Electronic Structure of Boron Flat Holeless Sheet

Abstract
1. Introduction
1.1. Why should Boron Sheets be Formed?
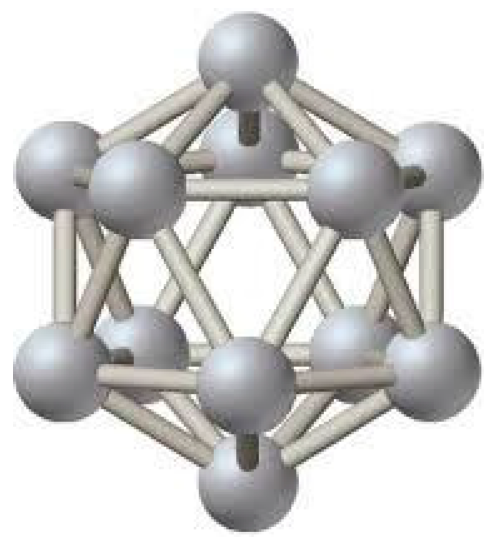
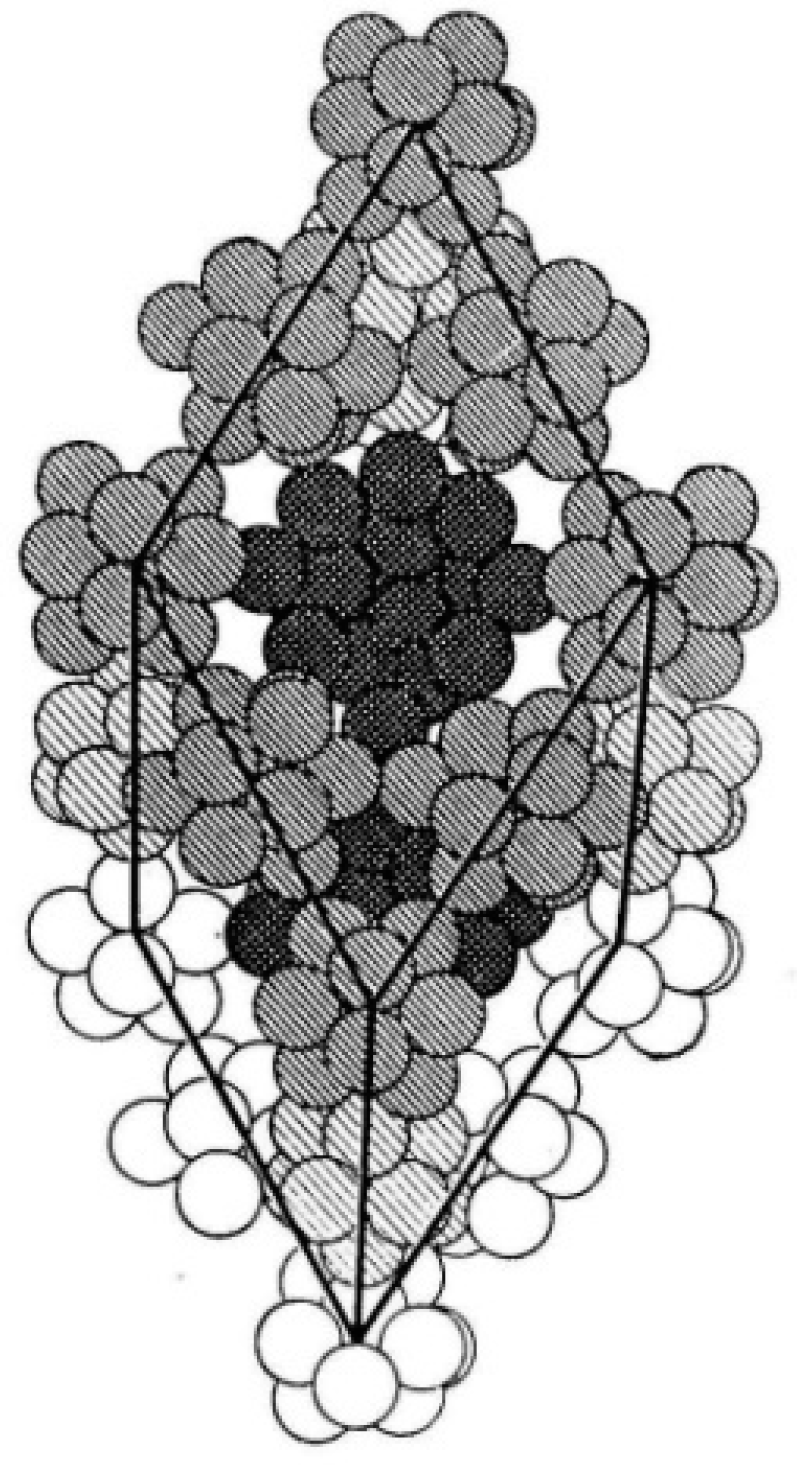
1.1.1. 3-D All-Boron Structures
1.1.2. Boron Quasi-Planar Clusters
1.1.3. Liquid Boron Structure
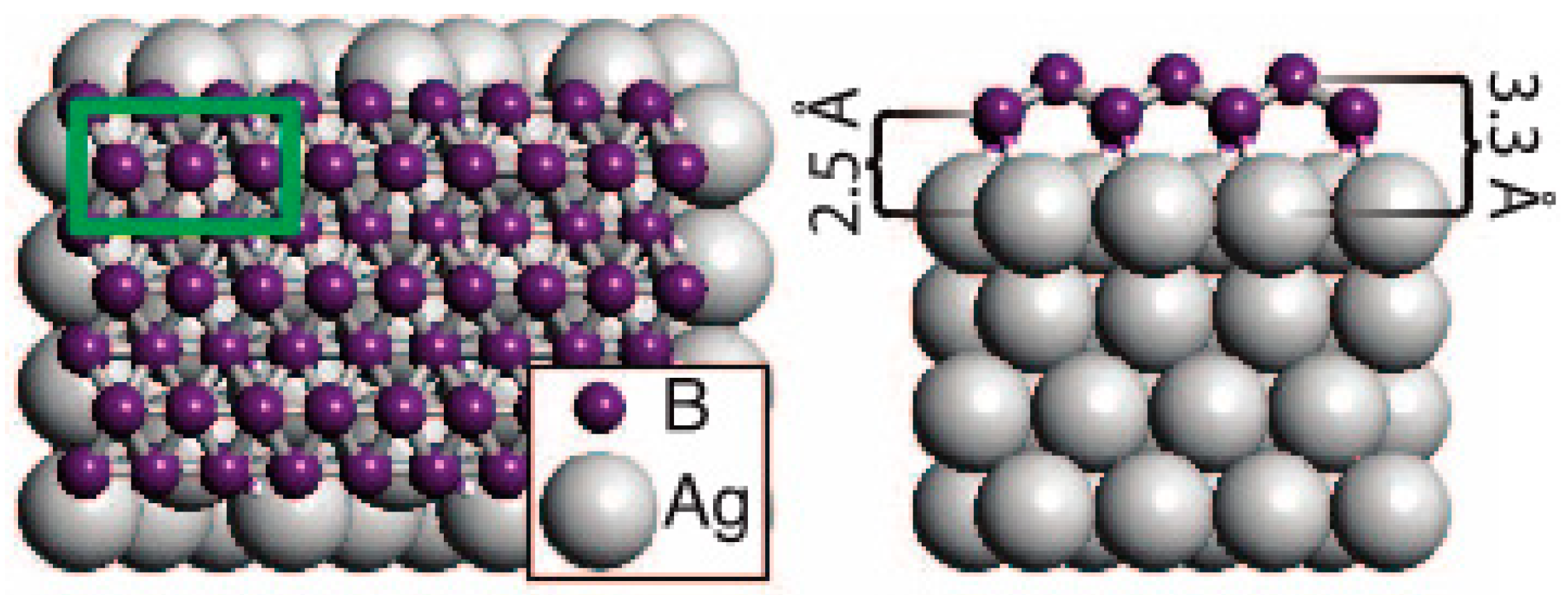
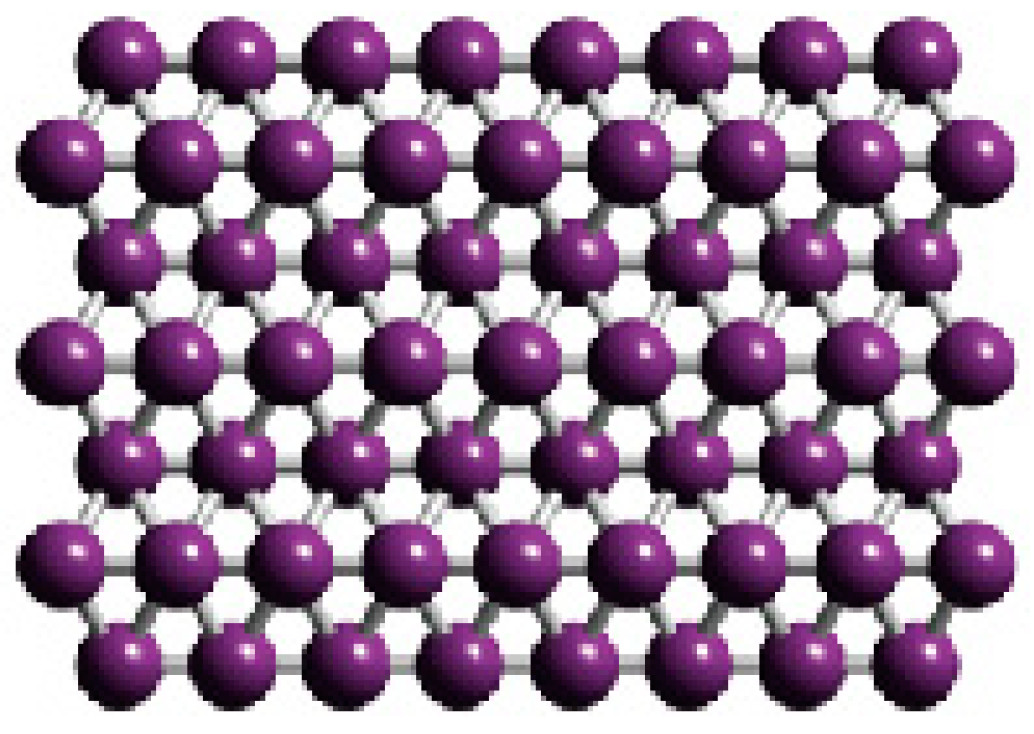
1.1.4. Growing of Boron Sheets
1.2. Applications
1.3. Available Electron Structure Calculations
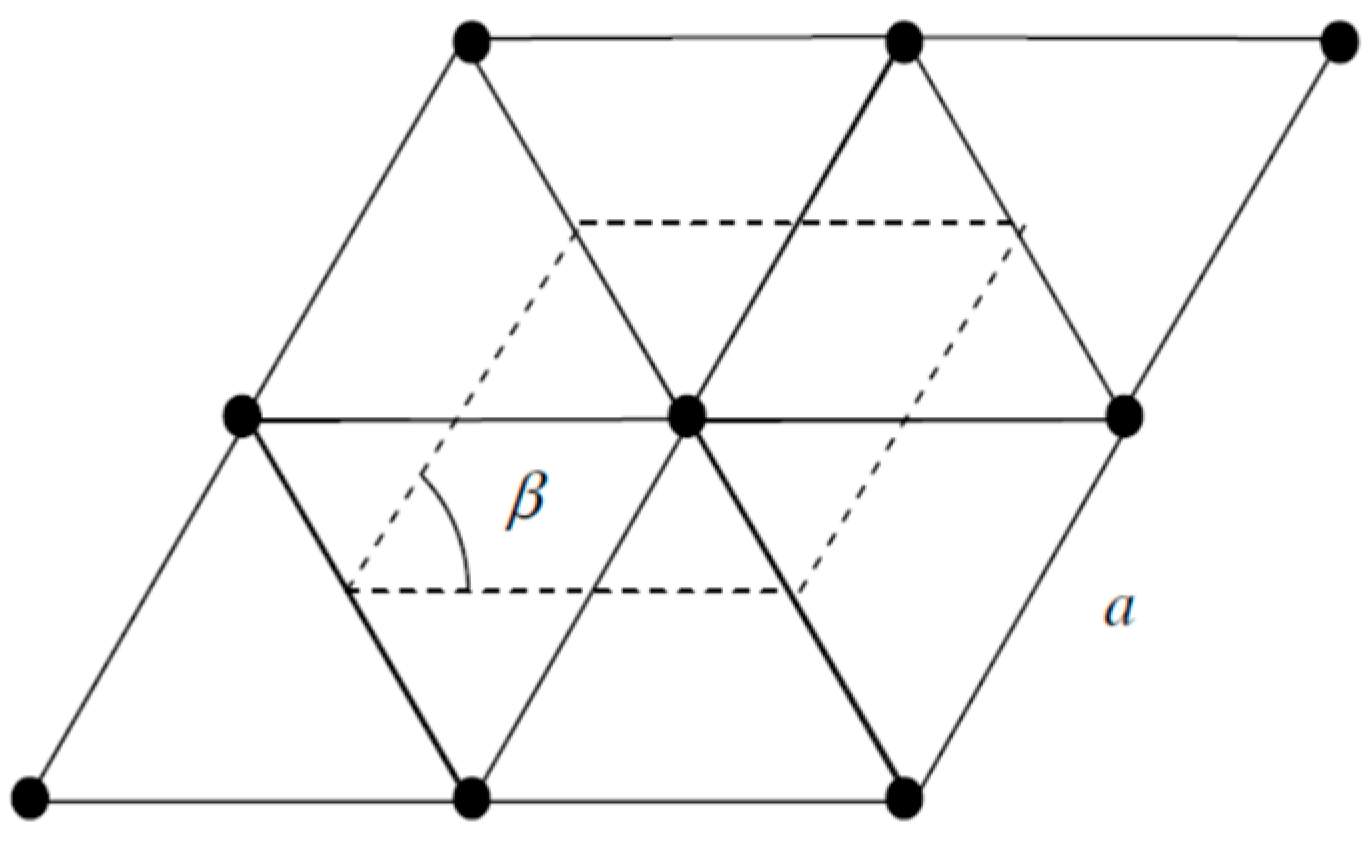
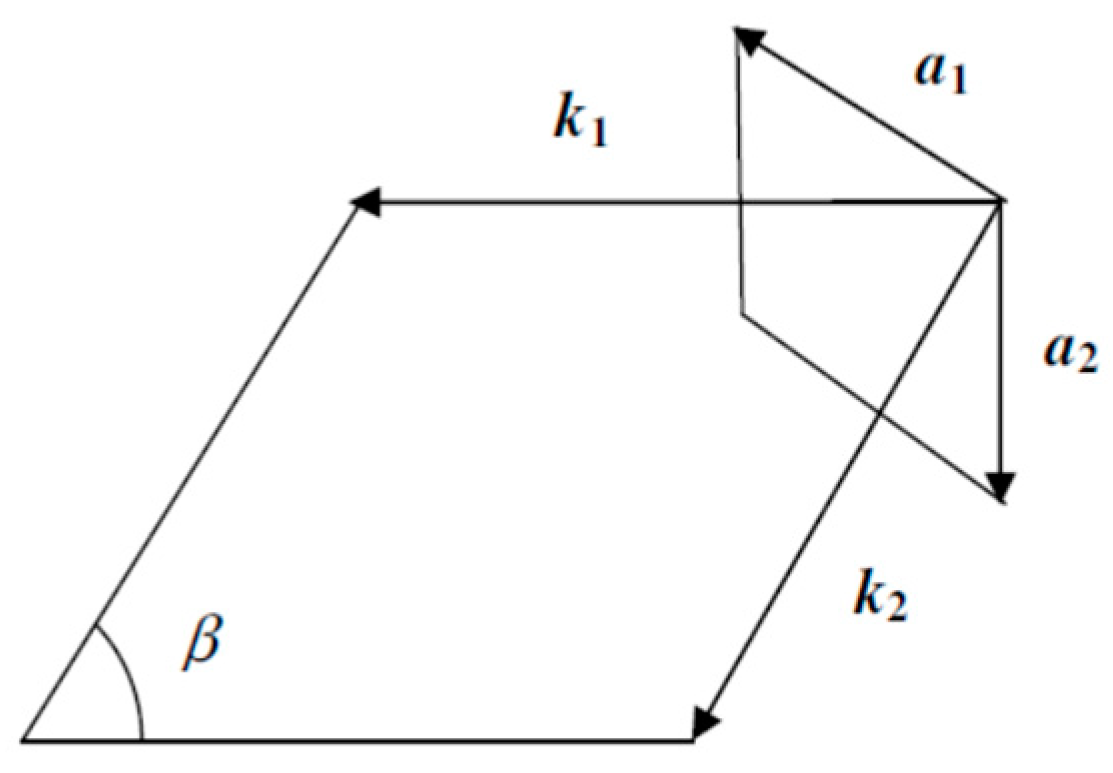
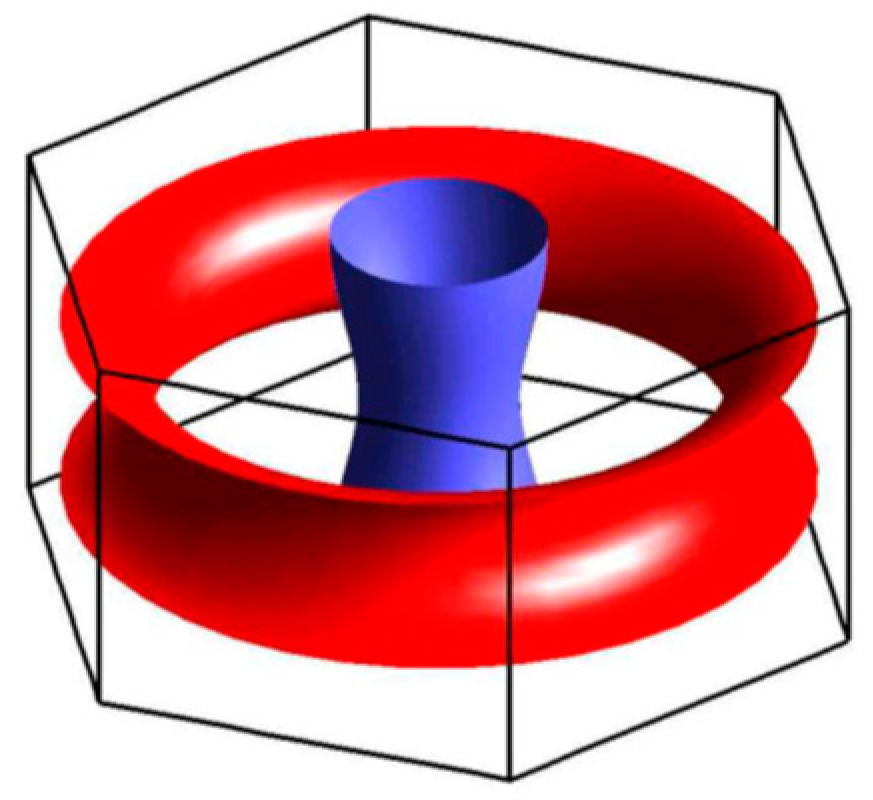
2. Theoretical Approach
- The construction of matrix elements for secular equation, which, within the initial quasi-classical approximation, reduces to a geometric task of determining the volume of the intersection of three spheres [89], and
- The solving of the secular equation, which determines the crystalline electronic energy spectrum [90].
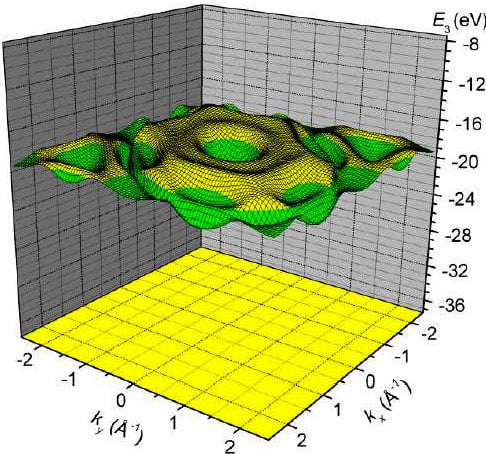
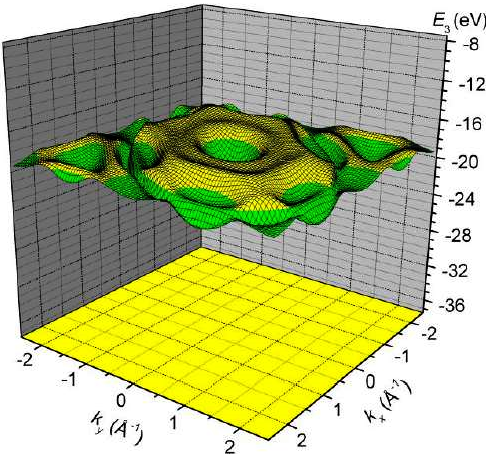
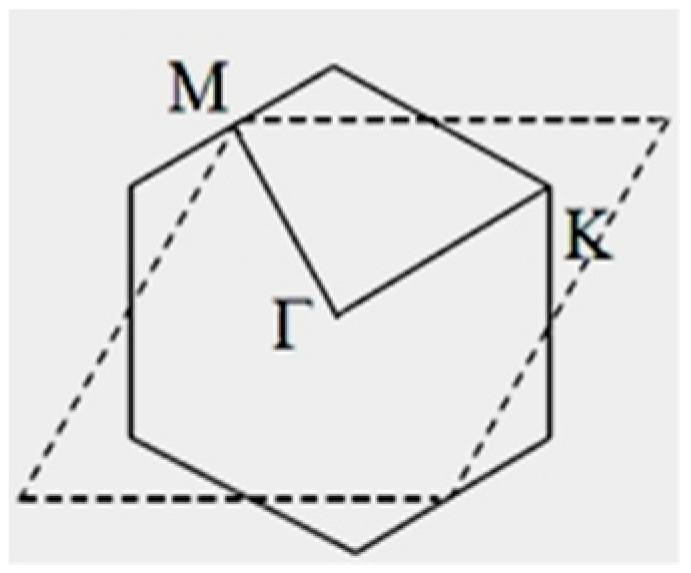
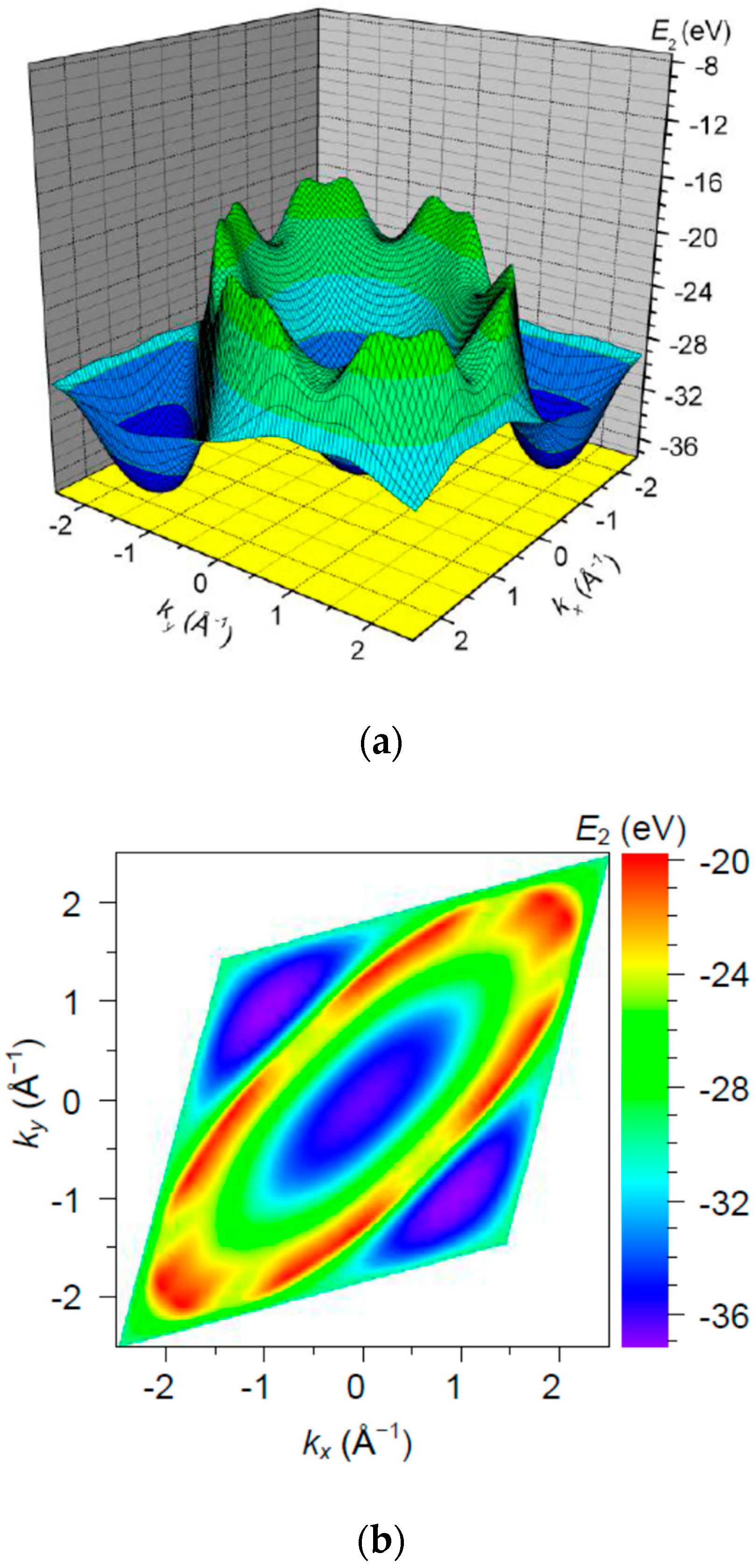
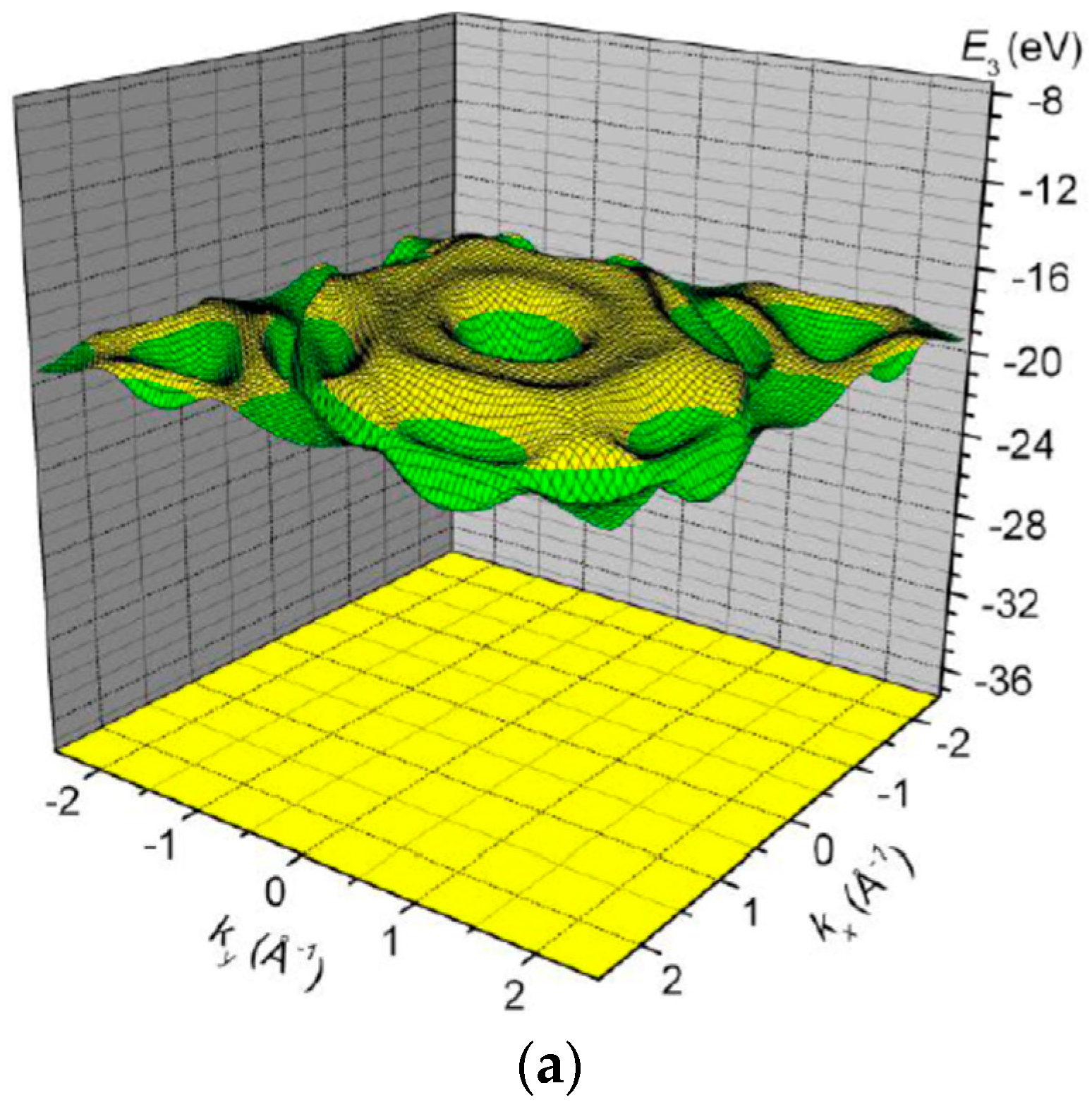
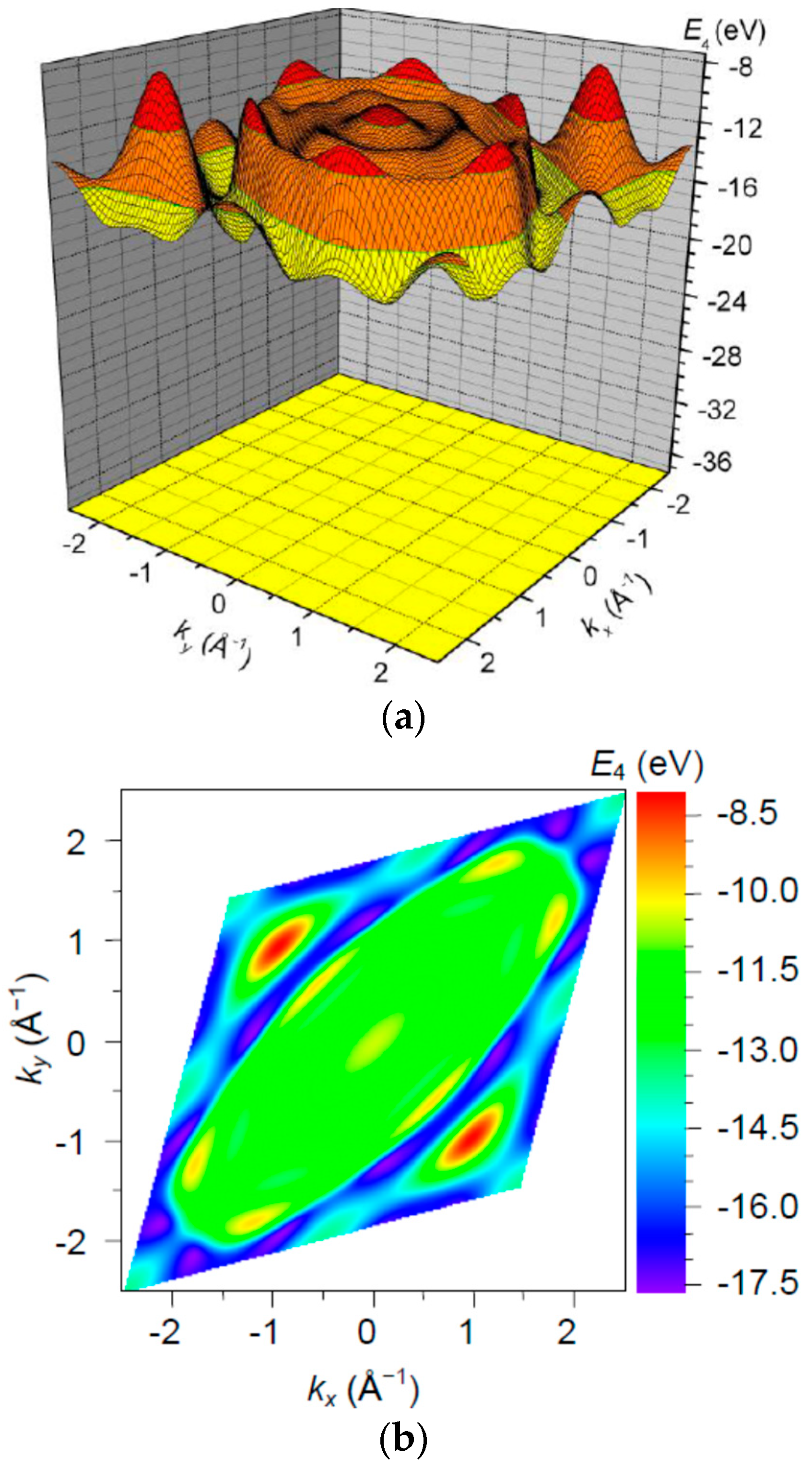
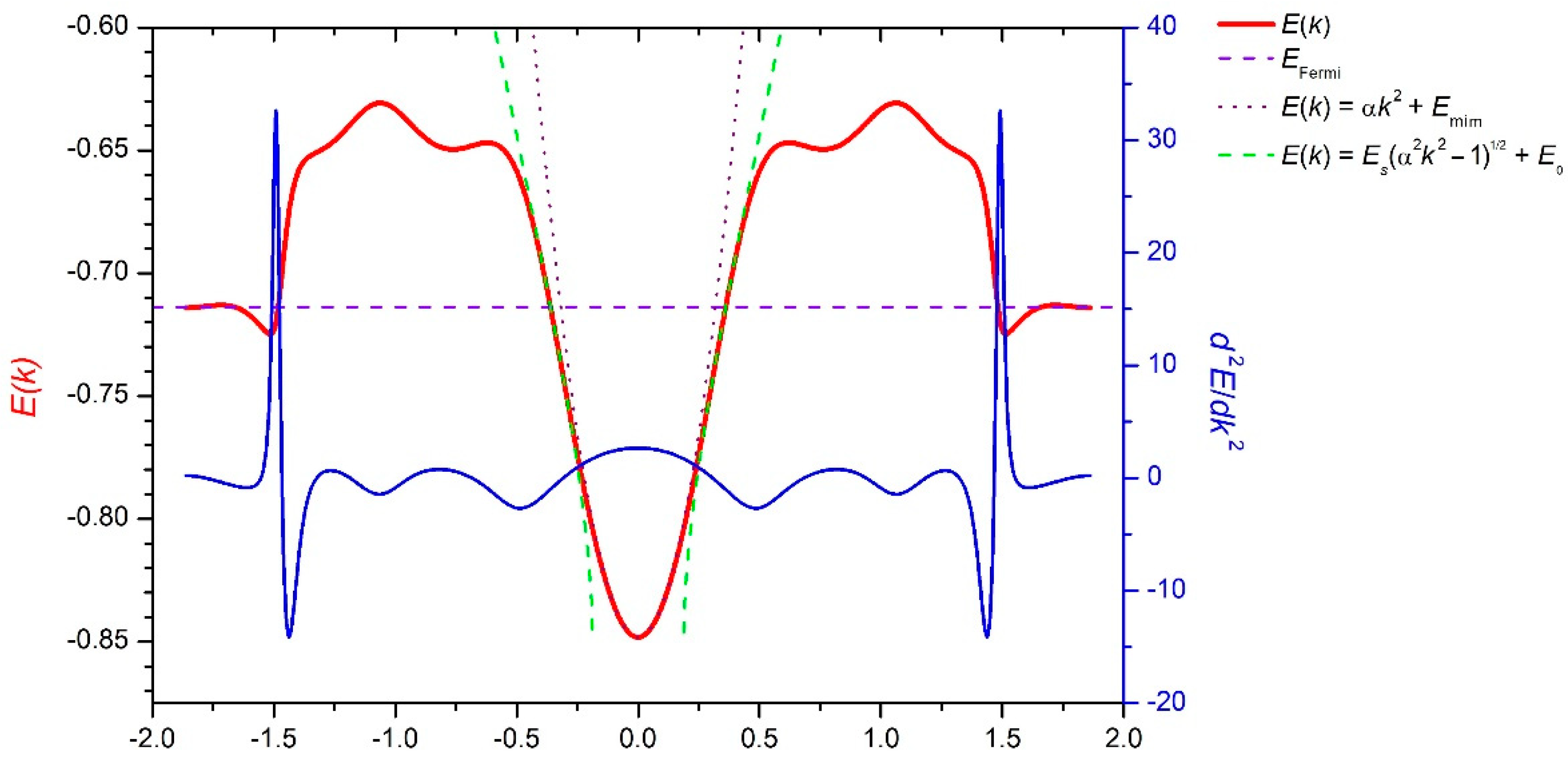
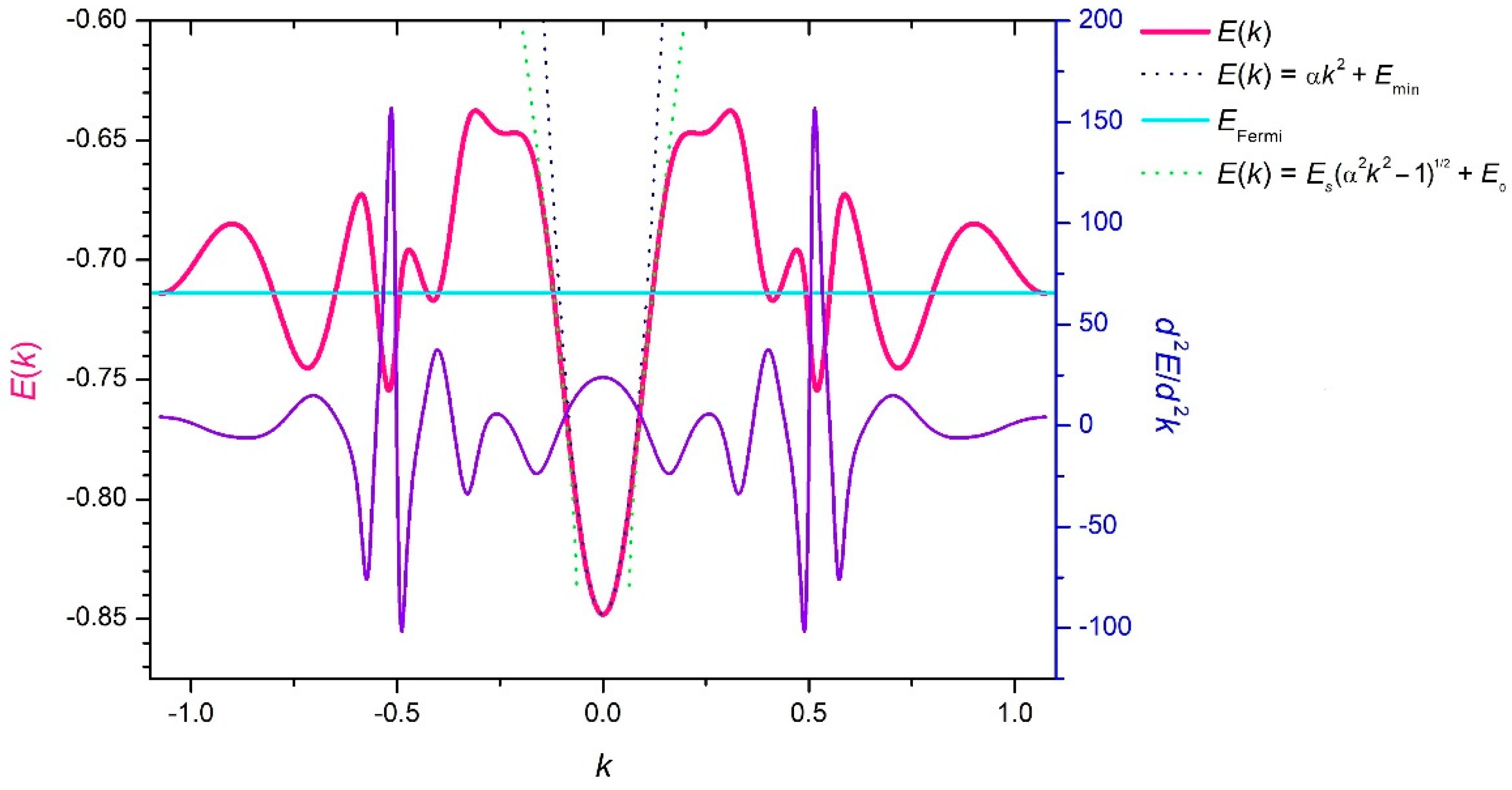
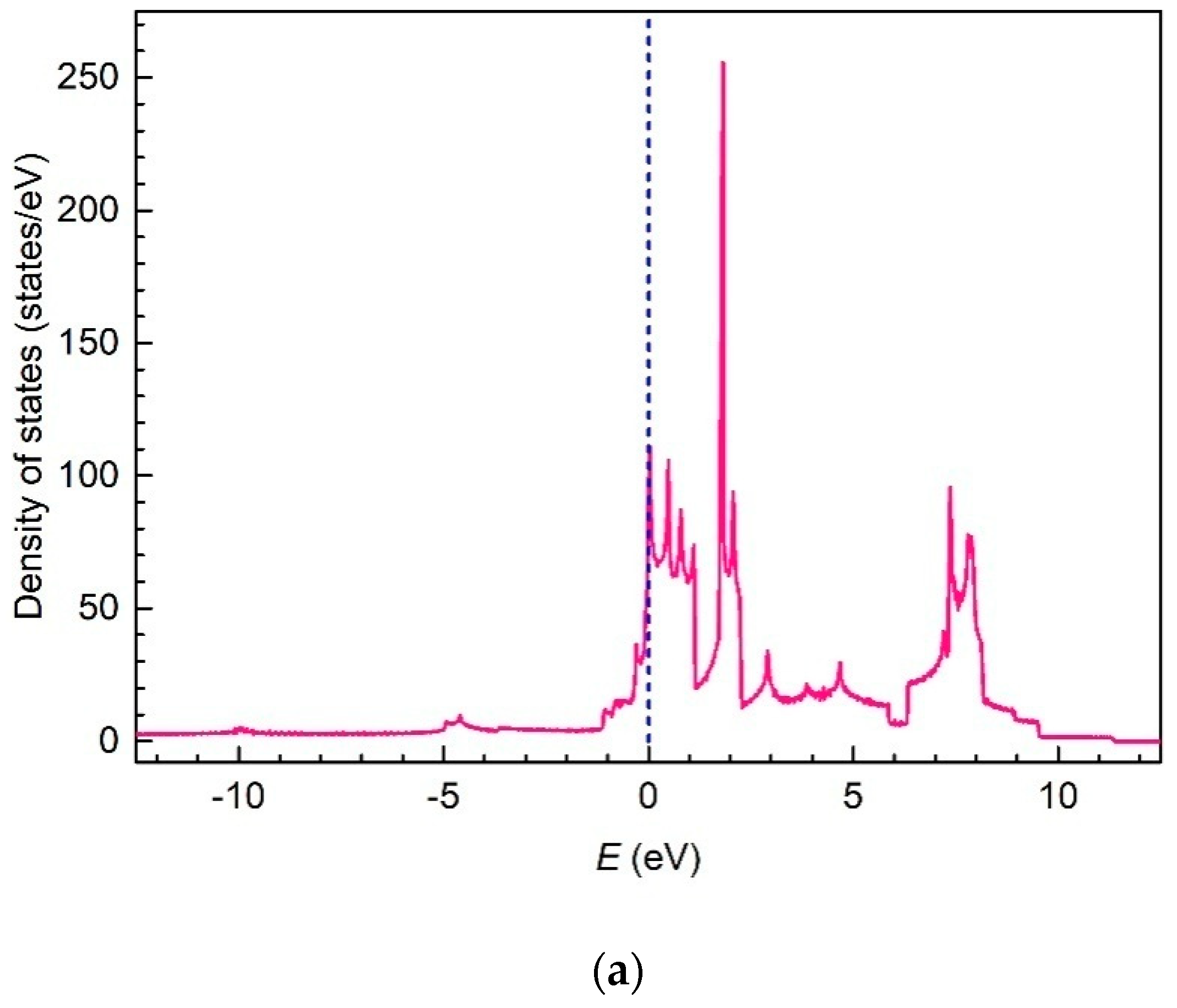
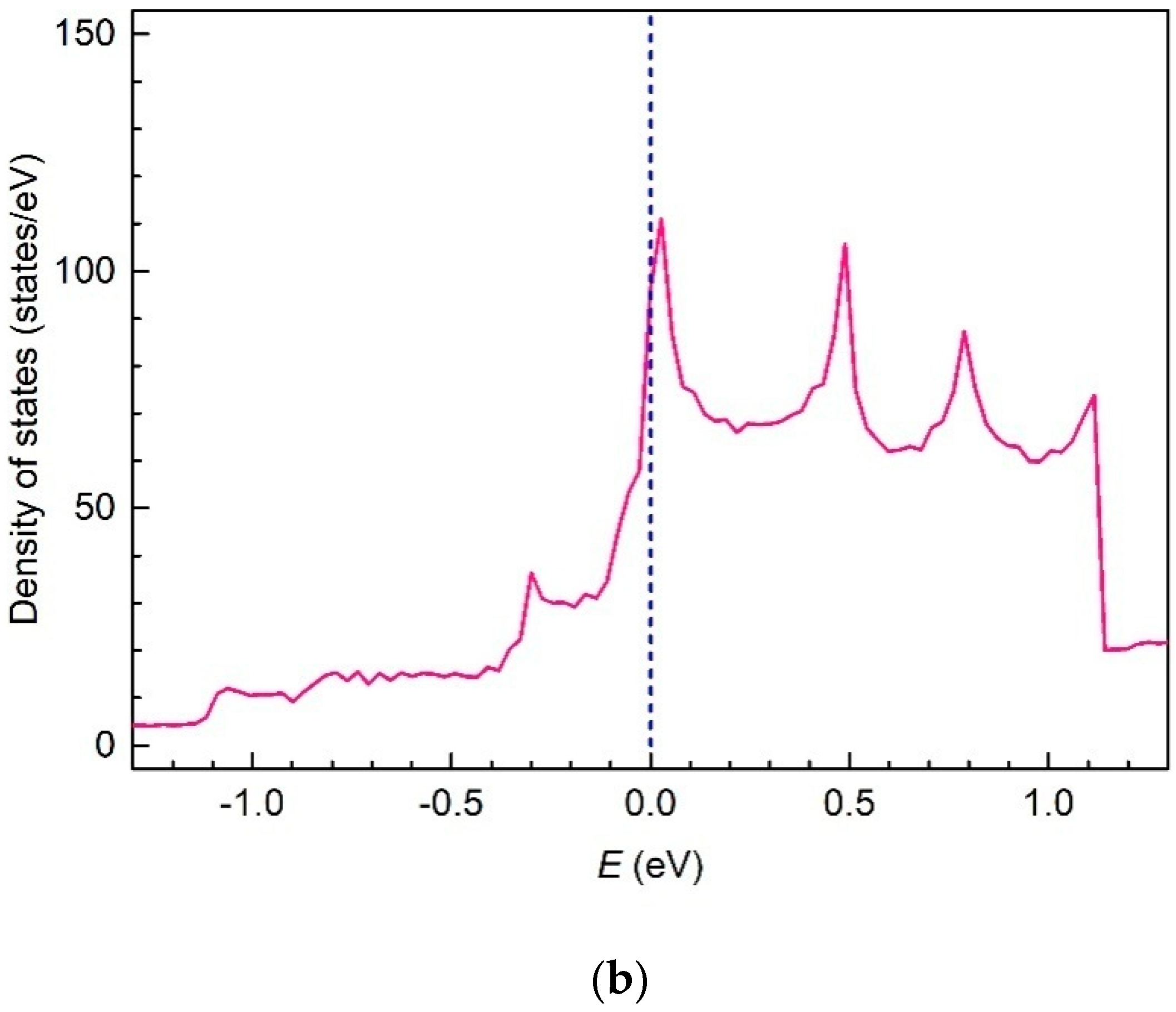
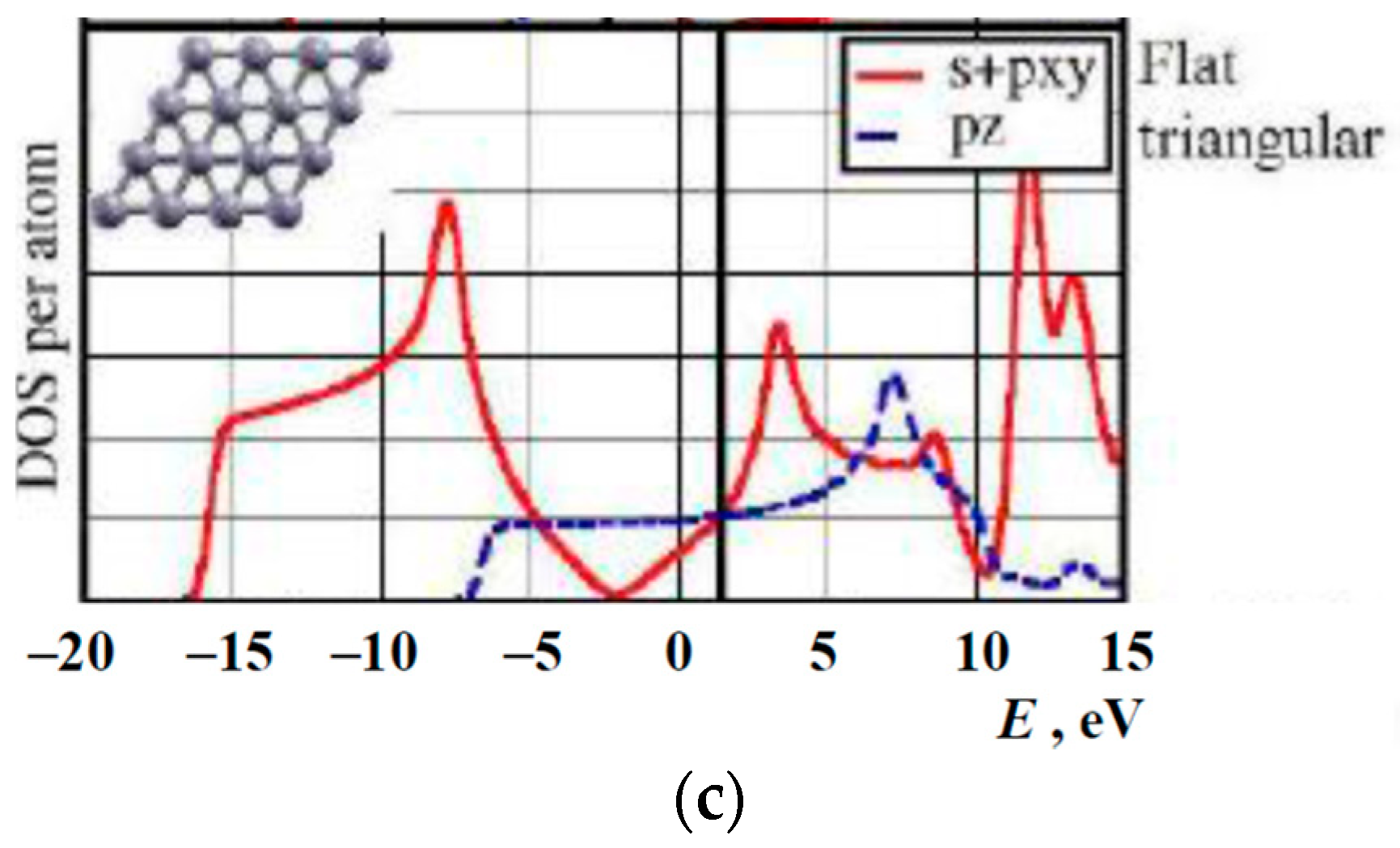
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A
References
- Becker, R.; Chkhartishvili, L.; Martin, P. Boron, the new graphene? Vac. Technol. Coat. 2015, 16, 38–44. [Google Scholar]
- Boustani, I. Systematic ab initio investigation of bare boron clusters: Determination of the geometry and electronic structures of Bn (n=12–14). Phys. Rev. B 1997, 55, 16426–16438. [Google Scholar] [CrossRef]
- Boustani, I.; Rubio, A.; Alonso, J.A. Ab initio study of B32 clusters: Competition between spherical, quasiplanar and tubular isomers. Chem. Phys. Lett. 1999, 311, 21–28. [Google Scholar] [CrossRef]
- Boustani, I.; Quandt, A.; Hernandez, E.; Rubio, A. New boron based nanostructured materials. J. Chem. Phys. 1999, 110, 3176–3185. [Google Scholar] [CrossRef]
- Boustani, I.; Quandt, A.; Rubio, A. Boron quasicrystals and boron nanotubes: Ab initio study of various B96 isomers. J. Solid State Chem. 2000, 154, 269–274. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; He, H.; Pandey, R.; Yap, Y.K.; Boustani, I. Novel spherical boron clusters and structural transition from 2D quasi-planar structures to 3D double-rings. J. Phys. Conf. Ser. 2009, 176, 012028. [Google Scholar] [CrossRef]
- Ozdogan, C.; Mukhopadhyay, S.; Hayami, W.; Guvenc, Z.B.; Pandey, R.; Boustani, I. The unusually stable B100 fullerene, structural transitions in boron nanostructures, and a comparative study of α- and γ-boron and sheets. J. Phys. Chem. C 2010, 114, 4362–4375. [Google Scholar] [CrossRef]
- Zhai, H.-J.; Kiran, B.; Li, J.; Wang, L.-S. Hydrocarbon analogues of boron clusters – Planarity, aromaticity and antiaromaticity. Nat. Mater. 2003, 2, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Piazza, Z.A.; Hu, H.-S.; Li, W.-L.; Zhao, Y.-F.; Li, J.; Wang, L.-S. Planar hexagonal B36 as a potential basis for extended single-atom layer boron sheets. Nat. Commun. 2014, 5, 3113–3118. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.-J.; Zhao, Y.-F.; Li, W.-L.; Chen, Q.; Bai, H.; Hu, H.-S.; Piazza, Z.A.; Tian, W.-J.; Lu, H.-G.; Wu, Y.-B.; et al. Observation of an all-boron fullerene. Nat. Chem. 2014, 6, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Ciuparu, D.; Klie, R.F.; Zhu, Y.; Pfefferle, L. Synthesis of pure boron single-wall nanotubes. J. Phys. Chem. B 2004, 108, 3967–3969. [Google Scholar] [CrossRef]
- Kiran, B.; Bulusu, S.; Zhai, H.-J.; Yoo, S.; Zeng, X.C.; Wang, L.-S. Planar-to-tubular structural transition in boron clusters: B20 as the embryo of single-walled boron nanotubes. Proc. Natl. Acad. Sci. USA 2005, 102, 961–964. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Shen, C.; Su, Z.; Ding, X.; Deng, S.; Chen, J.; Xu, N.; Gao, H. Metal-like single crystalline boron nanotubes: Synthesis and in situ study on electric transport and field emission properties. J. Mater. Chem. 2010, 20, 2197–2205. [Google Scholar] [CrossRef]
- Oger, E.; Crawford, N.R.M.; Kelting, R.; Weis, P.; Kappes, M.M.; Ahlrichs, R. Boron cluster cations: Transition from planar to cylindrical structures. Angew. Chem. Int. Ed. 2007, 46, 8503–8506. [Google Scholar] [CrossRef] [PubMed]
- Oger, E. Strukturaufklärung durch Mobilitätsmessungen an massenselektierten Clusterionen in der Gasphase. Ph.D. Thesis, University of Karlsruhe—Karlsruher Institute of Technology, Karlsruher, Germany, 2010. [Google Scholar]
- Mannix, A.J.; Zhou, X.-F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Parakhonskiy, G.; Dubrovinskaia, N.; Bykova, E.; Wirth, R.; Dubrovinsky, L. Experimental pressure-temperature phase diagram of boron: Resolving the long-standing enigma. Sci. Rep. 2011, 1, 96. [Google Scholar] [CrossRef] [PubMed]
- McGrady, J.W.; Papaconstantopoulos, D.A.; Mehl, M.J. Tight-binding study of boron structures. J. Phys. Chem. Solids 2014, 75, 1106–1112. [Google Scholar] [CrossRef]
- Shirai, K. Phase diagram of boron crystals. Jpn. J. Appl. Phys. 2017, 56, 05FA06. [Google Scholar] [CrossRef]
- Shirai, K.; Uemura, N.; Dekura, H. Structure and stability of pseudo-cubic tetragonal boron. Jpn. J. Appl. Phys. 2017, 56, 05FB05. [Google Scholar] [CrossRef]
- Imai, Y.; Mukaida, M.; Ueda, M.; Watanabe, A. Band-calculation of the electronic densities of states and the total energies of boron–silicon system. J. Alloys Comp. 2002, 347, 244–251. [Google Scholar] [CrossRef]
- Masago, A.; Shirai, K.; Katayama–Yoshida, H. Crystal stability of α- and β-boron. Phys. Rev. B 2006, 73, 104102. [Google Scholar] [CrossRef]
- Jemmis, E.D.; Prasad, D.L.V.K. Icosahedral B12, macropolyhedral boranes, β-rhombohedral boron and boron-rich solids. J. Solid State Chem. 2006, 179, 2768–2774. [Google Scholar] [CrossRef]
- Van Setten, M.J.; Uijttewaal, M.A.; DeWijs, G.A.; DeGroot, R.A. Thermodynamic stability of boron: The role of defects and zero point motion. J. Am. Chem. Soc. 2007, 129, 2458–2465. [Google Scholar] [CrossRef] [PubMed]
- Ogitsu, T.; Gygi, F.; Reed, J.; Motome, Y.; Schwegler, E.; Galli, G. Imperfect crystal and unusual semiconductor: Boron, a frustrated element. J. Am. Chem. Soc. 2009, 131, 1903–1909. [Google Scholar] [CrossRef] [PubMed]
- Widom, M.; Mihalkovic, M. Relative stability of α and β boron. J. Phys. Conf. Ser. 2009, 176, 012024. [Google Scholar] [CrossRef]
- Slack, G.A.; Hejna, C.I.; Garbauskas, M.F.; Kasper, J.S. The crystal structure and density of β-rhombohedral boron. J. Solid State Chem. 1988, 76, 52–63. [Google Scholar] [CrossRef]
- Boustani, I. New quasi-planar surfaces o fbare boron. Surf. Sci. 1997, 377, 355–363. [Google Scholar] [CrossRef]
- Boustani, I.; Quandt, A. Boronin ab initio calculations. Comput. Mater. Sci. 1998, 11, 132–137. [Google Scholar] [CrossRef]
- Evans, M.H.; Joannopoulos, J.D.; Pantelides, S.T. Electronic and mechanical properties of planar and tubular boron structures. Phys. Rev. B 2005, 72, 045434. [Google Scholar] [CrossRef]
- Cabria, I.; Lopez, M.J.; Alonso, J.A. Density functional calculations of hydrogen adsorption on boron nanotubes and boron sheets. Nanotechnology 2006, 17, 778–785. [Google Scholar] [CrossRef]
- Cabria, I.; Alonso, J.A.; Lopez, M.J. Buckling in boron sheets and nanotubes. Phys. Status Solidi A 2006, 203, 1105–1110. [Google Scholar] [CrossRef]
- Kunstmann, J.; Quandt, A. Broad boron sheets and boron nanotubes: An ab initio study of structural, electronic, and mechanical properties. Phys. Rev. B 2006, 74, 035413. [Google Scholar] [CrossRef]
- Lau, K.C.; Pati, R.; Pandey, R.; Pineda, A.C. First-principles study of the stability and electronic properties of sheets and nanotubes of elemental boron. Chem. Phys. Lett. 2006, 418, 549–554. [Google Scholar] [CrossRef]
- Lau, K.C.; Pandey, R. Stability and electronic properties of atomistically-engineered 2D boron sheets. J. Phys. Chem. C 2007, 111, 2906–2912. [Google Scholar] [CrossRef]
- Tang, H.; Ismail–Beigi, S. Novel precursors for boron nanotubes: The competition of two-center and three-center bonding in boron sheets. Phys. Rev. Lett. 2007, 99, 115501. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ding, Y.; Ni, J. Ab initio prediction of stable boron sheets and boron nanotubes: Structure, stability, and electronic properties. Phys. Rev. B 2008, 77, 041402(R). [Google Scholar] [CrossRef]
- Sebetci, A.; Mete, E.; Boustani, I. Freestanding double walled boron nanotubes. J. Phys. Chem. Solids 2008, 69, 2004–2012. [Google Scholar] [CrossRef]
- Singh, A.K.; Sadrzadeh, A.; Yakobson, B.I. Probing properties of boron α-tubes by ab initio calculations. Nano Lett. 2008, 8, 1314–1317. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Li, Y.-C. A new class of boron nanotubes. Chem. Phys. Chem. 2009, 10, 3119–3121. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Ismail–Beigi, S. First-principles study of boron sheets and nanotubes. Phys. Rev. B 2010, 82, 115412. [Google Scholar] [CrossRef]
- Zope, R.R.; Baruah, T. Snub boro nnanostructures: Chiral fullerenes, nanotubes and planar sheet. Chem. Phys. Lett. 2011, 501, 193–196. [Google Scholar] [CrossRef]
- Simsek, M.; Aydın, S. First-principles calculations of two dimensional boron sheets. In Abstracts of the 17th International Symposium on Boron, Borides and Related Materials, Istanbul, Turkey, 11–17 September 2011; Yucel, O., Ed.; BKM: Ankara, Turkey, 2011; p. 86. [Google Scholar]
- Kunstmann, J.; Boeri, L.; Kortus, J. Bonding in boron: Building high-pressure phases from boron sheets. In Abstracts of the 17th International Symposium on Boron, Borides and Related Materials, Istanbul, Turkey, 11–17 September 2011; Yucel, O., Ed.; BKM: Ankara, Turkey, 2011; p. 289. [Google Scholar]
- Bezugly, V.; Kunstmann, J.; Grundkotter-Stock, B.; Frauenheim, T.; Niehaus, T.; Cuniberti, G. Highly conductive boron nanotubes: Transport properties, work functions, and structural stabilities. ACS Nano 2011, 5, 4997–5005. [Google Scholar] [CrossRef] [PubMed]
- Tang, H. First-Principles Investigation on Boron Nanostructures. Ph.D. Thesis, Yale University, New Haven, CT, USA, 2011. [Google Scholar]
- Zhou, X.-F.; Dong, X.; Oganov, A.R.; Zhu, Q.; Tian, Y.; Wang, H.-T. Semimetallic two-dimensional boron allotrope with massless Dirac fermions. Phys. Rev. Lett. 2014, 112, 085502. [Google Scholar] [CrossRef]
- Wu, X.; Dai, J.; Zhao, Y.; Zhuo, Z.; Yang, J.; Zeng, X. Two-dimensional boron monolayer sheets. ACS Nano 2012, 6, 7443–7453. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Mu, Y.; Li, S.-D. Comment on “Two-dimensional boron monolayer sheets”. ACS Nano 2013, 7, 879. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dai, J.; Zhao, Y.; Zhuo, Z.; Yang, J.; Zeng, X.C. Reply to “Comment on ‘Two-dimensional boron monolayer sheets’”. ACS Nano 2013, 7, 880–881. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-F.; Oganov, A.R.; Shao, X.; Zhu, Q.; Wang, H.-T. Unexpected reconstruction of the α-boron (111) surface. Phys. Rev. Lett. 2014, 113, 176101. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Chen, Q.; Chen, N.; Lu, H.; Li, S.-D. A novel borophene featuring heptagonal holes: Common precursor of borospherenes. Phys. Chem. Chem. Phys. 2017, 30, 19890–19895. [Google Scholar] [CrossRef] [PubMed]
- Boroznina, E.V.; Davletova, O.A.; Zaporotskova, I.V. Boron monolayer γ3-type. Formation of the vacancy defect and pinhole. J. Nano-Electron. Phys. 2016, 8, 04054. [Google Scholar] [CrossRef]
- Chkhartishvili, L. All-boron nanostructures. In CRC Concise Encyclopedia of Nanotechnology; Kharisov, B.I., Kharissova, O.V., Ortiz–Mendez, U., Eds.; CRC Press: Boca Raton, FL, USA, 2016; pp. 53–69. [Google Scholar]
- Li, W.-L.; Pal, R.; Piazza, Z.A.; Zeng, X.C.; Wang, L.-S. B27−: Appearance of the smallest planar boron cluster containing a hexagonal vacancy. J. Chem. Phys. 2015, 142, 204305. [Google Scholar] [CrossRef] [PubMed]
- Vast, N.; Bernard, S.; Zerah, G. Structural and electronic properties of liquid boron from a molecular-dynamics simulation. Phys. Rev. B 1995, 52, 4123–4130. [Google Scholar] [CrossRef]
- Krishnan, S.; Ansell, S.; Felten, J.J.; Volin, K.J.; Price, D.L. Structure of liquid boron. Phys. Rev. Lett. 1998, 81, 586–589. [Google Scholar] [CrossRef]
- Price, D.L.; Alatas, A.; Hennet, L.; Jakse, N.; Krishnan, S.; Pasturel, A.; Pozdnyakova, I.; Saboungi, M.-L.; Said, A.; Scheunemann, R.; et al. Liquid boron: X-ray measurements and ab initio molecular dynamics simulations. Phys. Rev. B 2009, 79, 134201. [Google Scholar] [CrossRef]
- Butler, S.Z.; Hollen, S.M.; Cao, L.; Cui, Y.; Gupta, J.A.; Gutierrez, H.R.; Heinz, T.F.; Hong, S.S.; Huang, J.; Ismach, A.F.; et al. Progress, challenges, and opportunities in two-dimensional materials beyond graphene. ACS Nano 2013, 7, 2898–2926. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xu, C.; Su, H.; Liang, J.; Lin, S.; Gu, L.; Wang, X.; Chen, M.; Zheng, N. Single-crystalline rhodium nanosheets with atomic thickness. Adv. Sci. 2015, 2, 1500100. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chang, Y.; Gan, L.; Ma, Y.; Zhai, T. Ultra thin nanosheets: Ultrathin single-crystalline boron nanosheets for enhanced electro-optical performances. Adv. Sci. 2015, 2, 1500023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Z.; Yan, Q.B.; Du, S.X.; Su, G.; Gao, H.-J. Boron sheet adsorbed on metal surfaces: Structures and electronic properties. J. Phys. Chem. C 2012, 116, 18202–18206. [Google Scholar] [CrossRef]
- Penev, E.S.; Bhowmick, S.; Sadrzadeh, A.; Yakobson, B.I. Polymorphism of two-dimensional boron. Nano Lett. 2012, 12, 2441–2445. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Penev, E.S.; Yakobson, B.I. Probing the synthesis of two-dimensional boron by first-principles computations. Angew. Chem. Int. Ed. 2013, 52, 3156–3159. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gao, J.; Zhao, J. Fromboronclustertotwo-dimensionalboronsheetonCu (111) surface: Growth mechanism and hole formation. Sci. Rep. 2013, 3, 3238. [Google Scholar]
- Yi, W.-C.; Liu, W.; Botana, J.; Zhao, L.; Liu, Z.; Liu, J.-Y.; Miao, M.-S. Honeycomb boron allotropes with Dirac cones: A true analogue to graphene. J. Phys. Chem. Lett. 2017, 8, 2647–2653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Mannix, A.J.; Hu, Z.; Kiraly, B.; Guisinger, N.P.; Hersam, M.C.; Yakobson, B.I. Substrate-induced nanoscale undulations of borophene on silver. Nano Lett. 2016, 16, 6622–6627. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Drozdov, I.K.; Eltinge, S.; Zahl, P.; Ismail–Beigi, S.; Bozovic, I.; Gozar, A. Large-area single-crystal sheets of borophene on Cu(111) surfaces. Nat. Nanotechnol. 2019, 14, 44–49. [Google Scholar] [CrossRef] [PubMed]
- He, X.-L.; Weng, X.-J.; Zhang, Y.; Zhao, Z.; Wang, Z.; Xu, B.; Oganov, A.R.; Tian, Y.; Zhou, X.-F.; Wang, H.-T. Two-dimensional boron on Pb(110) surface. Flat Chem. 2018, 7, 34–41. [Google Scholar]
- Tarkowski, T.; Majewski, J.A.; Gonzalez Szwacki, N. Energy decomposition analysis of neutral and negatively charged borophenes. Flat Chem. 2018, 7, 42–47. [Google Scholar] [CrossRef]
- Gao, N.; Wu, X.; Jiang, X.; Bai, Y.; Zhao, J. Structure and stability of bilayer borophene: The roles of hexagonal holes and interlayer bonding. Flat Chem. 2018, 7, 48–54. [Google Scholar] [CrossRef]
- Liu, D.; Tomanek, D. Effect of net charge on the relative stability of different 2D boron allotropes (ContributedTalk). In Proceedings of the APS March Meeting 2019, Boston, MA, USA, March 4–8 2019. Abstract: X13.00003. [Google Scholar]
- Vangaveti, R. Boron Induced Surface Modification of Transition Metals. MSc Thesis, New Jersey’s Science & Technology University, Newark, NJ, USA, 2006. [Google Scholar]
- Chkhartishvili, L. Interaction between neutron-radiation and boron-containing materials. In Radiation Synthesis of Materials and Compounds; Kharisov, B.I., Kharissova, O.V., Ortiz–Méndez, U., Eds.; CRC Press—Taylor & Francis Group: Boca Raton, FL, USA, 2013; pp. 43–80. [Google Scholar]
- Chkhartishvili, L.; Murusidze, I. Band structure of all-boron 2D metallic crystals as a prospective electromagnetic shielding material. In Proceedings of the International Conference on Fundamental & Applied Nano Electro Magnetics, Minsk, Belarus, 22–25 May 2012; Belarusian State University: Minsk, Belarus, 2012; p. 11. [Google Scholar]
- Zhang, Z.; Yang, Y.; Penev, E.S.; Yakobson, B.I. Elasticity, flexibility, and ideal strength of borophenes. Adv. Funct. Mater. 2017, 27, 1605059. [Google Scholar] [CrossRef]
- Martin, P.M. Active thin films: Graphene-related materials graphene oxide and borophene. Vac. Technol. Coat. 2018, 19, 6–13. [Google Scholar]
- Martin, P.M. Active thin films: Applications for graphene and related materials. Vac. Technol. Coat. 2018, 19, 6–14. [Google Scholar]
- Zhao, Y.; Xu, Q.; Simpson, L.J.; Dillon, A.C. Prediction of diamond-like, metallic boron structures. Chem. Phys. Lett. 2010, 496, 280–283. [Google Scholar] [CrossRef]
- Zabolotsky, A.D.; Lozovik, Y.E. Strain-induced pseudomagnetic field in Dirac semimetal borophene. arXiv, 2016; arXiv:1607.02530v2. [Google Scholar] [CrossRef]
- Zhou, X.-F.; Oganov, A.R.; Wang, Z.; Popov, I.A.; Boldyrev, A.I.; Wang, H.-T. Two-Dimensional magnetic boron. Phys. Rev. B 2016, 93, 085406. [Google Scholar] [CrossRef]
- Chkhartishvili, L. Quasi-Classical Theory of Substance Ground State; Technical University Press: Tbilisi, Georgia, 2004. [Google Scholar]
- Chkhartishvili, L.; Mamisashvili, N.; Maisuradze, N. Single-Parameter model for multi-walled geometry of nanotubular boron. Solid State Sci. 2015, 47, 61–67. [Google Scholar] [CrossRef]
- Chkhartishvili, L. Boron quasi-planar clusters. A mini-review on diatomic approach. In Proceedings of the IEEE 7th International Conference on Nanomaterials: Applications and Properties, Part 4, Track: Nanomaterials for Electronics, Spintronics and Photonics, Zatoka, Ukraine, 10–15 September 2017; Pogrebnjak, A.D., Ed.; Sumy State University: Sumy, Ukraine, 2017; pp. 1–5. [Google Scholar]
- Chkhartishvili, L. Relative stability of planar clusters B11, B12, and B13 in neutral- and charged states. Char. Appl. Nanomater. 2018, 1, 3. [Google Scholar] [CrossRef]
- Chkhartishvili, L.; Murusidze, I. Frequencies of vibrations localized on interstitial metal impurities in beta-rhombohedral boron based materials. Am. J. Mater. Sci. 2014, 4, 103–110. [Google Scholar]
- Chkhartishvili, L.; Tsagareishvili, O.; Gabunia, D. Isotopic expansion of boron. J. Metall. Eng. 2014, 3, 97–103. [Google Scholar] [CrossRef]
- Chkhartishvili, L.S. Volume of the intersection of three spheres. Math. Notes 2001, 69, 421–428. [Google Scholar] [CrossRef]
- Chkhartishvili, L.S. Iterative solution of the secular equation. Math. Notes 2005, 77, 273–279. [Google Scholar] [CrossRef]
- Chkhartishvili, L.; Lezhava, D.; Tsagareishvili, O. Quasi-classical determination of electronic energies and vibration frequencies in boron compounds. J. Solid State Chem. 2000, 154, 148–152. [Google Scholar] [CrossRef]
- Chkhartishvili, L. Quasi-classical approach: Electronic structure of cubic boron nitride crystals. J. Solid State Chem. 2004, 177, 395–399. [Google Scholar] [CrossRef]
- Chkhartishvili, L.S. Quasi-classical estimates of the lattice constant and bandgap of a crystal: Two-dimensional boron nitride. Phys. Solid State 2004, 46, 2126–2133. [Google Scholar] [CrossRef]
- Chkhartishvili, L. Density of electron states in wurtzite-like boron nitride: A quasi-classical calculation. Mater. Sci. Ind. J. 2006, 2, 18–23. [Google Scholar]
- Chkhartishvili, L.; Murusidze, I.; Darchiashvili, M.; Tsagareishvili, O.; Gabunia, D. Metal impurities in crystallographic voids of beta-rhombohedral boron lattice: Binding energies and electron levels. Solid State Sci. 2012, 14, 1673–1682. [Google Scholar] [CrossRef]
- Chkhartishvili, L.; Berberashvili, T. Intra-atomic electric field radial potentials in step-like presentation. J. Electr. Magn. Anal. Appl. 2010, 2, 205–243. [Google Scholar] [CrossRef]
- Froese–Fischer, C. Th eHartree–Fock Method for Atoms. A Numerical Approach; Wiley: New York, NY, USA, 1977. [Google Scholar]
- Radtsig, A.A.; Smirnov, B.M. Parameters of Atoms and Atomic Ions. Reference Book; Energoatomizdat: Moscow, Russia, 1986. [Google Scholar]
- Galvez, F.J.; Buendia, E.; Sarsa, A. Excited states of boron isoelectronic series from explicitly correlated wave functions. J. Chem. Phys. 2005, 122, 154307. [Google Scholar] [CrossRef] [PubMed]
- Dahm, T.; Schopohl, N. Fermi surface topology and the upper critical field in two-band superconductors—Application to MgB2. Phys. Rev. Lett. 2003, 91, 017001. [Google Scholar] [CrossRef] [PubMed]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Orbital | State | (a.u.) | (a.u.) | (a.u.) |
|---|---|---|---|---|
| 1 | 1s | 7.695335 | 0 | 0.509802 |
| 2 | 2s | 0.494706 | 0 | 4.021346 |
| 3 | 2p | 0.309856 | 0.744122 | 4.337060 |
| 4 | 2p | 0.214595 | 0.894159 | 5.211538 |
| (a.u.) | (a.u.) | |
|---|---|---|
| 0 | 0 | – |
| 1 | 0.027585 | 210.5468 |
| 2 | 0.509802 | 8.882329 |
| 3 | 0.744122 | 3.65292 |
| 4 | 4.021346 | 0.206072 |
| 5 | 4.33706 | 0.000614 |
| Band | eV | eV |
|---|---|---|
| 1 | 0 | |
| 2 | 17.36 | 239 |
| 3 | 5.92 | −3.23 |
| 4 | 9.57 | −0.49 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chkhartishvili, L.; Murusidze, I.; Becker, R. Electronic Structure of Boron Flat Holeless Sheet. Condens. Matter 2019, 4, 28. https://doi.org/10.3390/condmat4010028
Chkhartishvili L, Murusidze I, Becker R. Electronic Structure of Boron Flat Holeless Sheet. Condensed Matter. 2019; 4(1):28. https://doi.org/10.3390/condmat4010028
Chicago/Turabian StyleChkhartishvili, Levan, Ivane Murusidze, and Rick Becker. 2019. "Electronic Structure of Boron Flat Holeless Sheet" Condensed Matter 4, no. 1: 28. https://doi.org/10.3390/condmat4010028
APA StyleChkhartishvili, L., Murusidze, I., & Becker, R. (2019). Electronic Structure of Boron Flat Holeless Sheet. Condensed Matter, 4(1), 28. https://doi.org/10.3390/condmat4010028