
Transcriptomic Profiling of Misgurnus anguillicaudatus Reveals the Anti-Inflammatory Action of Lonicera japonica Extract in Response to Lipopolysaccharide Challenge


Abstract
1. Introduction
2. Materials and Methods
2.1. Material and Experimental Procedure
2.2. RNA Sequencing
2.3. Transcript Splicing and Analysis of DEGs
2.4. Analysis by Quantitative Real-Time PCR
3. Results
3.1. Transcript Splicing Result
3.2. Function Annotation
3.2.1. Result of Annotation
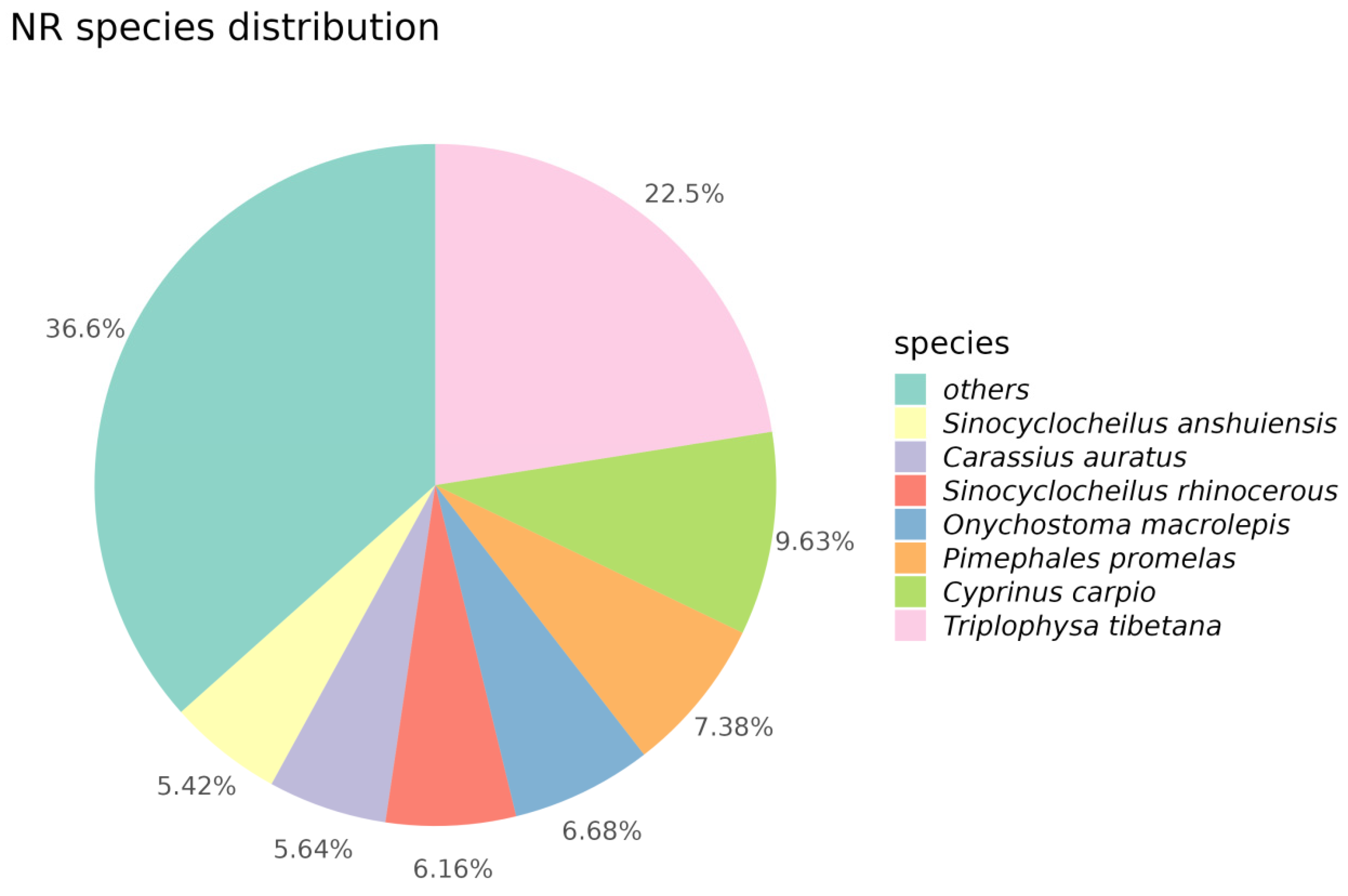
3.2.2. NR Annotation
3.2.3. GO Annotation
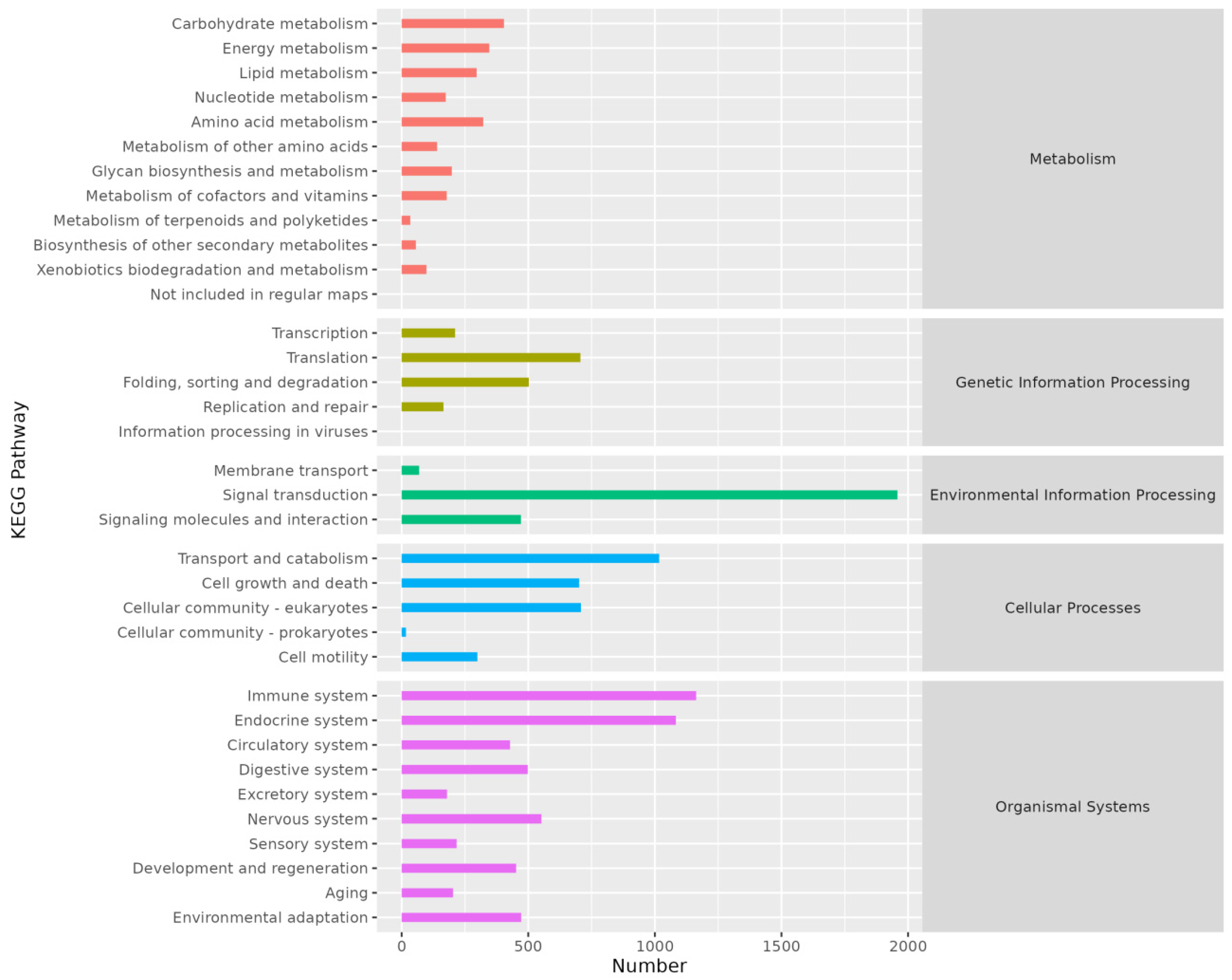
3.2.4. KEGG Annotation
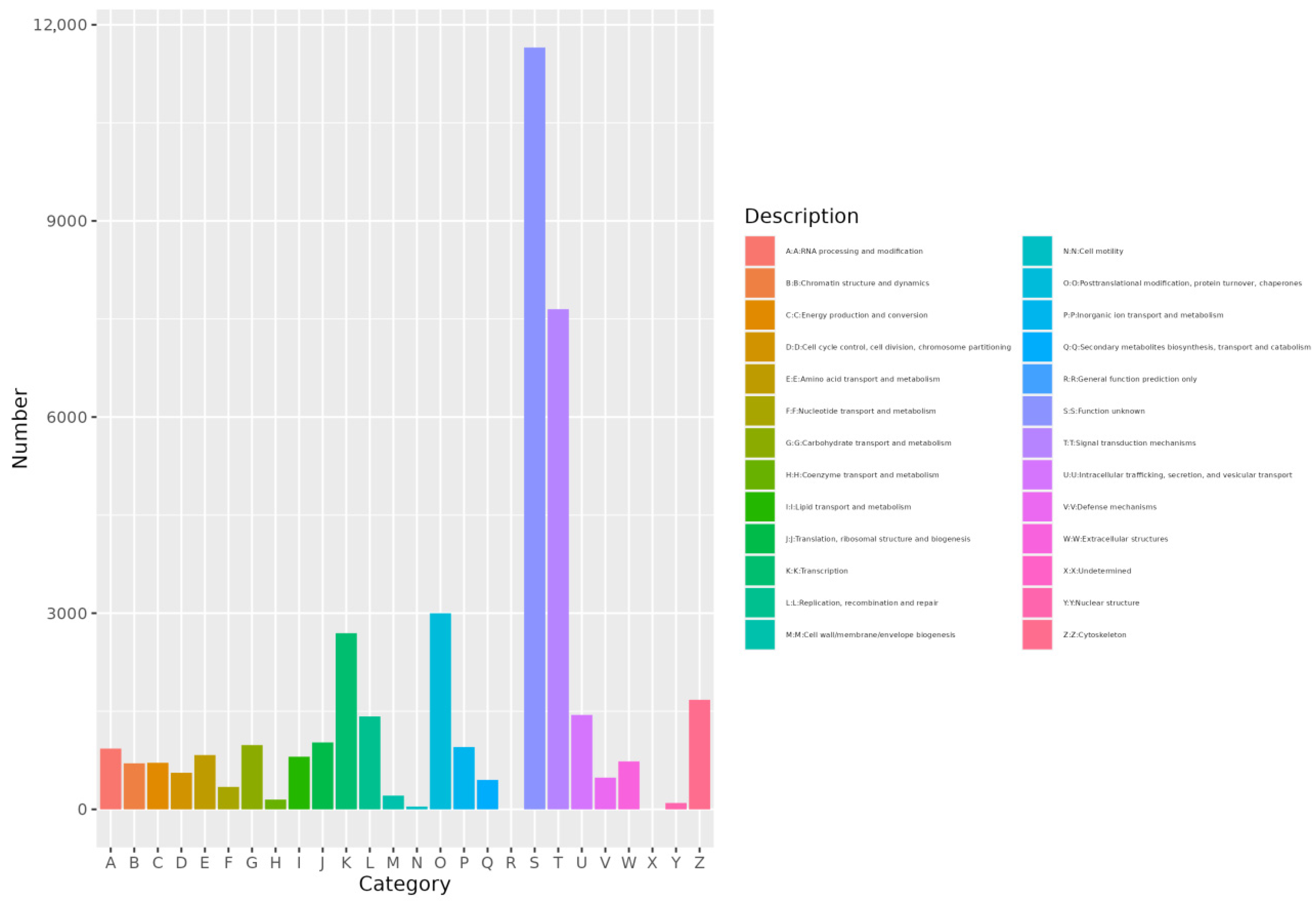
3.2.5. EggNOG Annotation
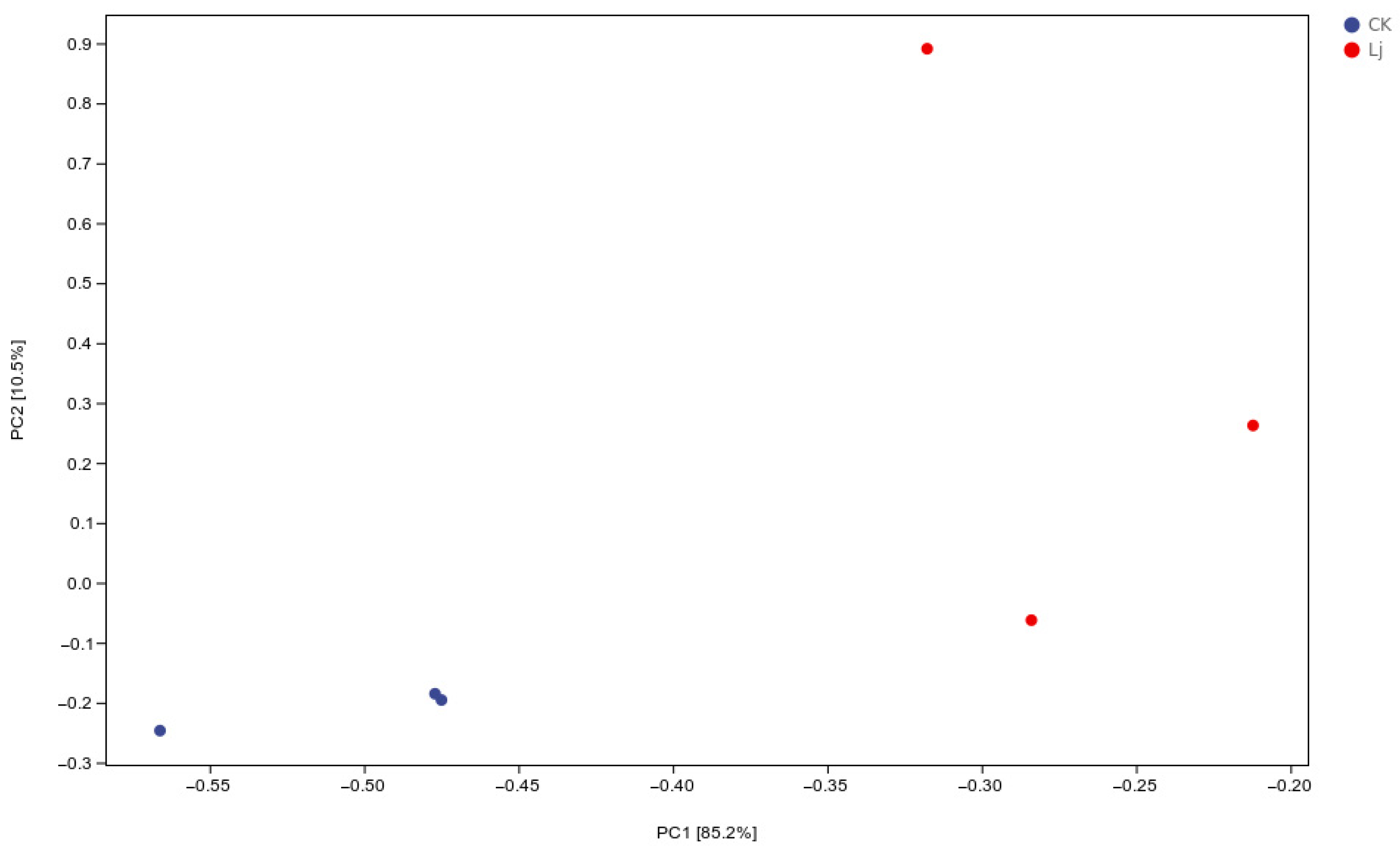
3.3. Analysis of PCA
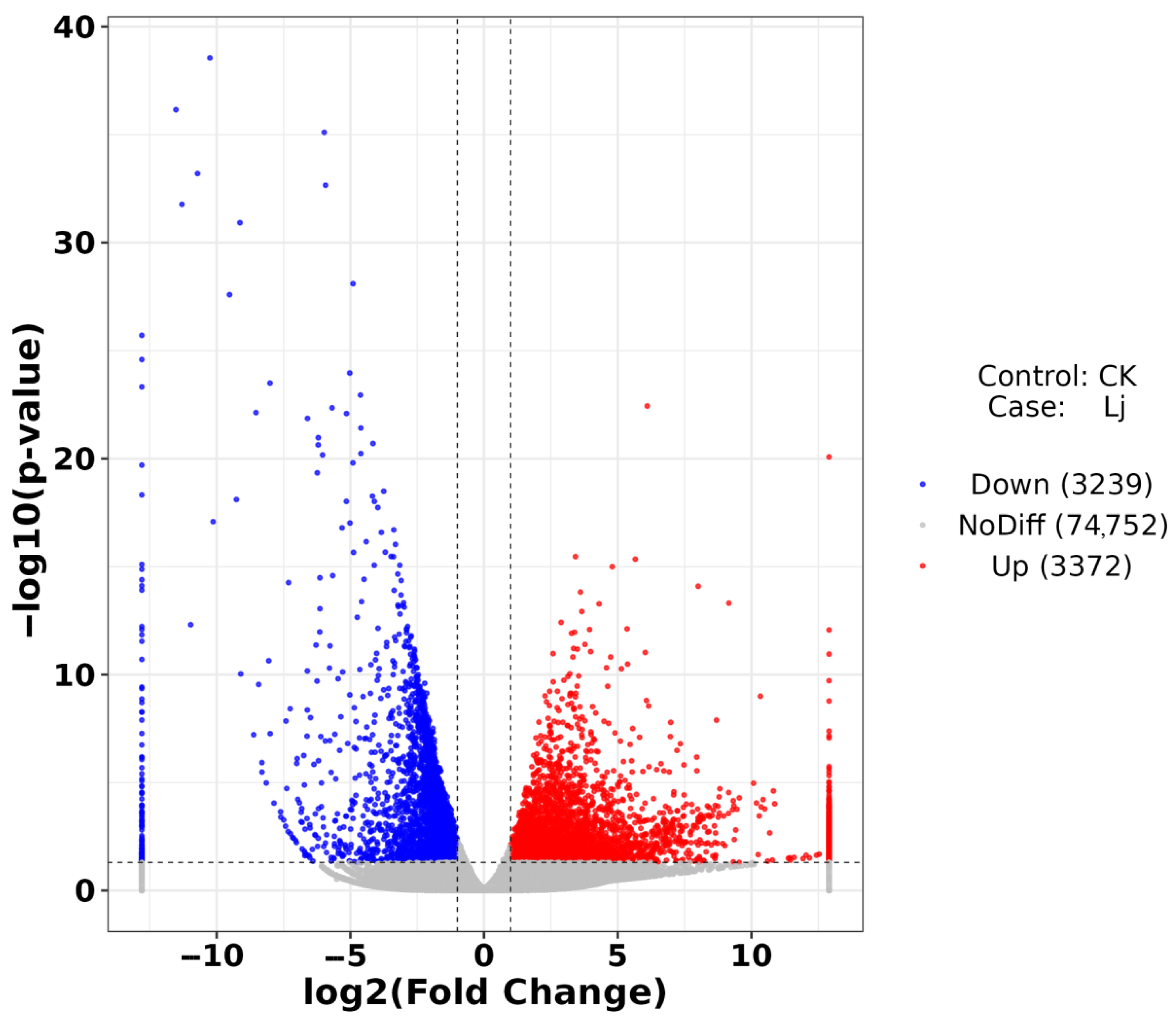
3.4. Analysis of Difference
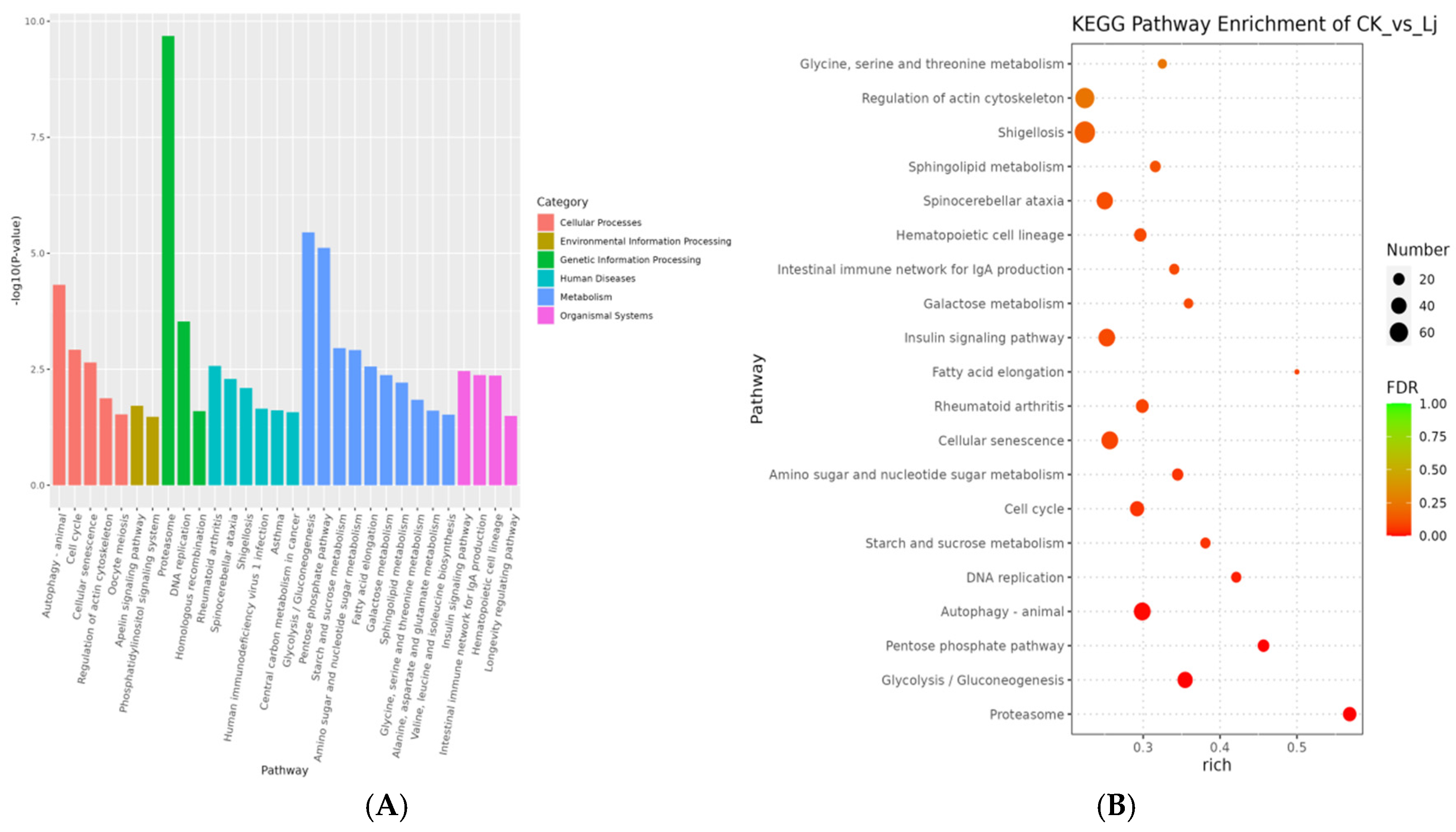
3.5. Enrichment Analysis of DEGs
3.5.1. GO Enrichment Analysis
3.5.2. KEGG Enrichment Analysis
3.5.3. KO Analysis
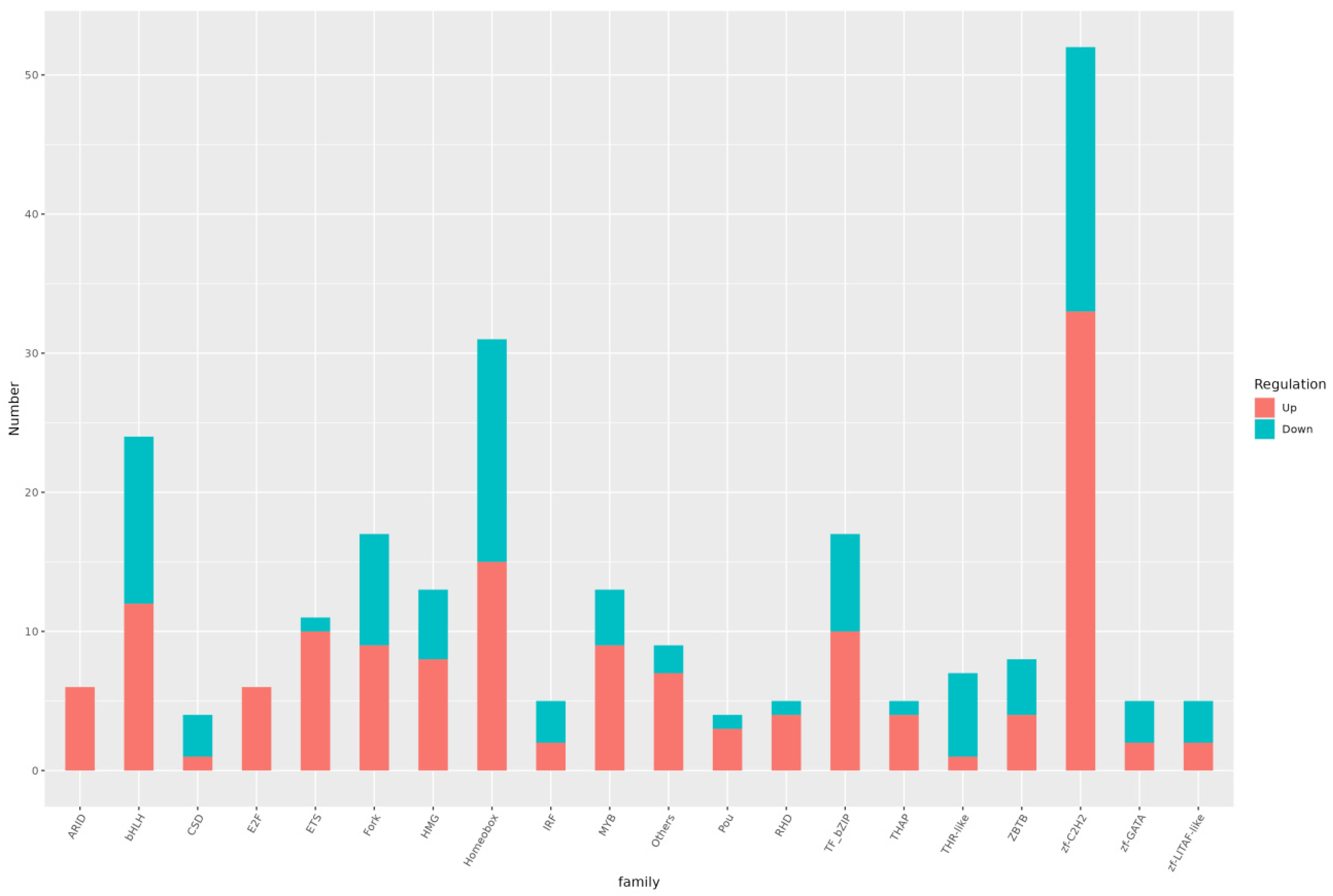
3.6. Differential TF Analysis
3.7. Validation of DEGs by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, E.J.; Kim, J.S.; Kim, H.P.; Lee, J.-H.; Kang, S.S. Phenolic constituents from the flower buds of Lonicera japonica and their 5-lipoxygenase inhibitory activities. Food Chem. 2010, 120, 134–139. [Google Scholar] [CrossRef]
- Chen, X.; Guo, D.; Gong, X.; Wan, N.; Wu, Z. Optimization of Steam Distillation Process for Volatile Oils from Forsythia suspensa and Lonicera japonica according to the Concept of Quality by Design. Separations 2023, 10, 25. [Google Scholar] [CrossRef]
- Tang, X.; Liu, X.; Zhong, J.; Fang, R. Potential Application of Lonicera japonica Extracts in Animal Production: From the Perspective of Intestinal Health. Front. Microbiol. 2021, 12, 719877. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Zhu, Z.H.; Zhang, L.; Wang, Q.; Xu, M.M.; Lu, C.; Zhu, Y.; Zeng, J.; Duan, J.A.; Zhao, M. Anti-inflammatory property and functional substances of Lonicerae Japonicae Caulis. J. Ethnopharmacol. 2021, 267, 113502. [Google Scholar] [CrossRef]
- Liu, C.; Yin, Z.; Feng, T.; Zhang, M.; Zhou, Z.; Zhou, Y. An integrated network pharmacology and RNA-Seq approach for exploring the preventive effect of Lonicerae japonicae flos on LPS-induced acute lung injury. J. Ethnopharmacol. 2021, 264, 113364. [Google Scholar] [CrossRef]
- Xiao, O.; Li, M.; Chen, D.; Chen, J.; Simal-Gandara, J.; Dai, X.; Kong, Z. The dissipation, processing factors, metabolites, and risk assessment of pesticides in honeysuckle from field to table. J. Hazard. Mater. 2022, 431, 128519. [Google Scholar] [CrossRef]
- Liu, Z.; Cheng, Y.; Chao, Z. A Comprehensive Quality Analysis of Different Colors of Medicinal and Edible Honeysuckle. Foods 2023, 12, 3126. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, X.; Li, D.; Qian, K.; Liu, Y.; Xu, T.; Dai, L.; Cheng, J. The transcriptome sequencing analysis reveals immune mechanisms of soybean fermented powder on the loach (Misgurnus anguillicaudatus) in response to Lipopolysaccharide (LPS) infection. Front. Immunol. 2023, 14, 1247038. [Google Scholar] [CrossRef]
- Qin, C.G.; Huang, K.X.; Xu, H.B. Protective effect of polysaccharide from the loach on the in vitro and in vivo peroxidative damage of hepatocyte. J. Nutr. Biochem. 2002, 13, 592–597. [Google Scholar] [CrossRef]
- Zhou, X.Y.; Gao, Z.X.; Luo, S.S.; Su, J.X.; Yi, S.K. Evidence for the Growth Superiority and Delayed Ovarian Development in Tetraploid Loach Misgurnus anguillicaudatus. Fishes 2022, 7, 322. [Google Scholar] [CrossRef]
- Ma, B.H.; Tao, Z.Y.; Wu, Z.B.; Wang, H.H.; Xu, X.D.; Chen, C.Y. Artificial Propagation and Embryonic Development in New Strain of Bigscale Loach Paramisgurnus dabryanus. Jiangxi Fish. Res. Inst. 2020, 6, 1003–1111. [Google Scholar]
- Mao, S.Q.; Yan, J.R.; Xu, P.; Zhang, Y.Y.; Song, L.P.; Hu, B.; Wu, J.; Wang, B.L. Comparative study on body index, nutrient composition, and digestive enzyme activity of Misgurnus anguillicaudatus, Paramisgurnus dabryanus, and Paramisgurnus dabryanus ssp. Isr. J. Aquac. 2022, 74, 1724798. [Google Scholar] [CrossRef]
- Guo, X.; Yu, X.; Zheng, B.; Zhang, L.; Zhang, F.; Zhang, Y.; Li, J.; Pu, G.; Zhang, L.; Wu, H. Network Pharmacology-Based Identification of Potential Targets of Lonicerae japonicae Flos Acting on Anti-Inflammatory Effects. BioMed Res. Int. 2021, 2021, 5507003. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.L.; Lin, T.L.; Chang, C.J.; Wu, T.R.; Lai, W.F.; Lu, C.C.; Lai, H.C. Probiotics, prebiotics and amelioration of diseases. J. Biomed. Sci. 2019, 26, 3. [Google Scholar] [CrossRef]
- Shang, X.; Pan, H.; Li, M.; Miao, X.; Ding, H. Lonicera japonica Thunb.: Ethnopharmacology, phytochemistry and pharmacology of an important traditional Chinese medicine. J. Ethnopharmacol. 2011, 138, 1–21. [Google Scholar] [CrossRef]
- Kwon, S.H.; Ma, S.X.; Hong, S.I.; Lee, S.Y.; Jang, C.G. Lonicera japonica THUNB. Extract Inhibits Lipopolysaccharide-Stimulated Inflammatory Responses by Suppressing NF-κB Signaling in BV-2 Microglial Cells. J. Med. Food 2015, 18, 762–775. [Google Scholar] [CrossRef]
- Malgorzata-Miller, G.; Heinbockel, L.; Brandenburg, K.; van der Meer, J.W.; Netea, M.G.; Joosten, L.A. Bartonella quintana lipopolysaccharide (LPS): Structure and characteristics of a potent TLR4 antagonist for in-vitro and in-vivo applications. Sci. Rep. 2016, 6, 34221. [Google Scholar] [CrossRef]
- Medina, C.; Royo, J.L. Zebrafish as a model organism to study host-pathogen interactions. Methods 2013, 62, 241–245. [Google Scholar] [CrossRef]
- Pan, M.; Liu, J.; Huang, D.; Guo, Y.; Luo, K.; Yang, M.; Gao, W.; Xu, Q.; Zhang, W.; Mai, K. FoxO3 Modulates LPS-Activated Hepatic Inflammation in Turbot (Scophthalmus maximus L.). Front. Immunol. 2021, 12, 679704. [Google Scholar] [CrossRef]
- Chai, M.; Liu, X.; Wei, L.; Li, J.; Gou, M.; Zhu, T.; Han, Y.; Liu, X. Molecular Characterization of a B Cell Adaptor for Phosphoinositide 3-Kinase Homolog in Lamprey (Lampetra japonica) and Its Function in the Immune Response. Int. J. Mol. Sci. 2022, 23, 14449. [Google Scholar] [CrossRef]
- Pickart, C.M.; Cohen, R.E. Proteasomes and their kin: Proteases in the machine age. Nat. Rev. Mol. Cell Biol. 2004, 5, 177–187. [Google Scholar] [CrossRef] [PubMed]
- Raffeiner, M.; Zhu, S.; González-Fuente, M.; Üstün, S. Interplay between autophagy and proteasome during protein turnover. Trends Plant Sci. 2023, 28, 698–714. [Google Scholar] [CrossRef] [PubMed]
- Wojcik, S. Crosstalk between autophagy and proteasome protein degradation systems: Possible implications for cancer therapy. Folia Histochem. Cytobiol. 2013, 51, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef]
- Russell, R.C.; Guan, K.L. The multifaceted role of autophagy in cancer. EMBO J. 2022, 41, e110031. [Google Scholar] [CrossRef]
- Yue, A.C.; Zhou, X.D.; Song, H.P.; Liu, X.H.; Bi, M.J.; Han, W.; Li, Q. Effect and molecular mechanism of Sulforaphane alleviates brain damage caused by acute carbon monoxide poisoning: Network pharmacology analysis, molecular docking, and experimental evidence. Environ. Toxicol. 2023, 39, 1140–1162. [Google Scholar] [CrossRef]
- Siddiqui, K.; On, K.F.; Diffley, J.F. Regulating DNA replication in eukarya. Cold Spring Harb. Perspect. Biol. 2013, 5, a012930. [Google Scholar] [CrossRef]
- Tanaka, S.; Araki, H. Helicase activation and establishment of replication forks at chromosomal origins of replication. Cold Spring Harb. Perspect. Biol. 2013, 5, a010371. [Google Scholar] [CrossRef]
- Le, T.K.C.; Dao, X.D.; Nguyen, D.V.; Luu, D.H.; Bui, T.M.H.; Le, T.H.; Nguyen, H.T.; Le, T.N.; Hosaka, T.; Nguyen, T.T.T. Insulin signaling and its application. Front. Endocrinol. 2023, 14, 1226655. [Google Scholar] [CrossRef]
- Miao, Y.; Zhang, Q.; Yuan, Z.; Wang, J.; Xu, Y.; Chai, Y.; Du, M.; Yu, Q.; Zhang, L.; Jiang, Z. Proteomics analysis reveals novel insights into the mechanism of hepatotoxicity induced by Tripterygium wilfordii multiglycoside in mice. Front. Pharmacol. 2022, 13, 1032741. [Google Scholar] [CrossRef]
- Mantis, N.J.; Rol, N.; Corthésy, B. Secretory IgA’s complex roles in immunity and mucosal homeostasis in the gut. Mucosal Immunol. 2011, 4, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Yao, Z.; Kong, J.; Zhang, X.; Li, H.; Chen, W.; Xie, Q. Transcriptomic analysis reveals the dynamic changes of transcription factors during early development of chicken embryo. BMC Genom. 2022, 23, 825. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Chen, Z.; Du, X.; Huang, Y.; Qin, J.; Wen, L.; Pan, X.; Lin, Y. SMRT Sequencing Reveals Candidate Genes and Pathways With Medicinal Value in Cipangopaludina chinensis. Front. Genet. 2022, 13, 881952. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, Y.; Peng, X.; Bo, L.; Wang, Z.; Song, Q. Characterization of cadmium-responsive transcription factors in wolf spider Pardosa pseudoannulata. Chemosphere 2021, 268, 129239. [Google Scholar] [CrossRef]
- Yu, M.; Zhan, J.; Zhang, H. HOX family transcription factors: Related signaling pathways and post-translational modifications in cancer. Cell Signal. 2020, 66, 109469. [Google Scholar] [CrossRef]
- Gorski, D.H.; Walsh, K. Control of vascular cell differentiation by homeobox transcription factors. Trends Cardiovasc. Med. 2003, 13, 213–220. [Google Scholar] [CrossRef]
- Parrillo, L.; Spinelli, R.; Longo, M.; Zatterale, F.; Santamaria, G.; Leone, A.; Campitelli, M.; Raciti, G.A.; Beguinot, F. The Transcription Factor HOXA5: Novel Insights into Metabolic Diseases and Adipose Tissue Dysfunction. Cells 2023, 12, 2090. [Google Scholar] [CrossRef]
- Surendran, H.; Palaniyandi, T.; Natarajan, S.; Hari, R.; Viwanathan, S.; Baskar, G.; Wahab, M.R.A.; Ravi, M.; Rajendran, B.K. Role of homeobox d10 gene targeted signaling pathways in cancers. Pathol. Res. Pract. 2023, 248, 154643. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, Q.; Wang, H.; Zhang, X.; Tian, M.; Liu, D.; Yang, X. Effects of HOXC8 on the Proliferation and Differentiation of Porcine Preadipocytes. Animals 2023, 13, 2615. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway ID | Pathway | Level1 | Level2 | p-Value | FDR |
|---|---|---|---|---|---|
| (A) | |||||
| ko04110 | Cell cycle | Cellular Processes | Cell growth and death | 1.04698 × 10−6 | 0.000217338 |
| ko03030 | DNA replication | Genetic Information Processing | Replication and repair | 1.31322 × 10−6 | 0.000217338 |
| ko04810 | Regulation of actin cytoskeleton | Cellular Processes | Cell motility | 3.41205 × 10−5 | 0.003764626 |
| ko04530 | Tight junction | Cellular Processes | Cellular community-eukaryotes | 0.00013942 | 0.011092812 |
| ko05130 | Pathogenic Escherichia coli infection | Human Diseases | Infectious disease: bacterial | 0.000167565 | 0.011092812 |
| ko00030 | Pentose phosphate pathway | Metabolism | Carbohydrate metabolism | 0.00036239 | 0.019991875 |
| ko04670 | Leukocyte transendothelial migration | Organismal Systems | Immune system | 0.00059778 | 0.028266442 |
| ko04640 | Hematopoietic cell lineage | Organismal Systems | Immune system | 0.000768791 | 0.031808743 |
| ko00010 | Glycolysis/Gluconeogenesis | Metabolism | Carbohydrate metabolism | 0.000976408 | 0.034237145 |
| ko00052 | Galactose metabolism | Metabolism | Carbohydrate metabolism | 0.001034355 | 0.034237145 |
| (B) | |||||
| ko03050 | Proteasome | Genetic Information Processing | Folding, sorting and degradation | 9.61422 × 10−18 | 2.99964 × 10−15 |
| ko04140 | Autophagy-animal | Cellular Processes | Transport and catabolism | 2.70307 × 10−9 | 4.21679 × 10−7 |
| ko05017 | Spinocerebellar ataxia | Human Diseases | Neurodegenerative disease | 6.21069 × 10−7 | 6.45912 × 10−5 |
| ko04137 | Mitophagy-animal | Cellular Processes | Transport and catabolism | 2.87134 × 10−6 | 0.000223964 |
| ko04120 | Ubiquitin mediated proteolysis | Genetic Information Processing | Folding, sorting and degradation | 1.5252 × 10−5 | 0.000951723 |
| ko04211 | Longevity regulating pathway | Organismal Systems | Aging | 6.09401 × 10−5 | 0.003168886 |
| ko00062 | Fatty acid elongation | Metabolism | Lipid metabolism | 7.82992 × 10−5 | 0.003489909 |
| ko04136 | Autophagy—other | Cellular Processes | Transport and catabolism | 0.0002324 | 0.009063607 |
| ko04213 | Longevity regulating pathway-multiple species | Organismal Systems | Aging | 0.001998418 | 0.069278488 |
| ko00010 | Glycolysis/Gluconeogenesis | Metabolism | Carbohydrate metabolism | 0.003074493 | 0.095924173 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, Y.; Wang, C.; Liu, Q. Transcriptomic Profiling of Misgurnus anguillicaudatus Reveals the Anti-Inflammatory Action of Lonicera japonica Extract in Response to Lipopolysaccharide Challenge. Fishes 2025, 10, 333. https://doi.org/10.3390/fishes10070333
Zhao Y, Wang C, Liu Q. Transcriptomic Profiling of Misgurnus anguillicaudatus Reveals the Anti-Inflammatory Action of Lonicera japonica Extract in Response to Lipopolysaccharide Challenge. Fishes. 2025; 10(7):333. https://doi.org/10.3390/fishes10070333
Chicago/Turabian StyleZhao, Yue, Chen Wang, and Qiuning Liu. 2025. "Transcriptomic Profiling of Misgurnus anguillicaudatus Reveals the Anti-Inflammatory Action of Lonicera japonica Extract in Response to Lipopolysaccharide Challenge" Fishes 10, no. 7: 333. https://doi.org/10.3390/fishes10070333
APA StyleZhao, Y., Wang, C., & Liu, Q. (2025). Transcriptomic Profiling of Misgurnus anguillicaudatus Reveals the Anti-Inflammatory Action of Lonicera japonica Extract in Response to Lipopolysaccharide Challenge. Fishes, 10(7), 333. https://doi.org/10.3390/fishes10070333