
Integrated Transcriptome and Microbiome Analyses Reveal Growth- and Stress-Response-Related Genes and Microbes in Mandarin Fish (Siniperca chuatsi)

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Materials and Rearing Conditions
2.2. RNA Isolation, Library Construction, and Illumina Sequencing
2.3. Transcriptome Assembly and DEG Screening
2.4. Functional Annotation and Enrichment Analyses
2.5. DNA Extraction and 16S rRNA Gene Amplification
2.6. Illumina MiSeq and Sequence Processing
2.7. Bioinformatic Analysis of the 16S rRNA
2.8. Association Analysis Between the Intestinal Microbiota and DEGs
2.9. Statistical Analysis
3. Results
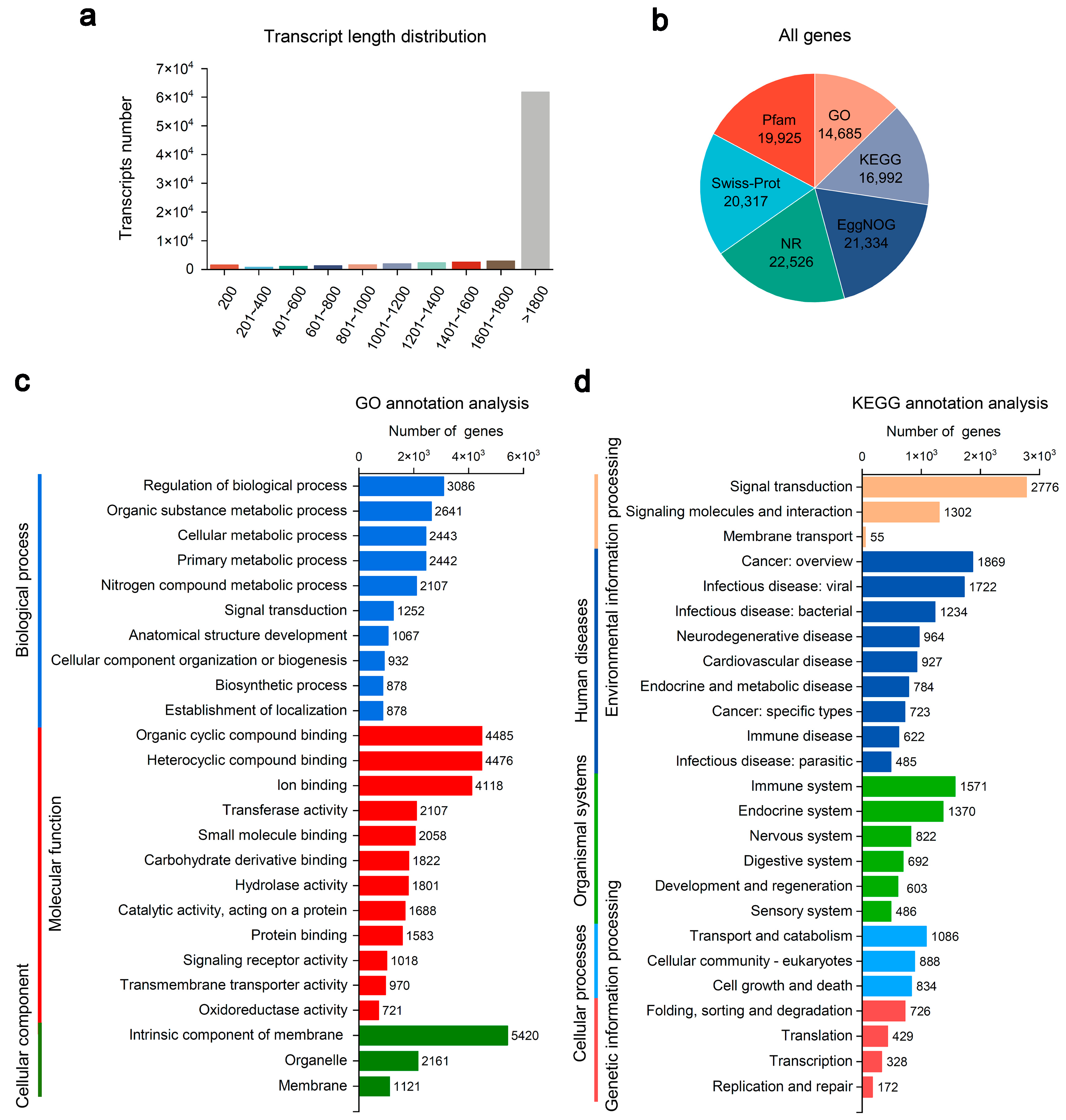
3.1. Transcriptome Overview of Mandarin Fish from Different Groups
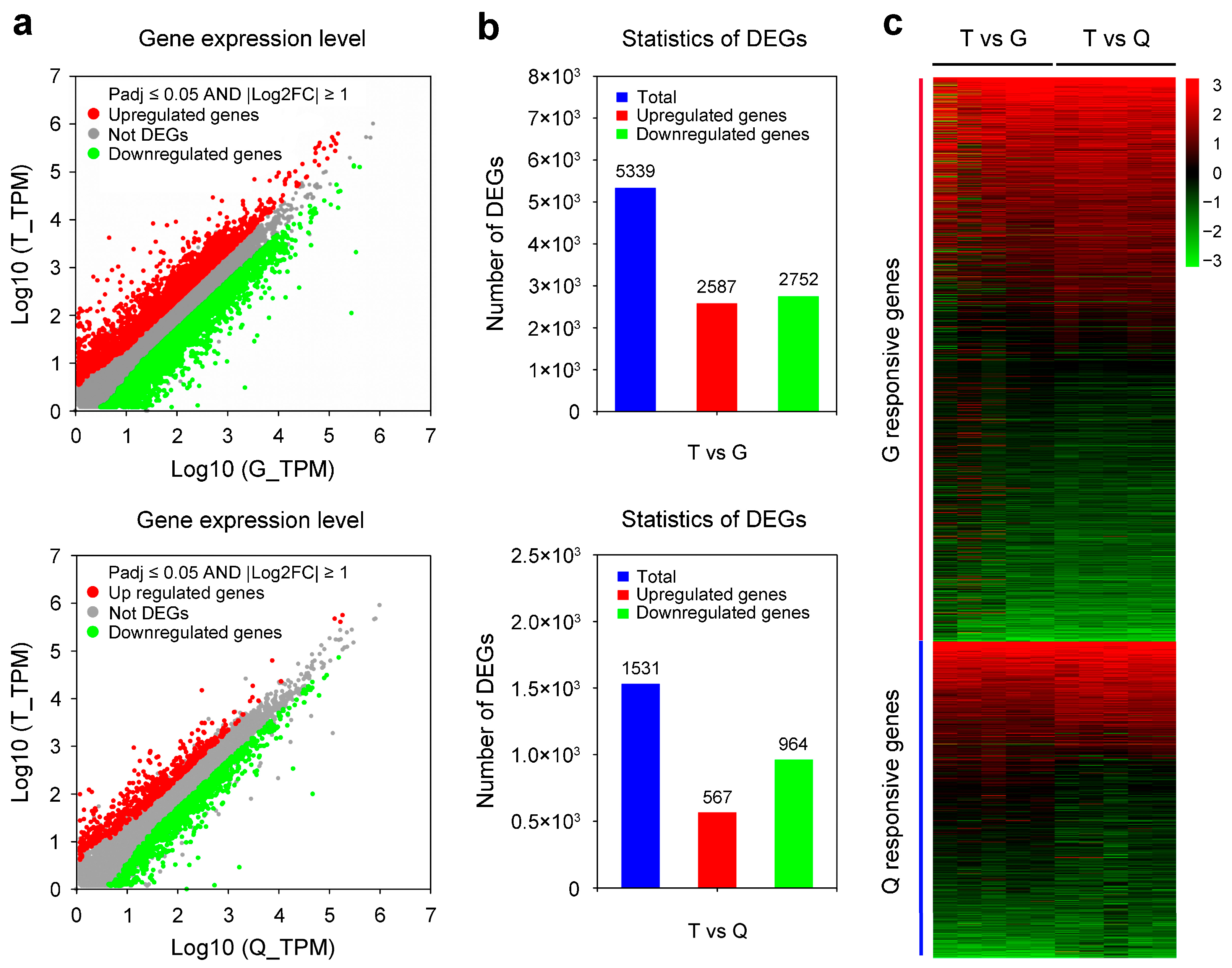
3.2. Scanning of DEGs in T Group Compared with the G and Q Groups
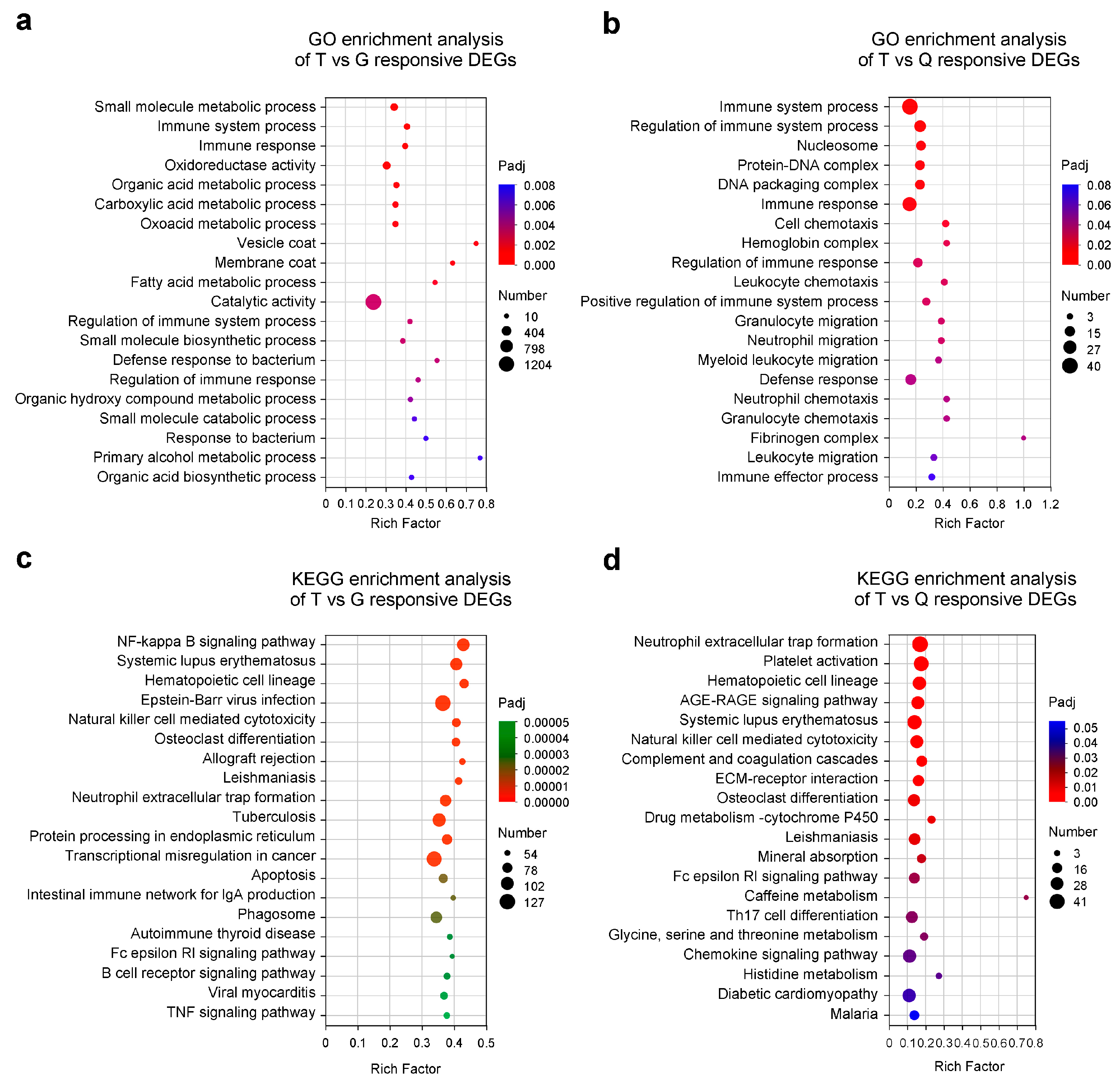
3.3. GO and KEGG Enrichment Analyses of the DEGs
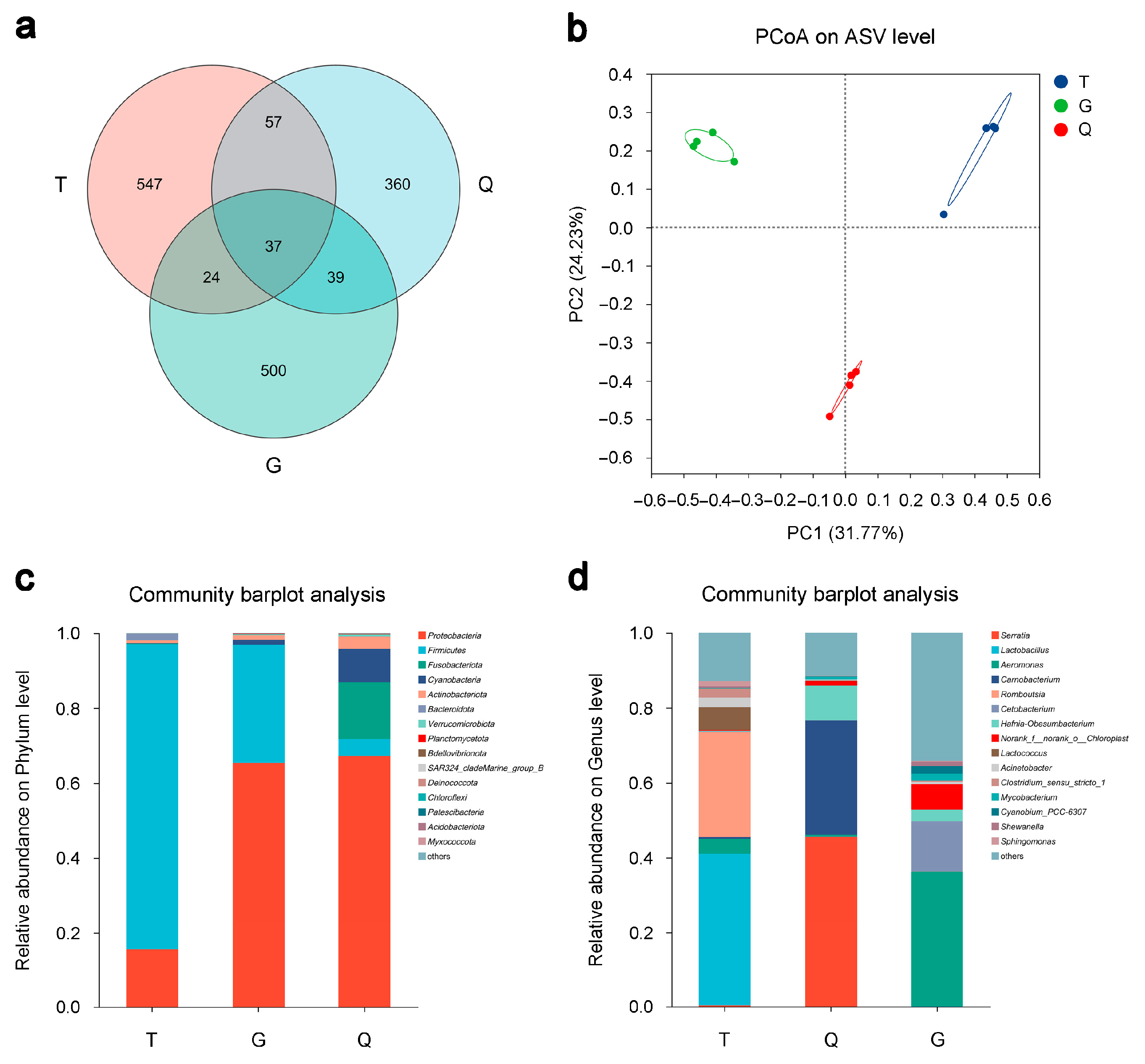
3.4. Overview of the Intestinal Microbiomes of Mandarin Fish from the T, G, and Q Groups
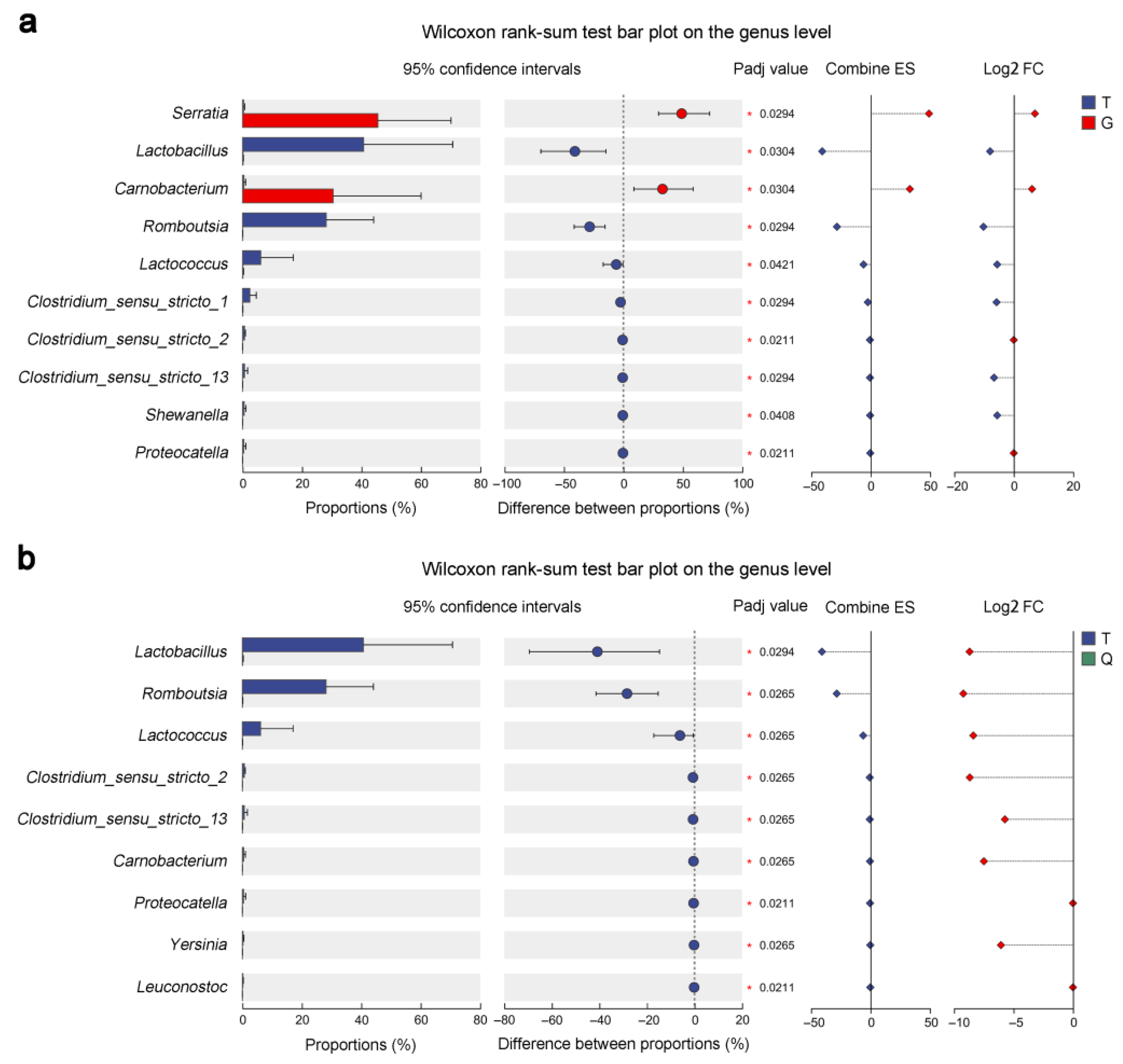
3.5. Compositional Changes in the Intestinal Microbiota of Mandarin Fish from the T, G, and Q Groups
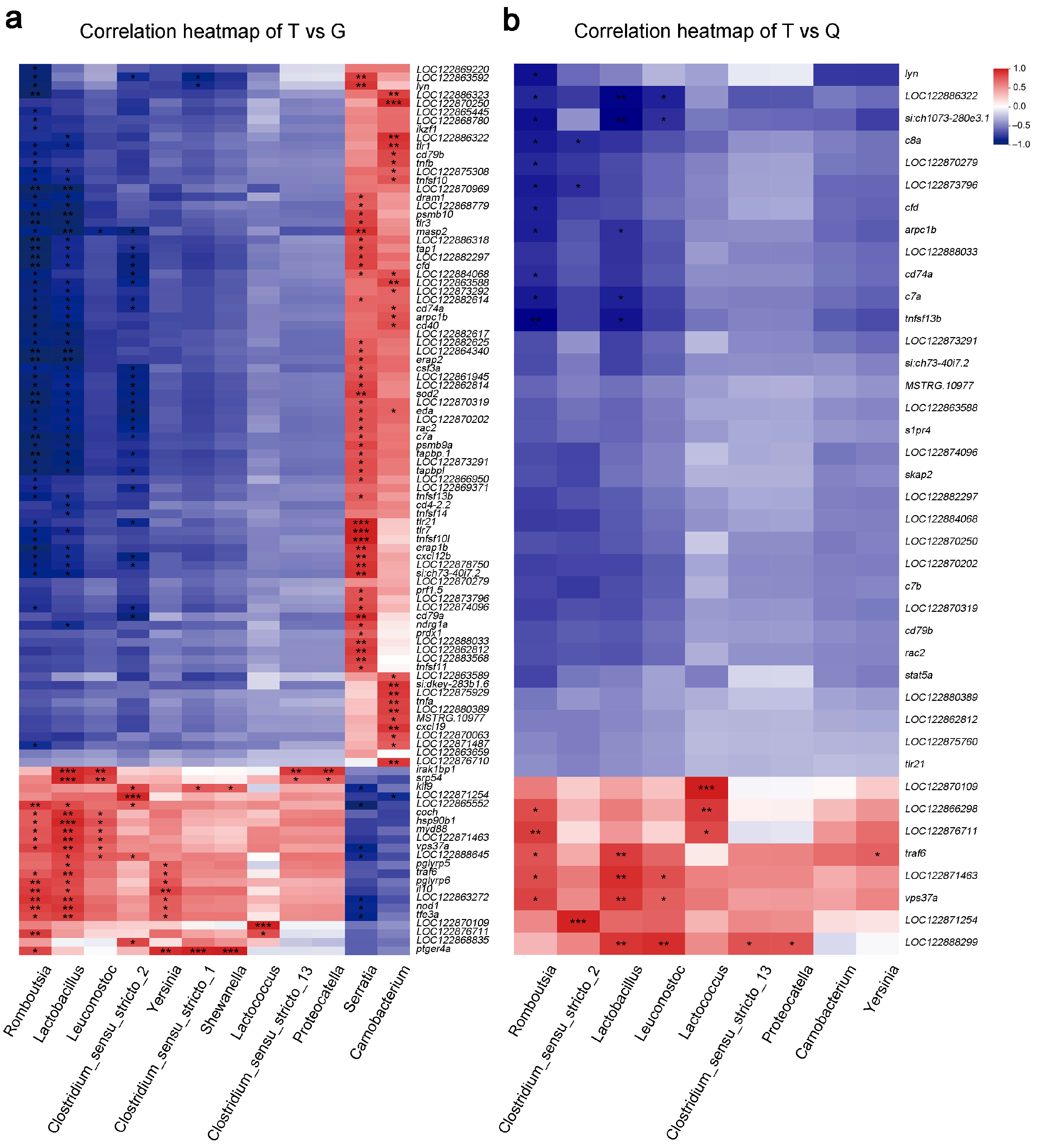
3.6. Bipartite Network of Microbe–Gene Interactions in T, G, and Q Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fiorella, K.J.; Okronipa, H.; Baker, K.; Heilpern, S. Contemporary aquaculture: Implications for human nutrition. Curr. Opin. Biotechnol. 2021, 70, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Boyd, C.E.; McNevin, A.A.; Davis, R.P. The contribution of fisheries and aquaculture to the global protein supply. Food Secur. 2022, 14, 805–827. [Google Scholar] [CrossRef]
- Ding, W.; Zhang, X.; Zhao, X.; Jing, W.; Cao, Z.; Li, J.; Huang, Y.; You, X.; Wang, M.; Shi, Q.; et al. A chromosome-level genome assembly of the mandarin fish (Siniperca chuatsi). Front. Genet. 2021, 12, 671650. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Qin, X.; Liang, M.; Luo, Z.; Zhan, Z.; Weng, S.; Guo, C.; He, J. Genome-wide identification, characterization, and expression analysis of the transient receptor potential gene family in mandarin fish Siniperca chuatsi. BMC Genom. 2024, 25, 848. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Lai, H.; Wang, G.; Guo, D.; Liu, S.; Chen, X.; Zhao, X.; Liu, X.; Li, G. Triploidy induction by hydrostatic pressure shock in mandarin fish (Siniperca chuatsi). Aquaculture 2020, 520, 734979. [Google Scholar] [CrossRef]
- Wang, Y.; Nie, S.; Li, C.; Xiang, H.; Zhao, Y.; Chen, S.; Li, L.; Wu, Y. Application of untargeted metabolomics to reveal the taste-related metabolite profiles during mandarin fish (Siniperca chuatsi) fermentation. Foods 2022, 11, 944. [Google Scholar] [CrossRef]
- Wu, P.; Chen, L.; Cheng, J.; Pan, Y.; Zhu, X.; Bao, L.; Chu, W.; Zhang, J. The miRNA expression profile directly reflects the energy metabolic differences between slow and fast muscle with nutritional regulation of the Chinese perch (Siniperca chuatsi). Comp. Biochem. Physiol. Mol. Integr. Physiol. 2021, 259, 111003. [Google Scholar] [CrossRef]
- Wu, P.; Zeng, Y.; Qin, Q.; Ji, W.; Wu, C.; Zhou, Y.; Zhao, R.; Tao, M.; Zhang, C.; Tang, C.; et al. Formation and identification of artificial gynogenetic mandarin fish (Siniperca chuatsi) induced by inactivated sperm of largemouth bass (Micropterus salmoides). Aquaculture 2023, 577, 739969. [Google Scholar] [CrossRef]
- Yao, G.C.; Li, W. Mandarin fish culture: Status and development prospects. In Aquaculture in China: Success Stories and Modern Trends, 1st ed.; Gui, J.F., Tang, Q., Li, Z., Liu, J., De Silva, S.S., Eds.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2018; pp. 256–269. [Google Scholar]
- Liu, X.; Shao, Z.; Cheng, G.; Lu, S.; Gu, Z.; Zhu, H.; Shen, H.; Wang, J.; Chen, X. Ecological engineering in pond aquaculture: A review from the whole-process perspective in China. Rev. Aquac. 2021, 13, 1060–1076. [Google Scholar] [CrossRef]
- Gu, J.; Li, S.; Shen, X.; Liang, Q.; Xu, T.; Shi, W. Effects of different fermenters on the quality and flavour of fermented mandarin fish (Siniperca chuatsi). Int. J. Food Sci. Technol. 2024, 59, 4992–5007. [Google Scholar] [CrossRef]
- Huang, J.; Liao, S.; Su, Y.; Li, M.; Hu, J.; Han, L.; Jiang, Y.; Yang, M.; Zhang, Y.; Li, S.; et al. Effects of temperature on ovarian development of mandarin fish (Siniperca chuatsi) and hormone therapy to induce its ovulation out of breeding season. Aquacult. Rep. 2024, 37, 102271. [Google Scholar] [CrossRef]
- Cui, R.; Huang, J.; Wang, S.; Zhang, X. Whole-genome resequencing reveals genetic diversity and selection signatures in five populations of mandarin fish (Siniperca chuatsi). Aquacult. Rep. 2024, 39, 102406. [Google Scholar] [CrossRef]
- Li, W.; Lin, M.; Ye, S.; Liu, J.; Gozlan, R.E.; Li, Z.; Zhang, T. Comparative growth, feeding and reproduction of hatchery-reared and wild mandarin fish Siniperca chuatsi in a shallow Yangtze lake, China. Aquacult. Environ. Interact. 2021, 13, 413–423. [Google Scholar] [CrossRef]
- Ding, L.; Zhang, Y.; Chen, J.; Chen, W.; Xie, S.; Chen, Q. Growth, muscle nutrition composition, and digestive enzyme activities of the juvenile and adult Siniperca chuatsi fed on live baits and a formulated diet. Fishes 2022, 7, 379. [Google Scholar] [CrossRef]
- Hu, F.; Zhong, H.; Wu, C.; Wang, S.; Guo, Z.; Tao, M.; Zhang, C.; Gong, D.; Gao, X.; Tang, C.; et al. Development of fisheries in China. Reprod. Breed. 2021, 1, 64–79. [Google Scholar] [CrossRef]
- Gong, J.; Pan, X.; Lin, L.; Zhu, Y.; Yao, J.; Wang, C.; Yin, W.; Huang, L.; Liu, Y.; Chen, F.; et al. Establishment and characterization of a spinal cord tissue cell line from mandarin fish, Siniperca chuatsi and its susceptibility to several viruses. J. Fish Dis. 2022, 45, 1419–1427. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, N.; Li, B.; Zhu, Q.; Ji, C. Smartphone-based agricultural extension services and farm incomes: Evidence from Zhejiang Province in China. Rev. Dev. Econ. 2023, 27, 1383–1402. [Google Scholar] [CrossRef]
- Li, S.M.; Zhou, Q.; Liu, W.; Hu, D.Y.; Qi, M.; Yao, G.H.; Wu, R.F.; Wang, Y.; Ding, X.Y.; Zhou, F. Evaluation of muscle nutritional quality of 3 kinds of different geographic groups of Siniperca chuatsi. J. Food Saf. Qual. 2024, 15, 124–133. [Google Scholar]
- Wang, Z.; Yang, L.; Zhou, F.; Li, J.; Wu, X.; Zhong, X.; Lv, H.; Yi, S.; Gao, Q.; Yang, Z.; et al. Integrated comparative transcriptome and weighted gene co-expression network analysis provide valuable insights into the response mechanisms of crayfish (Procambarus clarkii) to copper stress. J. Hazard. Mater. 2023, 448, 130820. [Google Scholar] [CrossRef]
- Rasheed, A.; Al-Huqail, A.A.; Ali, B.; Alghanem, S.M.S.; Shah, A.A.; Azeem, F.; Rizwan, M.; Al-Qthanin, R.N.; Soudy, F.A. Molecular characterization of genes involved in tolerance of cadmium in Triticum aestivum (L.) under Cd stress. J. Hazard. Mater. 2024, 464, 132955. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Wang, K.; Li, Q.; Yan, S.; Shi, H.; Liu, L.; Liang, S.; Yang, M.; Su, Z.; et al. RNA-Seq reveals pathways responsible for meat quality characteristic differences between two Yunnan indigenous chicken breeds and commercial broilers. Foods 2024, 13, 2008. [Google Scholar] [CrossRef]
- Muehlbauer, A.L.; Richards, A.L.; Alazizi, A.; Burns, M.B.; Gomez, A.; Clayton, J.B.; Petrzelkova, K.; Cascardo, C.; Resztak, J.; Wen, X.; et al. Interspecies variation in hominid gut microbiota controls host gene regulation. Cell Rep. 2021, 37, 110057. [Google Scholar] [CrossRef]
- He, H.; Fan, X.; Shen, H.; Gou, H.; Zhang, C.; Liu, Z.; Zhang, B.; Wuri, N.; Zhang, J.; Liao, M.; et al. Butyrate limits the replication of porcine epidemic diarrhea virus in intestine epithelial cells by enhancing GPR43-mediated IFN-III production. Front. Microbiol. 2023, 14, 1091807. [Google Scholar] [CrossRef]
- Zheng, J.; Yao, Y.; Rui, Q.; Zhou, Y.; Li, F.; Jiang, W.; Chi, M.; Liu, S.; Cheng, S.; Chen, J.; et al. Effect of different feeding regimens on physiological indicators, intestinal transcriptome, and bacterial flora of mandarin fish (Siniperca chuatsi). Comp. Biochem. Physiol. Part D Genom. Proteom. 2024, 52, 101301. [Google Scholar] [CrossRef] [PubMed]
- Kohl, K.D.; Yahn, J. Effects of environmental temperature on the gut microbial communities of tadpoles. Environ. Microbiol. 2016, 18, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.E.; Kueneman, J.G.; Nicholson, D.J.; Rosso, A.A.; Folfas, E.; Casement, B.; Gallegos-Koyner, M.A.; Neel, L.K.; Curlis, J.D.; McMillan, W.O.; et al. Sustained drought, but not short-term warming, alters the gut microbiomes of wild Anolis lizards. Appl. Environ. Microbiol. 2022, 88, e0053022. [Google Scholar] [CrossRef] [PubMed]
- Akbar, S.; Gu, L.; Sun, Y.; Zhang, L.; Lyu, K.; Huang, Y.; Yang, Z. Understanding host-microbiome-environment interactions: Insights from Daphnia as a model organism. Sci. Total Environ. 2022, 808, 152093. [Google Scholar] [CrossRef]
- Morshed, S.M.; Lee, T.H. The role of the microbiome on fish mucosal immunity under changing environments. Fish Shellfish Immunol. 2023, 139, 108877. [Google Scholar] [CrossRef]
- Wang, Z.; Li, J.; Zhao, P.; Yu, Z.; Yang, L.; Ding, X.; Lv, H.; Yi, S.; Sheng, Q.; Zhang, L.; et al. Integrated microbiome and metabolome analyses reveal the effects of low pH on intestinal health and homeostasis of crayfish (Procambarus clarkii). Aquat. Toxicol. 2024, 270, 106903. [Google Scholar] [CrossRef]
- Zhou, J.; Li, Q.; Huang, Z.; Zhang, L.; Mou, C.; Zhao, Z.; Zhao, H.; Du, J.; Yang, X.; Liang, X.; et al. Study on the adaptive regulation of light on the stress response of mandarin fish (Siniperca chuatsi) with re-feeding after starvation. Animals 2023, 13, 2610. [Google Scholar] [CrossRef]
- Lv, L.; Liang, X.F.; Huang, K.; He, S. Effect of agmatine on food intake in mandarin fish (Siniperca chuatsi). Fish Physiol. Biochem. 2019, 45, 1709–1716. [Google Scholar] [CrossRef]
- Nagothu, S.K.; Bindu Sri, P.; Anitha, G.; Vincent, S.; Kumar, O.P. Advancing aquaculture: Fuzzy logic-based water quality monitoring and maintenance system for precision aquaculture. Aquacult. Int. 2025, 33, 32. [Google Scholar] [CrossRef]
- Luo, J.X.; Gao, X.T.; Rong, Z.; Zhang, L.H.; Sun, Y.F.; Qi, Z.L.; Yu, Q.; Waiho, K.; Zhao, W.X.; Xu, Y.H.; et al. Transcriptome sequencing reveals effects of artificial feed domestication on intestinal performance and gene expression of carnivorous mandarin fish (Siniperca chuatsi) and related mechanisms. Mar. Biotechnol. 2025, 27, 41. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, Z.; Yang, M.; Yang, F.; Wang, G.; Liu, D.; Li, X.; Yang, L.; Wang, Z. Copper-induced oxidative stress, transcriptome changes, intestinal microbiota, and histopathology of common carp (Cyprinus carpio). Ecotoxicol. Environ. Saf. 2022, 246, 114136. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Kovaka, S.; Zimin, A.V.; Pertea, G.M.; Razaghi, R.; Salzberg, S.L.; Pertea, M. Transcriptome assembly from long-read RNA-Seq alignments with StringTie2. Genome Biol. 2019, 20, 278. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Batista, S.J.; Still, K.M.; Johanson, D.; Thompson, J.A.; O’Brien, C.A.; Lukens, J.R.; Harris, T.H. Gasdermin-D-dependent IL-1α release from microglia promotes protective immunity during chronic Toxoplasma gondii infection. Nat. Commun. 2020, 11, 3687. [Google Scholar] [CrossRef]
- Li, C.; Jiang, J.; Xie, J.; Yang, W.; Wang, Y. Transcriptome profiling and differential expression analysis of the immune-related genes during the acute phase of infection with Mycobacterium marinum in the goldfish (Carassius auratus L.). Aquaculture 2021, 533, 736198. [Google Scholar] [CrossRef]
- Zhou, F.; Qi, M.; Li, J.; Huang, Y.; Chen, X.; Liu, W.; Yao, G.; Meng, Q.; Zheng, T.; Wang, Z.; et al. Comparative transcriptomic analysis of largemouth bass (Micropterus salmoides) livers reveals response mechanisms to high temperatures. Genes 2023, 14, 2096. [Google Scholar] [CrossRef] [PubMed]
- Klopfenstein, D.V.; Zhang, L.; Pedersen, B.S.; Ramírez, F.; Warwick Vesztrocy, A.; Naldi, A.; Mungall, C.J.; Yunes, J.M.; Botvinnik, O.; Weigel, M.; et al. GOATOOLS: A python library for gene ontology analyses. Sci. Rep. 2018, 8, 10872. [Google Scholar] [CrossRef] [PubMed]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Yu, G.; Shi, C.; Liu, L.; Guo, Q.; Han, C.; Zhang, D.; Zhang, L.; Liu, B.; Gao, H.; et al. Majorbio Cloud: A one-stop, comprehensive bioinformatic platform for multiomics analyses. iMeta 2022, 1, e12. [Google Scholar] [CrossRef]
- Qi, R.; Zhang, Z.; Wang, J.; Qiu, X.; Wang, Q.; Yang, F.; Huang, J.; Liu, Z. Introduction of colonic and fecal microbiota from an adult pig differently affects the growth, gut health, intestinal microbiota and blood metabolome of newborn piglets. Front. Microbiol. 2021, 12, 623673. [Google Scholar] [CrossRef]
- Lin, X.; Zhang, C.; Cao, K.; Li, Z.; Zhao, Z.; Li, X.; Leng, X. Dietary sodium butyrate changed intestinal histology and microbiota of rainbow trout (Oncorhynchus mykiss), but did not promote growth and nutrient utilization. Aquac. Nutr. 2023, 2023, 3706109. [Google Scholar] [CrossRef]
- Zheng, J.; Guo, N.; Wagner, A. Mistranslation reduces mutation load in evolving proteins through negative epistasis with DNA mutations. Mol. Biol. Evol. 2021, 38, 4792–4804. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Symonds, J.E.; Walker, S.P.; Steiner, K.; Carter, C.G.; Bowman, J.P.; Nowak, B.F. Relationship between gut microbiota and Chinook salmon (Oncorhynchus tshawytscha) health and growth performance in freshwater recirculating aquaculture systems. Front. Microbiol. 2023, 14, 1065823. [Google Scholar] [CrossRef]
- Zhu, Z.; Xu, Y.M.; Liang, J.H.; Huang, W.; Chen, J.D.; Wu, S.T.; Huang, X.H.; Huang, Y.H.; Zhang, X.Y.; Sun, H.Y.; et al. Relationship of environmental factors in pond water and dynamic changes of gut microbes of sea bass Lateolabrax japonicus. Front. Microbiol. 2023, 14, 1086471. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Li, Z.; Li, X.; Xing, L.; Wang, S.; Sun, X.; Zhang, D. Evaluation of enrofloxacin in the Chinese soft-shelled turtle (Pelodiscus sinensis) based on the biochemical, histopathological and intestinal microbiota responses. Aquaculture 2024, 590, 741062. [Google Scholar] [CrossRef]
- Yuan, X.Y.; Zhang, X.T.; Xia, Y.T.; Zhang, Y.Q.; Wang, B.; Ye, W.W.; Ye, Z.F.; Qian, S.C.; Huang, M.M.; Yang, S.; et al. Transcriptome and 16S rRNA analyses revealed differences in the responses of largemouth bass (Micropterus salmoides) to early Aeromonas hydrophila infection and immunization. Aquaculture 2021, 541, 736759. [Google Scholar] [CrossRef]
- Yoon, J.B.; Hwang, S.; Yang, J.H.; Lee, S.; Bang, W.Y.; Moon, K.H. Dynamics of the gut microbiome and transcriptome in Korea native ricefish (Oryzias latipes) during chronic antibiotic exposure. Genes 2022, 13, 1243. [Google Scholar] [CrossRef]
- Pan, C.; Zhu, Y.; Cao, K.; Li, J.; Wang, S.; Zhu, J.; Zeng, X.; Zhang, H.; Qin, Z. Transcriptome, intestinal microbiome and histomorphology profiling of differences in the response of Chinese sea bass (Lateolabrax maculatus) to Aeromonas hydrophila infection. Front. Microbiol. 2023, 14, 1103412. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Zhang, Y.; Wen, Y.; Ji, W.; Zhou, Y.; Ji, Y.; Liu, X.; Wang, W.; Asim, M.; Liang, X.; et al. Analysis of the transcriptomic profilings of mandarin fish (Siniperca chuatsi) infected with Flavobacterium columnare with an emphasis on immune responses. Fish Shellfish Immunol. 2015, 43, 111–119. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, R.; Gao, T.; Wu, T.; Zhang, Q.; Shi, Y.; Ding, S.; Zhao, Z. Transcriptome analysis of immune response against Siniperca chuatsi rhabdovirus infection in mandarin fish Siniperca chuatsi. J. Fish Dis. 2021, 44, 675–687. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, C.; Zhang, Z.; Sun, W.; Zhang, X.; Liu, X. Analysis of the transcriptomic profiles of mandarin fish (Siniperca chuatsi) infected with red sea bream iridovirus (RSIV). Microb. Pathog. 2023, 174, 105921. [Google Scholar] [CrossRef]
- Gao, J.H.; Zhao, J.L.; Yao, X.L.; Tola, T.; Zheng, J.; Xue, W.B.; Wang, D.W.; Xing, Y. Identification of antimicrobial peptide genes from transcriptomes in mandarin fish (Siniperca chuatsi) and their response to infection with Aeromonas hydrophila. Fish Shellfish Immunol. 2024, 144, 109247. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Cao, L.; Cao, Z.; Bing, X. Transcriptomic responses of the liver of mandarin fish (Siniperca chuatsi) under hypoxic stress. J. Fish Biol. 2023, 103, 44–58. [Google Scholar] [CrossRef]
- Ding, W.; Cao, L.; Cao, Z.; Bing, X. Gill and brain transcriptomic analysis of mandarin fish (Siniperca chuatsi) reveals hypoxia-induced mitochondrial dysfunction and modulation of metabolism. Comp. Biochem. Physiol. Part D Genom. Proteom. 2025, 53, 101367. [Google Scholar] [CrossRef]
- He, J.; Yu, Y.; Qin, X.W.; Zeng, R.Y.; Wang, Y.Y.; Li, Z.M.; Mi, S.; Weng, S.P.; Guo, C.J.; He, J.G. Identification and functional analysis of the mandarin fish (Siniperca chuatsi) hypoxia-inducible factor-1α involved in the immune response. Fish Shellfish Immunol. 2019, 92, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, H.; Deng, N.; Xu, J.; Huang, J.; Han, C.; Liu, D.; Liu, S.; Yan, B.; Han, L.; Li, S.; et al. Effects of hyperosmotic stress on the intestinal microbiota, transcriptome, and immune function of mandarin fish (Siniperca chuatsi). Aquaculture 2023, 563, 738901. [Google Scholar] [CrossRef]
- Gasaly, N.; de Vos, P.; Hermoso, M.A. Impact of bacterial metabolites on gut barrier function and host immunity: A focus on bacterial metabolism and its relevance for intestinal inflammation. Front. Immunol. 2021, 12, 658354. [Google Scholar] [CrossRef] [PubMed]
- Sullam, K.E.; Essinger, S.D.; Lozupone, C.A.; O’Connor, M.P.; Rosen, G.L.; Knight, R.; Kilham, S.S.; Russell, J.A. Environmental and ecological factors that shape the gut bacterial communities of fish: A meta-analysis. Mol. Ecol. 2012, 21, 3363–3378. [Google Scholar] [CrossRef]
- Gao, Q.; Liu, P.; Li, Y.; Song, D.; Long, W.; Wang, Z.; Yi, S.; Jiang, L. Gut microbiota, host genetics and phenotypes in aquatic animals: A review. Aquacult. Rep. 2023, 31, 101648. [Google Scholar] [CrossRef]
- Jaramillo-Torres, A.; Rawling, M.D.; Rodiles, A.; Mikalsen, H.E.; Johansen, L.H.; Tinsley, J.; Forberg, T.; Aasum, E.; Castex, M.; Merrifield, D.L. Influence of dietary supplementation of probiotic Pediococcus acidilactici MA18/5M during the transition from freshwater to seawater on intestinal health and microbiota of atlantic salmon (Salmo salar L.). Front. Microbiol. 2019, 10, 2243. [Google Scholar] [CrossRef]
- Borges, N.; Keller-Costa, T.; Sanches-Fernandes, G.M.M.; Louvado, A.; Gomes, N.C.M.; Costa, R. Bacteriome structure, function, and probiotics in fish larviculture: The good, the bad, and the gaps. Annu. Rev. Anim. Biosci. 2021, 9, 423–452. [Google Scholar] [CrossRef]
- Fečkaninová, A.; Koščová, J.; Mudroňová, D.; Popelka, P.; Toropilová, J. The use of probiotic bacteria against Aeromonas infections in salmonid aquaculture. Aquaculture 2017, 469, 1–8. [Google Scholar] [CrossRef]
- Li, T.; Raza, S.H.A.; Yang, B.; Sun, Y.; Wang, G.; Sun, W.; Qian, A.; Wang, C.; Kang, Y.; Shan, X. Aeromonasveronii infection in commercial freshwater fish: A potential threat to public health. Animals 2020, 10, 608. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhu, X.; Li, Y.; Yang, P.; Wang, S.; Ning, K.; Chen, S. Intestinal microbiome-mediated resistance against vibriosis for Cynoglossus semilaevis. Microbiome 2022, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Priya, S.; Burns, M.B.; Ward, T.; Mars, R.A.T.; Adamowicz, B.; Lock, E.F.; Kashyap, P.C.; Knights, D.; Blekhman, R. Identification of shared and disease-specific host gene-microbiome associations across human diseases using multi-omic integration. Nat. Microbiol. 2022, 7, 780–795. [Google Scholar] [CrossRef]
- Yu, Y.Y.; Ding, L.G.; Huang, Z.Y.; Xu, H.Y.; Xu, Z. Commensal bacteria-immunity crosstalk shapes mucosal homeostasis in teleost fish. Rev. Aquac. 2021, 13, 2322–2343. [Google Scholar] [CrossRef]
- Zhang, B.; Yang, H.; Cai, G.; Nie, Q.; Sun, Y. The interactions between the host immunity and intestinal microorganisms in fish. Appl. Microbiol. Biotechnol. 2024, 108, 30. [Google Scholar] [CrossRef]
- Yukgehnaish, K.; Kumar, P.; Sivachandran, P.; Marimuthu, K.; Arshad, A.; Paray, B.A.; Arockiaraj, J. Gut microbiota metagenomics in aquaculture: Factors influencing gut microbiome and its physiological role in fish. Rev. Aquacult. 2020, 12, 1903–1927. [Google Scholar] [CrossRef]
- Diwan, A.D.; Harke, S.N.; Panche, A.N. Host-microbiome interaction in fish and shellfish: An overview. Fish Shellfish Immunol. Rep. 2023, 4, 100091. [Google Scholar] [CrossRef]
- Zhu, C.Z.; Li, D.; Chen, W.J.; Ban, S.N.; Liu, T.; Wen, H.; Jiang, M. Effects of dietary host-associated Lactococcus lactis on growth performance, disease resistance, intestinal morphology and intestinal microbiota of mandarin fish (Siniperca chuatsi). Aquaculture 2021, 540, 736702. [Google Scholar] [CrossRef]
- Wang, J.; Hao, Y.; Zhang, L.; Gao, X.; Xu, Y.; Wang, J.; Hanafiah, F.; Khor, W.; Sun, Y.; Wu, C. Profiling the gut structure and microbiota, and identifying two dominant bacteria belonging to the Weissella genus in mandarin fish (Siniperca chuatsi) fed an artificial diet. Front. Microbiol. 2024, 15, 1486501. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, Q.; Liu, H.; Jin, J.; Yang, Y.; Zhu, X.; Han, D.; Zhou, Z.; Xie, S. Potential functions of the gut microbiome and modulation strategies for improving aquatic animal growth. Rev. Aquacult. 2025, 17, e12959. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, F.; Liu, W.; Qi, M.; Liang, Q.; Yao, G.; Ma, C.; Ding, X.; Yu, Z.; Li, X.; Wang, Z. Integrated Transcriptome and Microbiome Analyses Reveal Growth- and Stress-Response-Related Genes and Microbes in Mandarin Fish (Siniperca chuatsi). Fishes 2025, 10, 341. https://doi.org/10.3390/fishes10070341
Zhou F, Liu W, Qi M, Liang Q, Yao G, Ma C, Ding X, Yu Z, Li X, Wang Z. Integrated Transcriptome and Microbiome Analyses Reveal Growth- and Stress-Response-Related Genes and Microbes in Mandarin Fish (Siniperca chuatsi). Fishes. 2025; 10(7):341. https://doi.org/10.3390/fishes10070341
Chicago/Turabian StyleZhou, Fan, Wei Liu, Ming Qi, Qianrong Liang, Gaohua Yao, Cheng Ma, Xueyan Ding, Zaihang Yu, Xinyu Li, and Zhanqi Wang. 2025. "Integrated Transcriptome and Microbiome Analyses Reveal Growth- and Stress-Response-Related Genes and Microbes in Mandarin Fish (Siniperca chuatsi)" Fishes 10, no. 7: 341. https://doi.org/10.3390/fishes10070341
APA StyleZhou, F., Liu, W., Qi, M., Liang, Q., Yao, G., Ma, C., Ding, X., Yu, Z., Li, X., & Wang, Z. (2025). Integrated Transcriptome and Microbiome Analyses Reveal Growth- and Stress-Response-Related Genes and Microbes in Mandarin Fish (Siniperca chuatsi). Fishes, 10(7), 341. https://doi.org/10.3390/fishes10070341