

Acceleration of CRISPR/Cas9-Mediated Editing at Multiple Sites in the Saccharomyces cerevisiae Genome

Abstract
1. Introduction
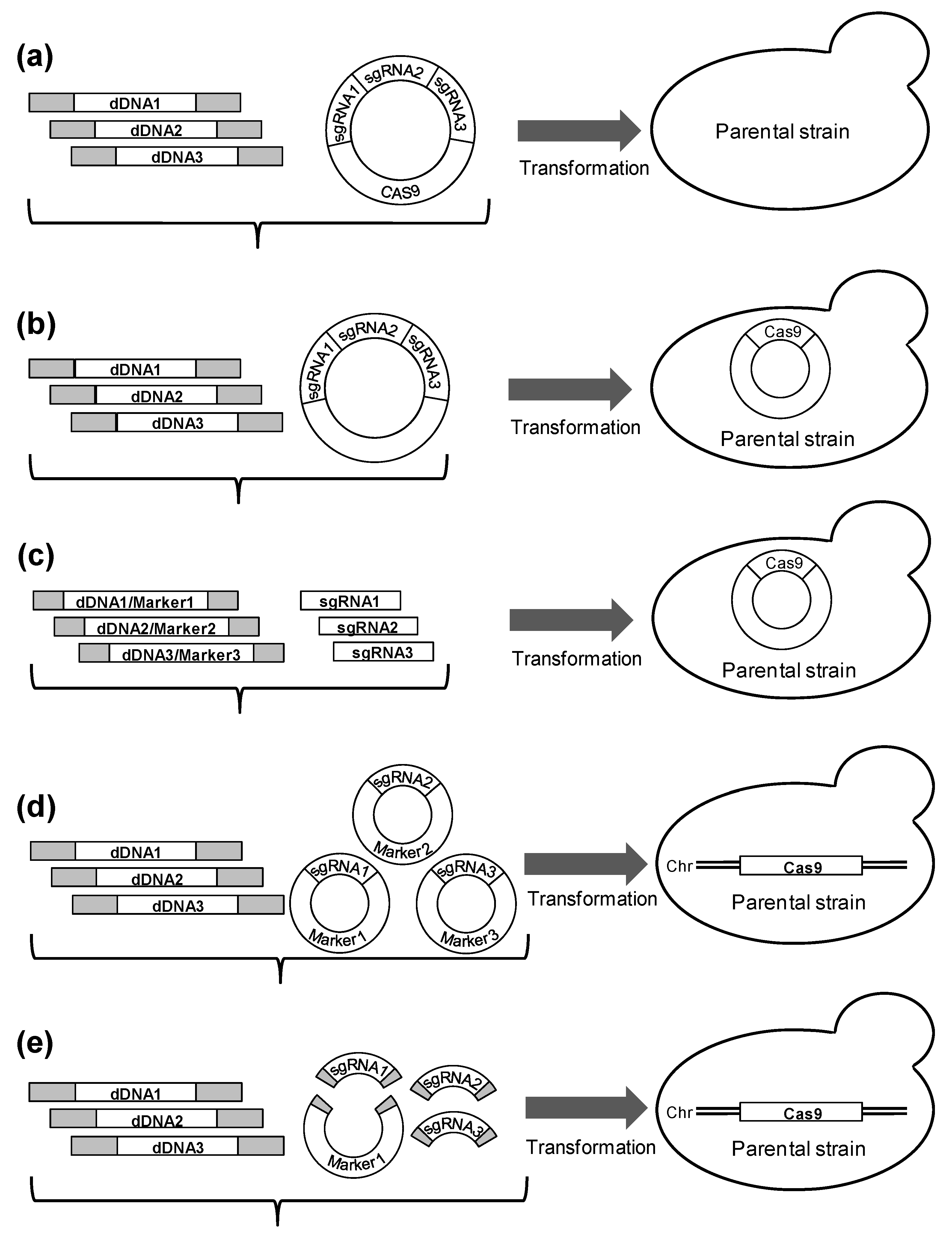
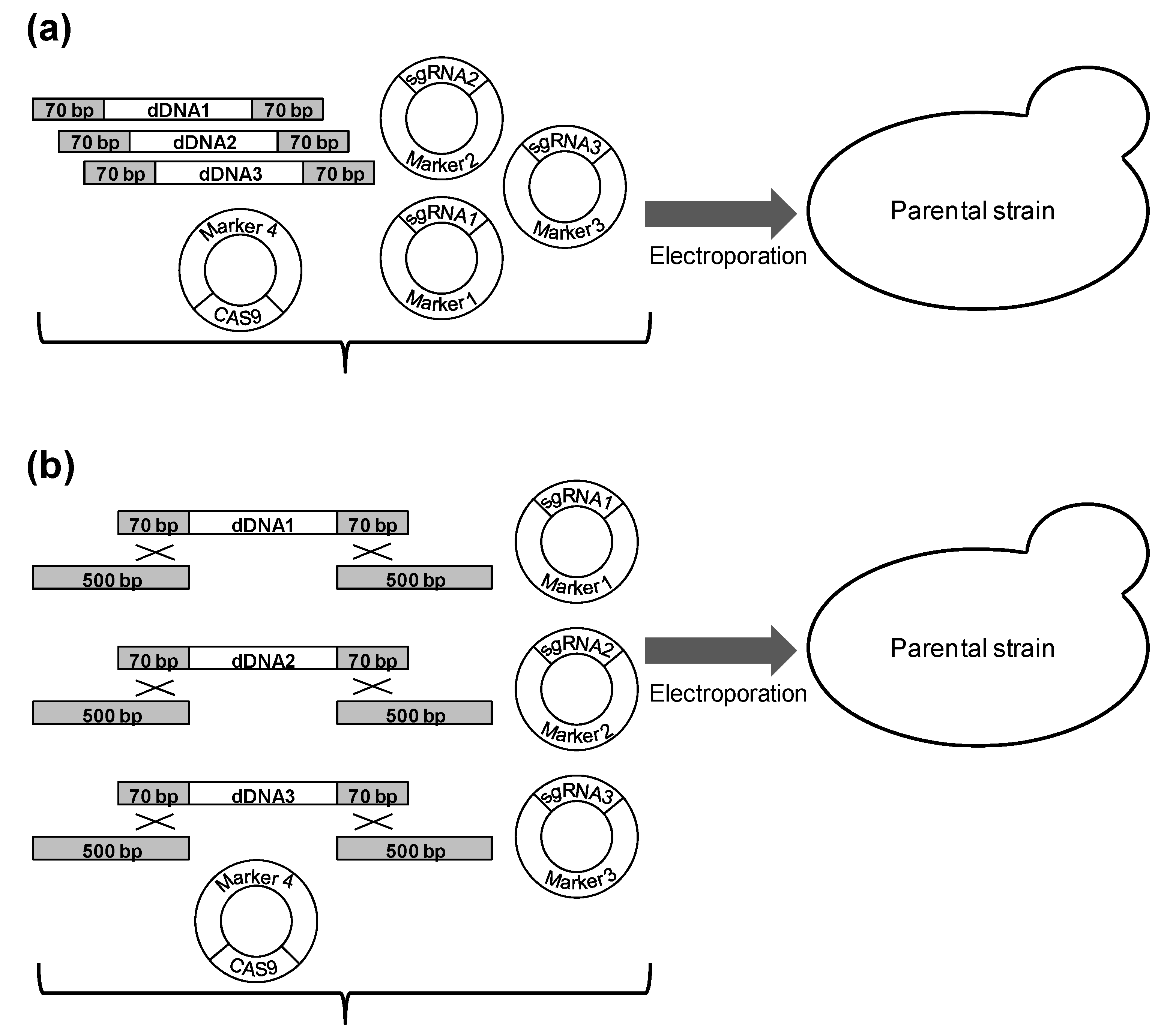
2. Materials and Methods
2.1. Strains and Media
2.2. Molecular Biological Techniques
2.3. Plasmids
2.4. Preparation of dDNA for Integration
2.5. Transformation Protocol
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jiang, F.; Doudna, J.A. CRISPR-Cas9 Structures and Mechanisms. Annu. Rev. Biophys. 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- Dudás, A.; Chovanec, M. DNA double-strand break repair by homologous recombination. Mutat. Res. 2004, 566, 131–167. [Google Scholar] [CrossRef] [PubMed]
- Easmin, F.; Hassan, N.; Sasano, Y.; Ekino, K.; Taguchi, H.; Harashima, S. gRNA-transient expression system for simplified gRNA delivery in CRISPR/Cas9 genome editing. J. Biosci. Bioeng. 2019, 128, 373–378. [Google Scholar] [CrossRef]
- DiCarlo, J.E.; Norville, J.E.; Mali, P.; Rios, X.; Aach, J.; Church, G.M. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013, 41, 4336–4343. [Google Scholar] [CrossRef] [PubMed]
- Ronda, C.; Maury, J.; Jakočiunas, T.; Jacobsen, S.A.; Germann, S.M.; Harrison, S.J.; Borodina, I.; Keasling, J.D.; Jensen, M.K.; Nielsen, A.T. CrEdit: CRISPR mediated multi-loci gene integration in Saccharomyces cerevisiae. Microb. Cell Fact. 2015, 14, 97. [Google Scholar] [CrossRef]
- Horwitz, A.A.; Walter, J.M.; Schubert, M.G.; Kung, S.H.; Hawkins, K.; Platt, D.M.; Hernday, A.D.; Mahatdejkul-Meadows, T.; Szeto, W.; Chandran, S.S.; et al. Efficient Multiplexed Integration of Synergistic Alleles and Metabolic Pathways in Yeasts via CRISPR-Cas. Cell Syst. 2015, 1, 88–96. [Google Scholar] [CrossRef]
- Jakočiūnas, T.; Rajkumar, A.S.; Zhang, J.; Arsovska, D.; Rodriguez, A.; Jendresen, C.B.; Skjødt, M.L.; Nielsen, A.T.; Borodina, I.; Jensen, M.K.; et al. CasEMBLR: Cas9-Facilitated Multiloci Genomic Integration of in Vivo Assembled DNA Parts in Saccharomyces cerevisiae. ACS Synth. Biol. 2015, 4, 1226–1234. [Google Scholar] [CrossRef]
- Ryan, O.W.; Skerker, J.M.; Maurer, M.J.; Li, X.; Tsai, J.C.; Poddar, S.; Lee, M.E.; DeLoache, W.; Dueber, J.E.; Arkin, A.P.; et al. Selection of chromosomal DNA libraries using a multiplex CRISPR system. eLife 2014, 3, e03703. [Google Scholar] [CrossRef]
- Bao, Z.; Xiao, H.; Liang, J.; Zhang, L.; Xiong, X.; Sun, N.; Si, T.; Zhao, H. Homology-integrated CRISPR-Cas (HI-CRISPR) system for one-step multigene disruption in Saccharomyces cerevisiae. ACS Synth. Biol. 2015, 4, 585–594. [Google Scholar] [CrossRef]
- Easmin, F.; Sasano, Y.; Kimura, S.; Hassan, N.; Ekino, K.; Taguchi, H.; Harashima, S. CRISPR-PCD and CRISPR-PCRep: Two novel technologies for simultaneous multiple segmental chromosomal deletion/replacement in Saccharomyces cerevisiae. J. Biosci. Bioeng. 2020, 129, 129–139. [Google Scholar] [CrossRef]
- Manivasakam, P.; Weber, S.C.; McElver, J.; Schiestl, R.H. Micro-homology mediated PCR targeting in Saccharomyces cerevisiae. Nucleic Acids Res. 1995, 23, 2799–2800. [Google Scholar] [CrossRef] [PubMed]
- Baudin, A.; Ozier-Kalogeropoulos, O.; Denouel, A.; Lacroute, F.; Cullin, C. A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic Acids Res. 1993, 21, 3329–3330. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, C.S. Preparation of yeast DNA. Curr. Protoc. Mol. Biol. 2001, 39, 13.11.1–13.11.4. [Google Scholar] [CrossRef]
- Jessop-Fabre, M.M.; Jakočiūnas, T.; Stovicek, V.; Dai, Z.; Jensen, M.K.; Keasling, J.D.; Borodina, I. EasyClone-MarkerFree: A vector toolkit for marker-less integration of genes into Saccharomyces cerevisiae via CRISPR-Cas9. Biotechnol. J. 2016, 11, 1110–1117. [Google Scholar] [CrossRef]
- Babaei, M.; Sartori, L.; Karpukhin, A.; Abashkin, D.; Matrosova, E.; Borodina, I. Expansion of EasyClone-MarkerFree toolkit for Saccharomyces cerevisiae genome with new integration sites. FEMS Yeast Res. 2021, 21, foab027. [Google Scholar] [CrossRef]
- Ma, H.; Kunes, S.; Schatz, P.J.; Botstein, D. Plasmid construction by homologous recombination in yeast. Gene 1987, 58, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Sheff, M.A.; Thorn, K.S. Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast 2004, 21, 661–670. [Google Scholar] [CrossRef]
- Goldstein, A.L.; McCusker, J.H. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast 1999, 15, 1541–1553. [Google Scholar] [CrossRef]
- Gueldener, U.; Heinisch, J.; Koehler, G.J.; Voss, D.; Hegemann, J.H. A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res. 2002, 30, e23. [Google Scholar] [CrossRef]
- Becker, D.M.; Lundblad, V. Introduction of DNA into yeast cells. Curr. Protoc. Mol. Biol. 2001, 27, 13.7.1–13.7.10. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.; Honigberg, S.M. Effect of chromosomal locus, GC content and length of homology on PCR-mediated targeted gene replacement in Saccharomyces. Nucleic Acids Res. 2001, 29, 5156–5162. [Google Scholar] [CrossRef]
- Shao, Z.; Zhao, H.; Zhao, H. DNA assembler, an in vivo genetic method for rapid construction of biochemical pathways. Nucleic Acids Res. 2009, 37, e16. [Google Scholar] [CrossRef] [PubMed]
- Flagfeldt, D.B.; Siewers, V.; Huang, L.; Nielsen, J. Characterization of chromosomal integration sites for heterologous gene expression in Saccharomyces cerevisiae. Yeast 2009, 26, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, N.G.; Chroumpi, S.; Vos, T.; Solis-Escalante, D.; Bosman, L.; Pronk, J.T.; Daran, J.M.; Daran-Lapujade, P. One-step assembly and targeted integration of multigene constructs assisted by the I-SceI meganuclease in Saccharomyces cerevisiae. FEMS Yeast Res. 2013, 13, 769–781. [Google Scholar] [CrossRef]
- Ryan, O.W.; Cate, J.H. Multiplex engineering of industrial yeast genomes using CRISPRm. Methods Enzymol. 2014, 546, 473–489. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Plasmid | Description | Reference |
|---|---|---|
| Plasmids for marker amplification | ||
| pAG26 | hphMX | [18] |
| pUG66 | bleMX | [19] |
| Cas9-carrying plasmid | ||
| pCfB2312 | CEN/ARS, TEF1p-Cas9-CYC1t, kanMX | [14] |
| sgRNA-expressing plasmids | ||
| pCfB9336 | 2μ, sgRNA II-1, natMX | [15] |
| pCfB9341 | 2μ, sgRNA IX-1, natMX | |
| pCfB9344 | 2μ, sgRNA XVI-1, natMX | |
| pCfB9341-hphMX | 2μ, sgRNA IX-1, hphMX | This work |
| pCfB9344-bleMX | 2μ, sgRNA XVI-1, bleMX | This work |
| Plasmid for dDNA amplification | ||
| pyEGFP | TPI1p–yEGFP–CYC1t | Lab collection |
| Primer | Sequence (5′ → 3′) | Application |
|---|---|---|
| Primers for replacement of selective markers in plasmids pCfB9341 and pCfB9344 | ||
| 1 | cttgctaggatacagttctcacatcacatccgaacataaacaaccatgggtaaaaagcctgaactc | For amplification of the ORF of hygromycin resistance gene (hph) |
| 2 | caaatgacaagttcttgaaaacaagaatctttttattgtcagtactgattattcctttgccctcggac | |
| 3 | cttgctaggatacagttctcacatcacatccgaacataaacaaccatggccgaccaagcgacg | For amplification of the ORF of phleomycin resistance gene (ble) |
| 4 | caaatgacaagttcttgaaaacaagaatctttttattgtcagtactgatcatgagatgcctgcaagcaattc | |
| 5 | gatggctgtgtagaagtactcgc | PCR screening for replacement of selective marker with hph |
| 6 | ctctgtctttcatttaagatgatcat | |
| 7 | cgcctgatacagaacgaattgc | PCR screening for replacement of selective marker with ble |
| 8 | tctcggtaacttgaatggtg | |
| Primers for amplification of dDNA fragments containing 70 bp recombination flanks | ||
| 9 | tctttactttgttttaagtagtctttccatctcccttcaacggaaacaagtgcacgcaacatgttattttcggatatttaacttacttagaataatgcc | Amplification of dDNA fragment for site II-1 |
| 10 | gcaaccgggtagggtgtcttctttgtaaagtttgtctaatgttggagaatggttgccacccggggaagaacttcgagcgtcccaaaac | |
| 11 | ctgtttaggaaataaagaagagaaacgtcacctaaaaagtttaaattgaagatgtcgttctatctcgcgacggatatttaacttacttagaataatgcc | Amplification of dDNA fragment for site IX-1 |
| 12 | ccacgctaaccttaatgtgcctagatatcatgggttatttaatgacaagtccgctagtgtcatcaacccacttcgagcgtcccaaaac | |
| 13 | tataagaaagtaaacgcaaaagataggctgactgccttcattcgactaggaggtgaggcgacatatttgtcggatatttaacttacttagaataatgcc | Amplification of dDNA fragment for site XVI-1 |
| 14 | ataaaatgggtagaaaaagcatcgttgctgcagtgtacaatcaagctcacccgagccatccacctctctacttcgagcgtcccaaaac | |
| Primers for amplification of dDNA fragments containing 500 bp recombination flanks | ||
| 15 | gttcacagttactcttttagaactcc | Amplification of dDNA fragment for site II-1 |
| 16 | atgccgtgatatgaacaaacac | |
| 17 | ctgtgatcttctaagataaaaaggc | Amplification of dDNA fragment for site IX-1 |
| 18 | agaaaactacccgtagaatacatattc | |
| 19 | gaagattgcaaactcaagtctac | Amplification of dDNA fragment for site XVI-1 |
| 20 | gttcagtagcaaagtattgtcg | |
| Primers for amplification of 500 bp flanks for recombination with desired sites | ||
| 21 | aaaataacatgttgcgtgcac | Amplification of 500 bp flank for “upstream” region of site II-1. Used with 15 |
| 22 | tcgcgagatagaacgacatcttc | Amplification of 500 bp flank for “upstream” region of site IX-1. Used with 17 |
| 23 | acaaatatgtcgcctcacctc | Amplification of 500 bp flank for “upstream” region of site XVI-1. Used with 19 |
| 24 | ttcttccccgggtggcaa | Amplification of 500 bp flank for “downstream” region of site II-1. Used with 16 |
| 25 | tgggttgatgacactagcgg | Amplification of 500 bp flank for “downstream” region of site IX-1. Used with 18 |
| 26 | tagagaggtggatggctcg | Amplification of 500 bp flank for “downstream” region of site XVI-1. Used with 20 |
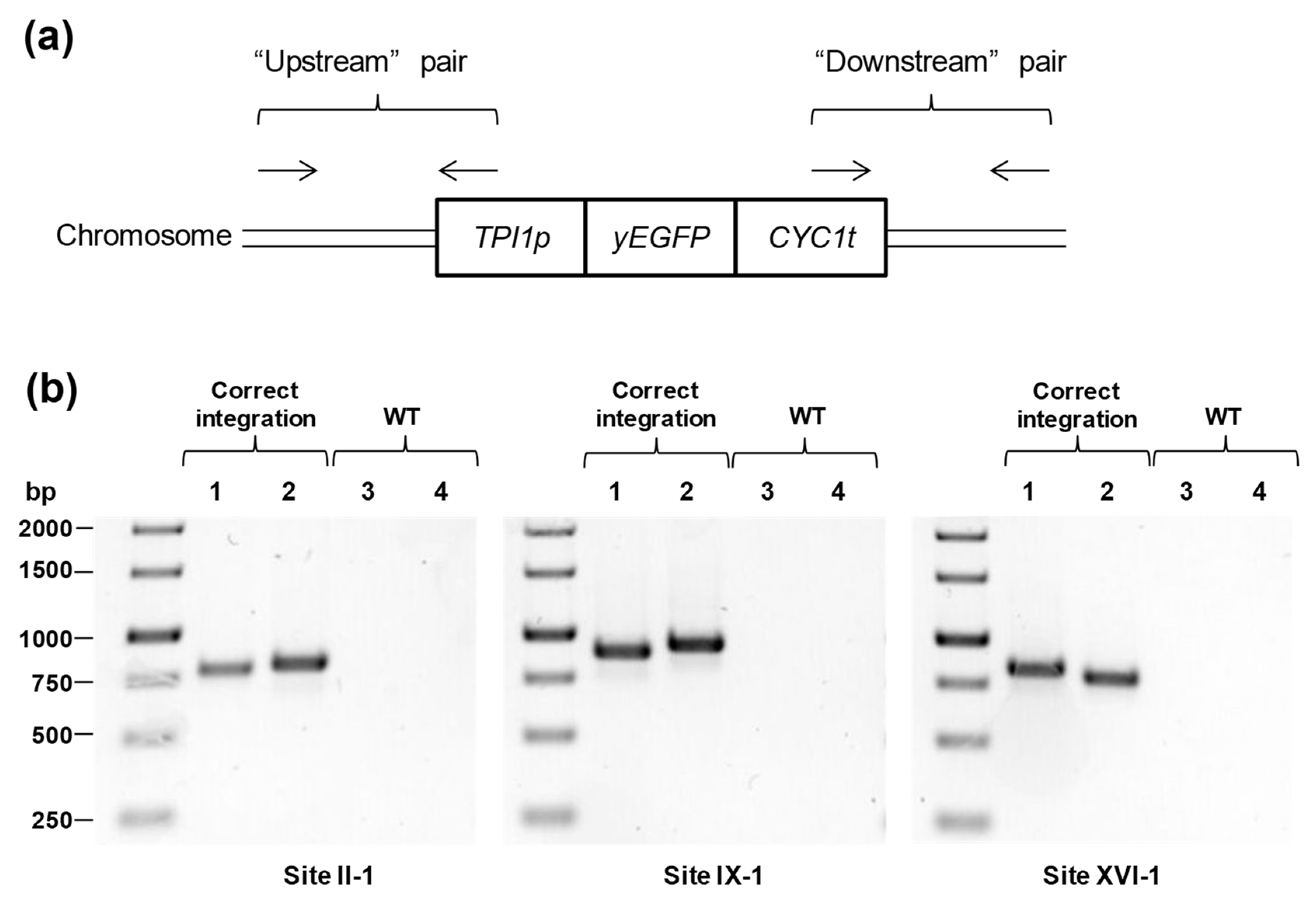
| Primers for testing integration into desired sites | ||
| 27 | gctgaaaagtcttagaacgggta | Checking of “upstream” region of dDNA fragment integrated into site II-1 |
| 28 | tccttatccattcagcttctc | |
| 29 | ccgctctaaccgaaaaggaagg | Checking of “downstream” region of dDNA fragment integrated into site II-1 |
| 30 | accctttttttccattccctc | |
| 31 | acagtgagagagtggcat | Checking of “upstream” region of dDNA fragment integrated into site IX-1. Used with 27 |
| 32 | acttaccaaggtgctgct | Checking of “downstream” region of dDNA fragment integrated into site IX-1. Used with 29 |
| 33 | acaagagacgaaaaggaccc | Checking of “upstream” region of dDNA fragment integrated into site XVI-1. Used with 27 |
| 34 | cacttgctttgctcactactc | Checking of “downstream” region of dDNA fragment integrated into site XVI-1. Used with 29 |
| Recipient Strain | Transforming Plasmids | Type of Transformation | Number of Transformants | Integration in 3 loci, % |
|---|---|---|---|---|
| with pCas9 | 3 sgRNA | LiAc | 9 ± 5 | 90 ± 17 |
| without pCas9 | 3 sgRNA, pCas9 | 3 ± 1 | 75 ± 25 | |
| with pCas9 | 3 sgRNA | Electroporation | 240 ± 57 | 95 ± 2 |
| without pCas9 | 3 sgRNA, pCas9 | 46 ± 8 | 77 ± 9 |
| Transforming Plasmids | Added Arms | Proportion of Transformants with the Correct Integration, % | Proportion of Transformants with the Incomplete Integration, % | |
|---|---|---|---|---|
| Into 1 Site | Into 2 Sites | |||
| 2 sgRNA, pCas9 (integration into 2 sites) | no | 45 ± 2 | 14 ± 7 | - |
| 3 sgRNA, pCas9 (integration into 3 sites) | no | 6 ± 3 | 2 ± 2 | 0 |
| 3 sgRNA, pCas9 (integration into 3 sites) | yes | 24 ± 5 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karpukhin, A.D.; Sabirzyanov, F.A.; Serebrianyi, V.A. Acceleration of CRISPR/Cas9-Mediated Editing at Multiple Sites in the Saccharomyces cerevisiae Genome. Methods Protoc. 2023, 6, 39. https://doi.org/10.3390/mps6020039
Karpukhin AD, Sabirzyanov FA, Serebrianyi VA. Acceleration of CRISPR/Cas9-Mediated Editing at Multiple Sites in the Saccharomyces cerevisiae Genome. Methods and Protocols. 2023; 6(2):39. https://doi.org/10.3390/mps6020039
Chicago/Turabian StyleKarpukhin, Alexey D., Fanis A. Sabirzyanov, and Vsevolod A. Serebrianyi. 2023. "Acceleration of CRISPR/Cas9-Mediated Editing at Multiple Sites in the Saccharomyces cerevisiae Genome" Methods and Protocols 6, no. 2: 39. https://doi.org/10.3390/mps6020039
APA StyleKarpukhin, A. D., Sabirzyanov, F. A., & Serebrianyi, V. A. (2023). Acceleration of CRISPR/Cas9-Mediated Editing at Multiple Sites in the Saccharomyces cerevisiae Genome. Methods and Protocols, 6(2), 39. https://doi.org/10.3390/mps6020039