and in vivo. Biomaterials 2007, 28, 595–608. [Google Scholar] [CrossRef]
- Longo, G.; Girasole, M.; Pompeo, G.; Cricenti, A.; Misiano, C.; Acclavio, A.; Tizzoni, A.C.; Mazzola, L.; Santini, P.; Politi, L.; et al. Effect of titanium carbide coating by ion plating plasma-assisted deposition on osteoblast response: A chemical, morphological and gene expression investigation. Surf. Coat. Technol. 2010, 204, 2605–2612. [Google Scholar] [CrossRef]
- Mazzola, L.; Bemporad, E.; Misiano, C.; Pepe, F.; Santini, P.; Scandurra, R. Surface analysis and osteoblasts response of a titanium oxi-carbide film deposited on titanium by Ion Plating Plasma Assisted (IPPA). J. Nanosci. Nanotechnol. 2011, 11, 8754–8762. [Google Scholar] [CrossRef]
- Zanoni, R.; Ioannidu, C.A.; Mazzola, L.; Politi, L.; Misiano, C.; Longo, G.; Falconieri, M.; Scandurra, R. Graphitic carbon in a nanostructured titanium oxycarbide thin film to improve implant osseointegration. Mater. Sci. Eng. C 2015, 46, 409–416. [Google Scholar] [CrossRef]
- Jaroch, K.; Jaroch, A.; Bojko, B. Cell cultures in drug discovery and development: The need of reliable in vitro-in vivo extrapolation for pharmacodynamics and pharmacokinetics assessment. J. Pharm. Biomed. Anal. 2018, 147, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Lopreiato, M.; Cocchiola, R.; Falcucci, S.; Leopizzi, M.; Cardone, M.; Di Maio, V.; Brocco, U.; D’Orazi, V.; Calvieri, S.; Scandurra, R.; et al. The Glucosamine-derivative NAPA Suppresses MAPK Activation and Restores Collagen Deposition in Human Diploid Fibroblasts Challenged with Environmental Levels of UVB. Photochem. Photobiol. 2020, 96, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Stoppoloni, D.; Politi, L.; Leopizzi, M.; Gaetani, S.; Guazzo, R.; Basciani, S.; Moreschini, O.; De Santi, M.; Scandurra, R.; Scotto d’Abusco, A. Effect of glucosamine and its peptidyl-derivative on the production of extracellular matrix components by human primary chondrocytes. Osteoarthr. Cartil. 2015, 23, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.C.; Larrouture, Q.C.; Li, Y.; Lin, H.; Beer-Stoltz, D.; Liu, L.; Tuan, R.S.; Robinson, L.J.; Schlesinger, P.H.; Nelson, D.J. Osteoblast differentiation and bone matrix formation in vivo and in vitro. Tissue Eng. Part B Rev. 2017, 23, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Contentin, R.; Demoor, M.; Concari, M.; Desancé, M.; Audigié, F.; Branly, T.; Galéra, P. Comparison of the Chondrogenic Potential of Mesenchymal Stem Cells Derived from Bone Marrow and Umbilical Cord Blood Intended for Cartilage Tissue Engineering. Stem Cell Rev. Rep. 2020, 16, 126–143. [Google Scholar] [CrossRef] [PubMed]
- Coumoul, X.; Deng, C.X. Roles of FGF Receptors in Mammalian Development and Congenital Diseases. Birth Defects Res. Part CEmbryo Today Rev. 2003, 69, 286–304. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.; McGrath, J.A.; Navsaria, H. The role of fibroblasts in tissue engineering and regeneration. Br. J. Dermatol. 2007, 156, 1149–1155. [Google Scholar] [CrossRef]
- Karsenty, G.; Ferron, M. The contribution of bone to whole-organism physiology. Nature 2012, 481, 314–320. [Google Scholar] [CrossRef]
- Abdian, N.; Ghasemi-Dehkordi, P.; Hashemzadeh-Chaleshtori, M.; Ganji-Arjenaki, M.; Doosti, A.; Amiri, B. Comparison of human dermal fibroblasts (HDFs) growth rate in culture media supplemented with or without basic fibroblast growth factor (bFGF). Cell Tissue Bank. 2015, 16, 487–495. [Google Scholar] [CrossRef]
- Zoch, M.L.; Clemens, T.L.; Riddle, R.C. New insights into the biology of osteocalcin. Bone 2016, 82, 42–49. [Google Scholar] [CrossRef]
- Hutter, J.L.; Bechhoefer, J. Calibration of atomic-force microscope tips. Rev. Sci. Instrum. 1993, 64, 1868–1873. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polystyrene | Poly-d-Lys | TiC | |
|---|---|---|---|
| Coll I in FBs | 1549 | 1653 | 2465 |
| Coll II in HPCs | 2236 | 2673 | 3641 |
| Coll I in hOBs | 949 | 2933 | 4381 |
| Polystyrene | Poly-d-Lys | TiC | |
|---|---|---|---|
| FBs | 1.60 ± 0.14 μm | 1.15 ± 0.21 μm | 0.78 ± 0.16 μm |
| HPCs | 1.21 ± 0.42 μm | 1.50 ± 0.48 μm | 0.69 ± 0.06 μm |
| hOBs | 1.1 ± 0.27 μm | 1.18 ± 0.30 μm | 0.79 ± 0.23 μm |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopreiato, M.; Mariano, A.; Cocchiola, R.; Longo, G.; Dalla Vedova, P.; Scandurra, R.; Scotto d’Abusco, A. Nanostructured TiC Layer is Highly Suitable Surface for Adhesion, Proliferation and Spreading of Cells. Condens. Matter 2020, 5, 29. https://doi.org/10.3390/condmat5020029
Lopreiato M, Mariano A, Cocchiola R, Longo G, Dalla Vedova P, Scandurra R, Scotto d’Abusco A. Nanostructured TiC Layer is Highly Suitable Surface for Adhesion, Proliferation and Spreading of Cells. Condensed Matter. 2020; 5(2):29. https://doi.org/10.3390/condmat5020029
Chicago/Turabian StyleLopreiato, Mariangela, Alessia Mariano, Rossana Cocchiola, Giovanni Longo, Pietro Dalla Vedova, Roberto Scandurra, and Anna Scotto d’Abusco. 2020. "Nanostructured TiC Layer is Highly Suitable Surface for Adhesion, Proliferation and Spreading of Cells" Condensed Matter 5, no. 2: 29. https://doi.org/10.3390/condmat5020029
APA StyleLopreiato, M., Mariano, A., Cocchiola, R., Longo, G., Dalla Vedova, P., Scandurra, R., & Scotto d’Abusco, A. (2020). Nanostructured TiC Layer is Highly Suitable Surface for Adhesion, Proliferation and Spreading of Cells. Condensed Matter, 5(2), 29. https://doi.org/10.3390/condmat5020029