

A Simple, Improved Method for Scarless Genome Editing of Budding Yeast Using CRISPR-Cas9


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Design
2.1. Materials
- 0.2 mL PCR tubes (USA Scientific, Ocala, FL, USA, Cat. no.: 1402-4708)
- 1.7 mL microcentrifuge tubes (VWR, Radnor, PA, USA, Cat. no.: 87003-294)
- Budding yeast strain to be genetically engineered (must NOT already contain a kanamycin resistance marker, conferring resistance to G418 in yeast)
- pCASB Plasmid (Addgene ID: 190175)
- Oligonucleotides encoding desired gRNA (described in Section 3.1)
- Oligonucleotide annealing buffer, such as TE (described below) containing 50 mM sodium chloride (Fisher Scientific, Hampton, NH, USA, Cat. no.: S271)
- BsaI-HF® v2 (New England Biolabs, Ipswich, MA, USA, Cat. no.: R3733)
- T4 DNA Ligase and buffer (New England Biolabs, Ipswich, MA, USA, Cat. no. M0202)
- Filtered water (such as Millipore), sterilized by means of autoclaving where indicated
- High-efficiency competent E. coli, such as One Shot TOP10 Chemically Competent E. coli (Thermo Fisher Scientific, Waltham, MA, USA, Cat. no.: C404010)
- 14 mL culture tubes for bacterial culture growth (Corning, Corning, NY, Cat. no.: 352051). Autoclaved to sterilize.
- Kanamycin Sulfate (Sigma-Aldrich, St. Louis, MO, USA, Cat. no.: 60615), prepared to 400 mg/mL (1000× stock solution) in sterile water
- 0.2-micron syringe filters (VWR, Radnor, PA, USA, Cat. no.: 76479-024), 50 mL conical tube vacuum filters (MilliporeSigma, Burlington, MA, USA, Cat. no.: SCGP00525), and 250–500 mL flask vacuum filters (Corning, Corning, NY, USA, Cat. no.: 430196-7) for sterilization of reagents
- LB liquid media
- ○
- LB Powder (IBI Scientific, Dubuque, IA, USA, Cat. no.: IB49020), 25 g/L in water. Autoclaved to sterilize.
- LB agar plates containing 400 μg/mL Kanamycin
- ○
- Petri dishes (Fisher Scientific, Hampton, NH, USA, Cat. no.: FB0875713)
- ○
- Agar A (Bio Basic, Amherst, NY, USA, Cat. no.: FB0010)
- ○
- Add 15 g/L Agar A to LB liquid media before autoclaving. Allow to cool to 55 °C before adding 1 mL/L of Kanamycin stock solution and pouring into Petri dishes.
- Glass beads or equivalent for plating (Zymo Research, Irvine, CA, Cat. no.: S1001). Autoclaved to sterilize.
- Plasmid miniprep kit (Zymo Research, Irvine, CA, USA, Cat. no.: D4016)
- Oligonucleotides or other DNA encoding template sequence for HDR (described in Section 3.3)
- High-fidelity DNA polymerase enzyme and buffer, such as Expand™ High Fidelity PCR System (EHIFI-RO Roche, Sigma Aldrich Cat. no.: 11732650001)
- 10 mM dNTPs (Promega, Madison, WI, USA, Cat. no.: U1515)
- 100% ethanol (VWR, Radnor, PA, USA, Cat. no.: 89125-186)
- 70% ethanol (diluted in filtered water)
- Sodium Acetate (Thermo Fisher Scientific, Waltham, MA, USA, Cat. no. BP333-500), 3 M solution in water
- TAE buffer (40 mM Tris-acetate, 1 mM EDTA)
- ○
- Tris Base (Thermo Fisher Scientific, Waltham, MA, USA, Cat. no.: BP152)
- ○
- Glacial acetic acid (Sigma-Aldrich, St. Louis, MO, USA, Cat. no.: A6283)
- ○
- For 1 L of 50× solution, combine 242 g Tris Base, 57.1 mL glacial acetic acid, and 100 mL 0.5 M EDTA, pH 8.0. Add water to 1 L)
- Quick dissolve agarose (Genesee Scientific, San Diego, CA, USA, Cat. no.: 20-102QD)
- ○
- To prepare DNA gel, mix 1% w/v agarose in TAE buffer, microwave to boil and dissolve agarose powder. Cool slightly before adding SYBR safe DNA gel stain (10,000×) and pouring into gel mold.
- SYBR™ Safe DNA Gel Stain (Thermo Fisher Scientific, Waltham, MA, USA, Cat. no.: S33102)
- DNA loading dye (New England Biolabs, Ipswich, MA, USA, Cat. no.: B7024)
- 100 bp DNA ladder (New England Biolabs, Ipswich, MA, USA, Cat. no.: N3231)
- TE Buffer (10 mM Tris-HCl pH 8.0, 0.1 mM EDTA)
- ○
- Tris Base (Thermo Fisher Scientific, Waltham, MA, USA, Cat. no.: BP152)
- ○
- EDTA (Caisson Laboratories, Smithfield, UT, USA, Cat. no.: E004)
- ○
- For 500 mL of 100× solution, combine 60.55 g Tris Base with 1.86 g EDTA Na2 • 2H2O. Add filtered water to under 500 mL, adjust pH to 7.8 with concentrated HCl (Thermo Fisher Scientific, (Waltham, MA, USA, Cat. no.: A144), then fill to 500 mL with filtered water. Use a 0.2-micron filter to sterilize.
- Yeast transformation buffer: 1× TE, 100 mM Lithium Acetate
- ○
- Lithium acetate (Sigma-Aldrich, St. Louis, MO, USA, Cat. No.: 517992), 1 M stock solution in water
- ○
- For 50 mL, mix 5 mL of 10× TE, 5 mL 1 M lithium acetate, and 40 mL of filtered water. Use a 0.2-micron filter to sterilize.
- Deoxyribonucleic acid, single stranded from salmon testes, 10 mg/mL (Sigma-Aldrich, St. Louis, MO, Cat. no.: D9156)
- PEG Solution: 40% Poly(ethylene) glycol in yeast transformation buffer
- ○
- Poly(ethylene) glycol (Sigma-Aldrich, St. Louis, MO, USA, Cat. no.: P4338)
- ○
- For 50 mL, combine 25 g poly(ethylene) glycol, 5 mL 1× TE, and 5 mL 1 M lithium acetate. Add warm (50–60 °C) filtered water to 50 mL. Mix until dissolved. Use a 0.2-micron filter to sterilize.
- Dimethyl Sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO, USA, Cat. no.: D8418)
- YPD liquid media
- ○
- 20 g/L Peptone (Research Products International, Mount Prospect, IL, USA, Cat. no.: P20250)
- ○
- 10 g/L Yeast Extract (Research Products International, Mount Prospect, IL, USA, Cat. no.: Y20025)
- ○
- 20 g/L Dextrose (Fisher Scientific, Hampton, NH, USA, Cat. no.: D16-1)
- ○
- Mix in filtered water, autoclave to sterilize.
- Glass tubes, 15 mL (Globe Scientific, Mahwah, NJ, USA, Cat. no.: 1517), and lids (DWK Life Sciences, Milville, NJ, Cat. no.: 73660-16) for yeast culture. Autoclave to sterilize.
- Geneticin (G418) sulfate (Santa Cruz Biotechnology, Dallas, TX, USA, Cat. no.: sc-29065), prepared to 200 mg/mL (1000× stock solution) in autoclaved filtered water.
- YPD Agar plates
- ○
- Add 15 g/L Agar A (Bio Basic, Amherst, NY, USA, Cat. no.: FB0010) to YPD liquid media before autoclaving.
- YPD Agar plates containing G418
- ○
- After autoclaving, allow liquid agar to cool to 55 °C before adding 1 mL/L of G418 stock solution.
- Wooden dowels (Puritan Medical Products, Guilford, ME, Cat. no.: 807), toothpicks, or pipette tips, for picking colonies. Autoclave to sterilize.
- Yeast genomic prep buffer (1% w/v SDS, 2% v/v Triton X-100, 100 mM NaCl, 10 mM Tris HCl pH 7.9, 1 mM EDTA)
- ○
- SDS (Promega, Madison, WI, USA, Cat. no.:H5113)
- ○
- Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA, Cat. no: X-100)
- ○
- Sodium chloride (Fisher Scientific, Hampton, NH, USA, Cat. no.: S271)
- 0.5 mm zirconia/silica beads (BioSpec Products, Bartlesville, OK, USA, Cat. No.: 11079105Z)
- Phenol:chloroform:isoamyl alcohol, 25:24:1 Saturated with 10 mM Tris, pH 8.0, 1 mM EDTA (Sigma-Aldrich, St. Louis, MO, USA, Cat. no.: P2069)
- Taq DNA polymerase (New England Biolabs, Ipswich, MA, USA, Cat. no.: M0273), or similar enzyme for low fidelity PCR
- PCR purification kit (Qiagen, Hilden, Germany, Cat. no.: 28104)
- 1 kb DNA ladder (New England Biolabs, Ipswich, MA, USA, Cat. no.: N3232)
- Glycerol (VWR, Radnor, PA, Cat. no.:0854), 80% v/v in filtered water. Autoclave to sterilize.
- Cryogenic tubes, 1.8 mL (Thermo Fisher Scientific, Waltham, MA, USA, Cat. no.: 377267)
2.2. Equipment
- Thermocycler
- Standard horizontal agarose gel electrophoresis tank, gel tray, comb and power supply
- Autoclave for sterilizing media, culture tubes, etc.
- 37 °C incubator for bacterial growth
- 37 °C shaking incubator for bacterial growth
- 30 °C incubator with a tube rotator for yeast growth
- Water bath or heat block set to 42 °C
- Multi-tube vortex (Scientific Industries, Bohemia, NY, USA, Cat. no.: SI-D238), kept at 4 °C in cold room or refrigerator
- Nanodrop Spectrophotometer
- Spectrophotometer to measure OD600 of cultures
- Microwave for preparing agarose gel
- Benchtop microfuge
- Access to a DNA sequencing facility
- Access to oligonucleotide synthesis for preparation of gRNA oligos, gRNA sequencing primer, HDR template DNA, and for PCR/sequencing of target gene
2.3. Software
- Benchling (https://benchling.com (accessed on 25 September 2022)), online resource for gRNA and HDR template design. Alternatively, another gRNA design software such as E-CRISP [25]
3. Procedure
3.1. Designing the gRNA Oligonucleotides
- Import the sequence of the gene to be edited as a DNA sequence. Search for the gene using the systematic open reading frame nomenclature (i.e., YDR007W rather than TRP1) to facilitate gene import.
- Select “Design and analyze guides” tool from the CRISPR menu.
- Set the parameters to design a 20-nucleotide guide, using the PAM sequence NGG, using the S. cerevisiae genome (used to calculate off-target scores).
- Select a region of the gene immediately adjacent to the desired mutation site (±50–100 bp is suggested).
- Select a gRNA sequence as close to the target mutation as possible (within 70 bp of the target mutation, as discussed earlier).
- Make note of the chosen gRNA sequence and proceed to gRNA oligonucleotide design.
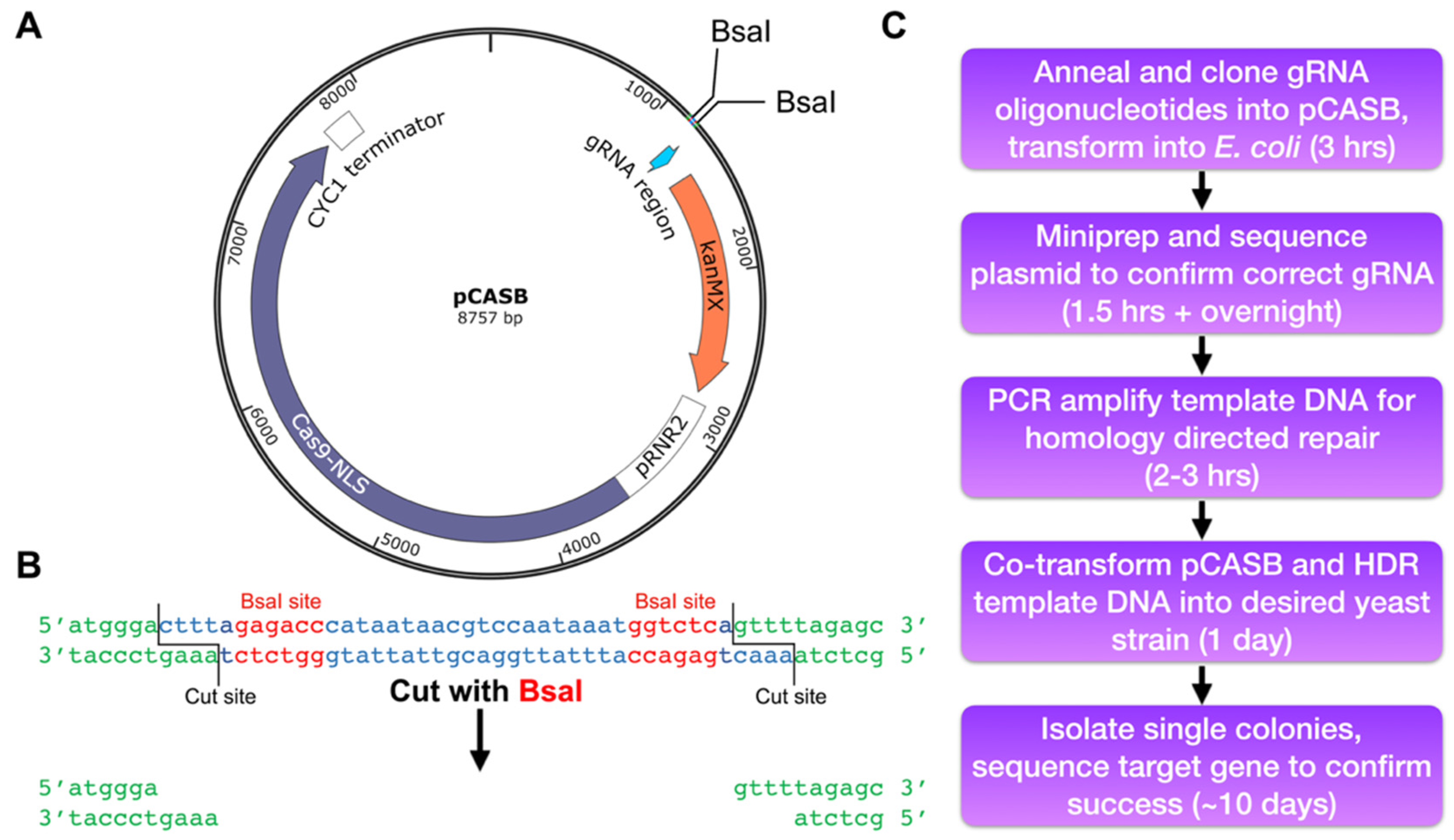
3.2. Clone the gRNA-Encoding Annealed Oligonucleotides into the pCASB Plasmid
3.2.1. Anneal the Complementary Oligonucleotides
- 8.
- Resuspend each oligonucleotide in filtered water to 100 mM.
- 9.
- In a PCR tube, mix 2.5 µL of each 100 mM oligonucleotide with 20 µL of oligonucleotide annealing buffer.
- 10.
- Using a thermocycler, heat oligonucleotides to 95 °C for 2 min then cool by 0.1 °C per second to 20 °C.
- 11.
- Use annealed oligonucleotides within a few hours or freeze at −20 °C for future use. Their final concentration is approximately 160 ng/µL.
3.2.2. Clone Annealed Oligonucleotides into pCASB Plasmid
- 12.
- Dilute annealed oligonucleotides 1:1000 in filtered water.
- 13.
- Assemble a 20 µL cloning reaction, adding reagents in the following order:
- Filtered water (to 20 µL final volume)
- 3 µL of annealed oligonucleotides (~480 pg)
- 75 ng of pCASB plasmid
- 2 µL of T4 DNA Ligase buffer
- 1 µL of T4 DNA Ligase enzyme
- 1 µL of BsaIHF-v2 enzyme
- 14.
- In a thermocycler, incubate reaction for 30 min at 37 °C, followed by 5 min at 60 °C. (While 30 min at 37 °C was chosen for the Golden Gate cloning (Section 3.2.2 step 3), as few as 10 min at 37 °C can lead to a successful reaction in a single-insert reaction. For troubleshooting an unsuccessful cloning, the time can be increased to 60 min)
3.2.3. Transform Golden Gate Cloning Reaction into E. coli and Confirm Successful Cloning
- 15.
- Transform 4 µL of ligation reaction into competent E. coli according to manufacturer protocol.
- 16.
- Grow all transformed cells overnight on LB + Kanamycin plates.
- 17.
- The next day, pick 6–10 colonies using sterile wooden sticks (or pipette tips) into 2 mL LB + Kanamycin (0.4 mg/mL diluted from 1000× stock) liquid cultures in 14 mL sterile plastic tubes.
- 18.
- Grow cultures overnight at 37 °C with shaking.
- 19.
- Isolate plasmid DNA from each culture using a Miniprep kit or similar method.
- 20.
- Measure concentration of each plasmid using a NanoDrop spectrophotometer, and send 800 ng (or as directed by the sequencing facility) of pCASB-based vector for Sanger sequencing of the gRNA site using the following sequencing primer [20]:
- 21.
- After obtaining sequencing results, save the samples containing successfully cloned plasmids. This DNA will be directly used for yeast transformation (step 41-i) and can be stored for many months at −20 °C.
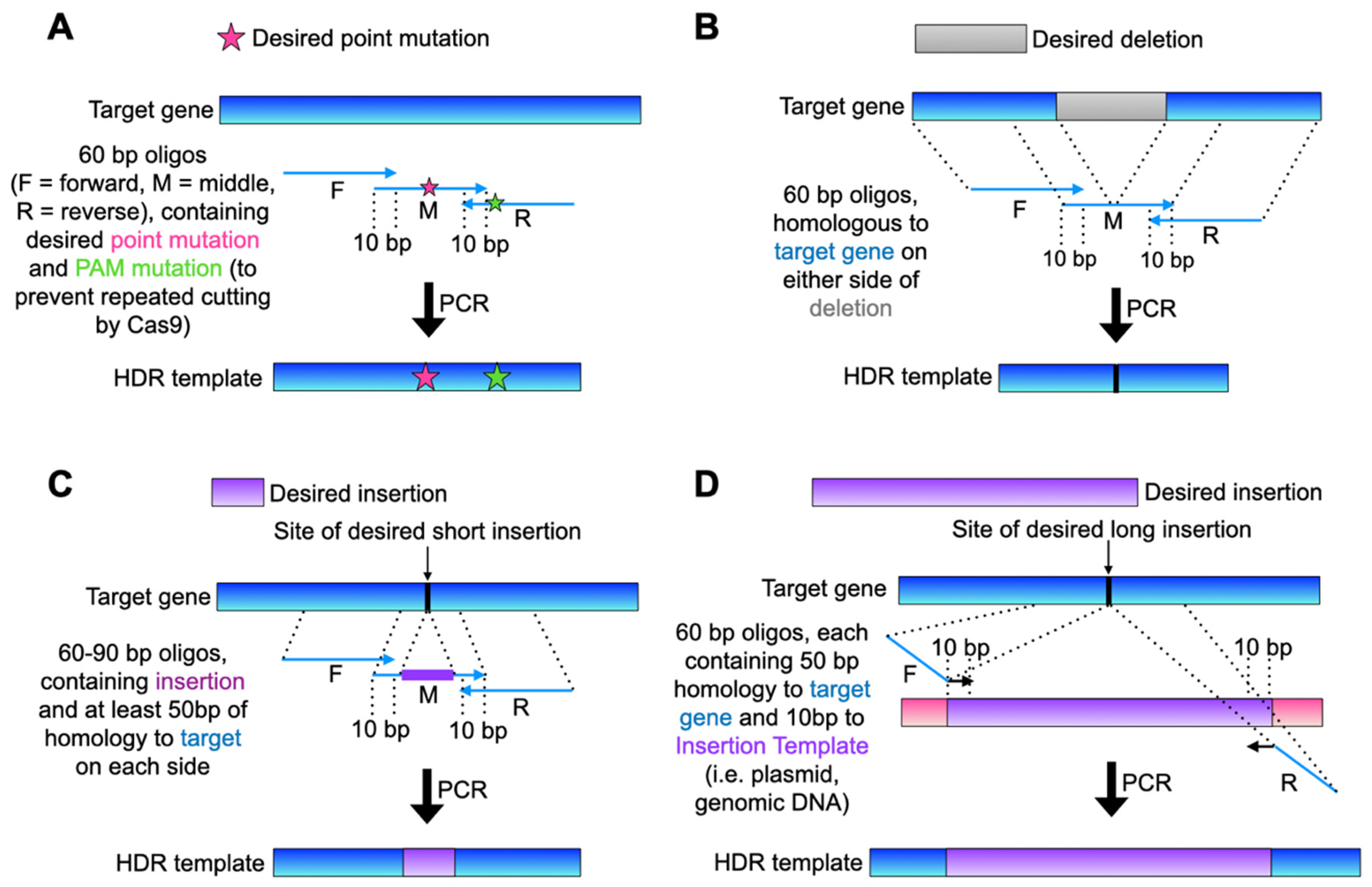
3.3. Design and Amplify HDR Template DNA
3.3.1. Design HDR Template
3.3.2. PCR Amplify the HDR Template (The following Is an Example Protocol Using Three 60–90-bp Oligos, Which Can Be Replaced with Any High-Fidelity PCR Reaction to Obtain the Desired Template)
- 22.
- Dilute the three oligonucleotides to 10 µM in filtered water
- 23.
- Set up PCR reaction (we have found that two 50 µL reactions is usually sufficient for one yeast transformation, but the efficiency of the PCR can vary for each set of oligos such that the number of PCR reactions can be increased as needed).
- 76 µL of filtered water
- 10 µL of Expand HF buffer
- 2 µL of 10 mM dNTPs
- 1 µL of “middle” template oligonucleotide
- 5 µL of “forward” primer oligonucleotide
- 5 µL of “reverse” primer oligonucleotide
- 1 µL of Expand HF enzyme
- 24.
- Split the reaction into two PCR tubes, 50 µL per tube.
- 25.
- Perform PCR with the following parameters:
- Initial denaturation: 2 min, 95 °C
- 20 * cycles:
- ○
- Denaturation: 30 s, 95 °C
- ○
- Annealing: 30 s, 55 °C *
- ○
- Elongation: 1 min *, 72 °C
- Final elongation: 7 min, 72 °C
- Parameters marked with an asterisk (*) (annealing temperature, elongation time, and number of cycles) are recommended for all PCR enzymes. Other parameters may vary.
- 26.
- Ethanol precipitate PCR product (recommended for short templates, longer templates can be concentrated using a PCR purification kit (such as Qiagen):
- Combine all PCR reactions into one 1.7 mL PCR tube.
- Add 3× volume of 100% ethanol (300 µL for two PCR reactions).
- Add 1/10th volume of 3 M Sodium Acetate (10 µL for two PCR reactions).
- OPTIONAL STEP: Incubate at −20 °C for 30 min or more (freezing DNA facilitates precipitation from ethanol but is not required).
- Centrifuge at maximum speed in a room temperature benchtop microfuge for 10 min, aspirate and discard supernatant while being careful not to disturb the clear/white DNA pellet.
- Wash pellet with 1 mL of 70% ethanol.
- Centrifuge at maximum speed in a room temperature benchtop microfuge for 5 min, aspirate supernatant.
- Repeat steps vi and vii, for two washes in total.
- Let pellet air dry for 15 min or until no ethanol remains in the tube.
- Resuspend pellet in 12 µL of filtered water: 10 µL for transformation reaction (below), plus 2 µL to run on an agarose gel.
- 27.
- Dilute 2 µL of resuspended PCR product in 8 µL of filtered water and 2 µL of 6× DNA loading dye.
- 28.
- Confirm that the PCR product is the correct size by visualizing on a 1% agarose/TAE gel containing 1× SYBR Safe DNA stain (or equivalent). Include 6 µL of 100 bp ladder).
- 29.
- Image the gel on a UV transilluminator. If the PCR product is brighter than the equivalently sized band of the 100-bp ladder, there is an adequate amount of PCR product. If the PCR band is noticeably dimmer, repeat PCR with more reactions. If the band is very bright (>10× the intensity of the ladder), consider diluting the remaining DNA with additional water or using less for each yeast transformation.
 PAUSE STEP: the HDR template DNA can be stored for many weeks at −20 °C.
PAUSE STEP: the HDR template DNA can be stored for many weeks at −20 °C.3.4. Transform Plasmid and HDR Template DNA into Desired Yeast Strain
- 30.
- The night before the transformation, use a wooden stick to inoculate 2–5 mL of liquid YPD media (It is not necessary to use YPD media. Other media, such as SC with amino acid dropout selections, can be used if necessary. Take care to properly prepare the media when G418 is necessary for plasmid selection. For example, ammonium sulfate present in SC media should be replaced with 1 mg/mL of monosodium glutamate) in a glass culture tube with a colony of the desired yeast strain. Grow overnight at 30 °C on a rotator.
- 31.
- The next morning, measure the OD600 on a spectrophotometer and dilute the culture to an OD600 of 0.2 in at least 5 mL of YPD. Grow diluted culture at 30 °C on a rotator.
- 32.
- Allow the culture to go through at least 2–3 more cell divisions, until it reaches mid-log phase (OD600 0.8–1.0).
- 33.
- In two 1.7 mL microcentrifuge tubes (one tube for the transformation, and one as a negative control), harvest 2 OD (~3 × 107) cells.
- 34.
- Centrifuge the tubes for 1 min at 13,000 RPM in a benchtop microcentrifuge at room temperature. Aspirate off and discard the supernatant.
- 35.
- Resuspend each pellet in 1 mL of yeast transformation buffer.
- 36.
- Vortex tubes at max speed for 30 s.
- 37.
- Centrifuge the tubes for 1 min at 13,000 RPM in a benchtop microcentrifuge at room temperature. Aspirate off and discard the supernatant.
- 38.
- Repeat steps 24–27.
- 39.
- Resuspend cell pellet in 100 µL yeast transformation buffer.
- 40.
- To each tube, add 10 µL of single-stranded DNA from salmon testes.
- 41.
- Add nothing else to the negative control tube. To the other add:
- 100–500 ng of cloned plasmid encoding Cas9 and correct gRNA (1 µL of miniprepped plasmid DNA is sufficient).
- 5–10 µL of prepared HDR template DNA PCR product, depending on brightness of visualized band.
- 42.
- Incubate tubes on the benchtop for 10 min.
- 43.
- Add 270 µL of 40% PEG solution to each tube, pipette to mix.
- 44.
- Place tubes in 30 °C incubator for 30 min (no shaking or rotating necessary).
- 45.
- Add 50 µL DMSO to each tube.
- 46.
- Heat shock cells for 15 min at 42 °C, in either a heat block or water bath.
- 47.
- Centrifuge cells for 1 min at 13,000 RPM in a benchtop microfuge, aspirate off and discard the supernatant.
- 48.
- Wash pellets once with 1 mL of sterile filtered water, centrifuge at 13,000 RPM in a benchtop microfuge. Aspirate off and discard the supernatant.
- 49.
- Resuspend cell pellets in 250 µL of YPD media.
- 50.
- Allow cells to recover for 2 h at 30 °C with rotating.
- 51.
- Spread each 250 µL culture on a YPD plate containing 400 µg/mL G418, allow to grow for 2–3 days at 30 °C for colonies to form. After this time, confirm that the negative control plate has no colonies and discard it.
3.5. Isolate New Strain and Confirm Gene Editing Success
3.5.1. Select for Cells That Have Lost the Cas9-Expression Plasmid to Allow for Future Re-Use of the Marker
- 52.
- Using a sterile wood stick, pick 12 individual colonies (isolates) from the transformation plate and re-streak (for single colonies) onto YPD plates.
- 53.
- Grow plates for 2 days at 30 °C until single colonies form.
- 54.
- From the YPD plates, pick one colony from each of the 12 isolates, and re-streak again onto:
- YPD plates, for single colonies
- G418 plates (all isolates can be plated as patches on a single plate, as individual colonies are not necessary) to check for plasmid loss. Yeast that has lost the pCASB plasmid should now not grow on G418-containing media.
- 55.
- Grow plates for 2 days at 30 °C.
- 56.
- Identify isolates which did not grow on G418.
- 57.
- From the corresponding isolate on the YPD plate, use a wood stick to choose a single colony. Use the same stick to both inoculate a fresh YPD plate and a glass culture tube containing 2 mL of YPD liquid media. Streak each isolate on the fresh YPD plates for single colonies.
- 58.
- Grow plates for 2 days at 30 °C and liquid cultures overnight at 30 °C on a rotator.
3.5.2. Isolate Genomic DNA from Transformants (Can Be Done by Any Method, Such as a Genomic DNA Isolation Kit. Below Are Instructions for a Phenol-Chloroform Extraction)
- 59.
- Harvest 1–1.5 mL of a saturated overnight yeast culture in a 1.7 mL microcentrifuge tube by centrifugation 1 min at 13,000 RPM in a benchtop microfuge.
- 60.
- Discard supernatant, resuspend cell pellet in 1 mL of filtered water.
- 61.
- Centrifuge cells 1 min at 13,000 RPM in a benchtop microfuge, aspirate off and discard supernatant.
- 62.
- To each tube, add 200 µL of the following:
- 1× TE buffer
- Yeast genomic prep buffer
- 0.5 mm zirconia/silica beads
- Phenol:chloroform:isoamyl alcohol
- 63.
- Vortex for 5 min at 4 °C.
- 64.
- Centrifuge 10 min at 13,000 RPM in a benchtop microfuge.
- 65.
- Tubes will have visible layers; transfer the top layer (should be 250–300 µL) to a fresh 1.7 mL tube. Discard other layers.
- 66.
- Precipitate DNA from the top layer by adding 750 µL (~3× volume) of 100% ethanol and 25 µL (1/10th volume) of 3M sodium acetate. Mix tubes by inverting several times.
- 67.
- OPTIONAL STEP: Freeze tubes at −20 °C for 30 min to facilitate precipitation.
- 68.
- Centrifuge cells at 13,000 RPM in a benchtop microfuge to pellet DNA.
- 69.
- Wash DNA pellets as described in steps vi–ix of step 15.
- 70.
- Resuspend air-dried pellets in desired amount of water (~300 µL will yield a DNA concentration that can be used directly as a template in the subsequent PCR reaction for screening).
3.5.3. Confirm Gene Editing Success by PCR and Sequencing
- 71.
- Design PCR primers flanking the region of interest, to amplify a region of at least 250 bp around the site of the mutation.
- 72.
- Design a sequencing primer at least 100 bp upstream of the desired mutation (Sanger sequencing will begin >50 bp downstream of the sequencing primer).
- 73.
- Using any standard PCR polymerase enzyme (high-fidelity not necessary), amplify the desired site.
- 74.
- OPTIONAL STEP: Run a small portion of each PCR product (~4 µL of a 25 µL reaction) on a 1% agarose/TAE gel with 1 kb ladder to visualize a product of the correct size).
- 75.
- Use a PCR purification kit to isolate the PCR products
- 76.
- Send PCR product from each isolate for Sanger sequencing to confirm gene editing success.
- 77.
- After obtaining sequencing results and identifying successful mutants, the yeast strains are complete. If desired, make glycerol stocks and store at −80 °C (Glycerol stocks should be made of multiple isolates due to the chance of off-target effects. While off-target mutations as a result of CRISPR-Cas9 in yeast have been found to be rare [14], we recommend performing initial assays on three (or more, if desired) isolates to screen for uniform phenotypes):
- Using a wood stick, pick a single colony of each desired isolate and inoculate 2 mL of YPD liquid media in a glass culture tube.
- Grow overnight at 30 °C on a rotator.
- The next day, in a cryogenic tube, combine 800 μL of the saturated overnight culture with 200 μL of 80% glycerol. Store at −80 °C.
4. Expected Results and Troubleshooting
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gardner, J.M.; Jaspersen, S.L. Manipulating the yeast genome: Deletion, mutation, and tagging by PCR. Methods Mol. Biol. 2014, 1205, 45–78. [Google Scholar] [PubMed]
- Storici, F.; Durham, C.L.; Gordenin, D.A.; Resnick, M.A. Chromosomal site-specific double-strand breaks are efficiently targeted for repair by oligonucleotides in yeast. Proc. Natl. Acad. Sci. USA 2003, 100, 14994–14999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wach, A.; Brachat, A.; Pohlmann, R.; Philippsen, P. New heterologous modules for classical or PCR-based gene disruptions in Saccharomyces cerevisiae. Yeast 1994, 10, 1793–1808. [Google Scholar] [CrossRef] [PubMed]
- Siewers, V. An overview on selection marker genes for transformation of Saccharomyces cerevisiae. Methods Mol. Biol. 2014, 1152, 3–15. [Google Scholar]
- Goldstein, A.L.; Pan, X.; McCusker, J.H. Heterologous URA3MX cassettes for gene replacement in Saccharomyces cerevisiae. Yeast 1999, 15, 507–511. [Google Scholar] [CrossRef]
- Longtine, M.S.; McKenzie, A., 3rd; Demarini, D.J.; Shah, N.G.; Wach, A.; Brachat, A.; Philippsen, P.; Pringle, J.R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998, 14, 953–961. [Google Scholar] [CrossRef]
- Alani, E.; Cao, L.; Kleckner, N. A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics 1987, 116, 541–545. [Google Scholar] [CrossRef]
- Rothstein, R. Targeting, disruption, replacement, and allele rescue: Integrative DNA transformation in yeast. Methods Enzymol. 1991, 194, 281–301. [Google Scholar] [PubMed]
- Cormack, B.; Castano, I. Introduction of point mutations into cloned genes. Methods Enzymol. 2002, 350, 199–218. [Google Scholar]
- Toulmay, A.; Schneiter, R. A two-step method for the introduction of single or multiple defined point mutations into the genome of Saccharomyces cerevisiae. Yeast 2006, 23, 825–831. [Google Scholar] [CrossRef]
- DiCarlo, J.E.; Norville, J.E.; Mali, P.; Rios, X.; Aach, J.; Church, G.M. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013, 41, 4336–4343. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Gietz, R.D.; Schiestl, R.H. High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc. 2007, 2, 31–34. [Google Scholar] [CrossRef]
- Ryan, O.W.; Skerker, J.M.; Maurer, M.J.; Li, X.; Tsai, J.C.; Poddar, S.; Lee, M.E.; DeLoache, W.; Dueber, J.E.; Arkin, A.P.; et al. Selection of chromosomal DNA libraries using a multiplex CRISPR system. Elife 2014, 3, e03703. [Google Scholar] [CrossRef]
- Laughery, M.F.; Hunter, T.; Brown, A.; Hoopes, J.; Ostbye, T.; Shumaker, T.; Wyrick, J.J. New vectors for simple and streamlined CRISPR-Cas9 genome editing in Saccharomyces cerevisiae. Yeast 2015, 32, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Giersch, R.M.; Finnigan, G.C. Yeast Still a Beast: Diverse Applications of CRISPR/Cas Editing Technology in S. cerevisiae. Yale J. Biol. Med. 2017, 90, 643–651. [Google Scholar] [PubMed]
- Jessop-Fabre, M.M.; Jakociunas, T.; Stovicek, V.; Dai, Z.; Jensen, M.K.; Keasling, J.D.; Borodina, I. EasyClone-MarkerFree: A vector toolkit for marker-less integration of genes into Saccharomyces cerevisiae via CRISPR-Cas9. Biotechnol. J. 2016, 11, 1110–1117. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef] [PubMed]
- Edskes, H.K.; Kryndushkin, D.; Shewmaker, F.; Wickner, R.B. Prion Transfection of Yeast. Cold Spring Harb. Protoc. 2017, 2017, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Ryan, O.W.; Poddar, S.; Cate, J.H. CRISPR-Cas9 Genome Engineering in Saccharomyces cerevisiae Cells. Cold Spring Harb. Protoc. 2016, 2016, 525–533. [Google Scholar] [CrossRef]
- Engler, C.; Kandzia, R.; Marillonnet, S. A one pot, one step, precision cloning method with high throughput capability. PLoS ONE 2008, 3, e3647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhmetov, A.; Laurent, J.M.; Gollihar, J.; Gardner, E.C.; Garge, R.K.; Ellington, A.D.; Kachroo, A.H.; Marcotte, E.M. Single-step Precision Genome Editing in Yeast Using CRISPR-Cas9. Bio Protoc. 2018, 8, e2765. [Google Scholar] [CrossRef] [PubMed]
- Levi, O.; Arava, Y. Expanding the CRISPR/Cas9 Toolbox for Gene Engineering in S. cerevisiae. Curr. Microbiol. 2020, 77, 468–478. [Google Scholar] [CrossRef]
- Flick, J.S.; Johnston, M. Two systems of glucose repression of the GAL1 promoter in Saccharomyces cerevisiae. Mol. Cell. Biol. 1990, 10, 4757–4769. [Google Scholar]
- Heigwer, F.; Kerr, G.; Boutros, M. E-CRISP: Fast CRISPR target site identification. Nat. Methods 2014, 11, 122–123. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.D.; Scott, D.A.; Weinstein, J.A.; Ran, F.A.; Konermann, S.; Agarwala, V.; Li, Y.; Fine, E.J.; Wu, X.; Shalem, O.; et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013, 31, 827–832. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aguilar, R.R.; Shen, Z.-J.; Tyler, J.K. A Simple, Improved Method for Scarless Genome Editing of Budding Yeast Using CRISPR-Cas9. Methods Protoc. 2022, 5, 79. https://doi.org/10.3390/mps5050079
Aguilar RR, Shen Z-J, Tyler JK. A Simple, Improved Method for Scarless Genome Editing of Budding Yeast Using CRISPR-Cas9. Methods and Protocols. 2022; 5(5):79. https://doi.org/10.3390/mps5050079
Chicago/Turabian StyleAguilar, Rhiannon R., Zih-Jie Shen, and Jessica K. Tyler. 2022. "A Simple, Improved Method for Scarless Genome Editing of Budding Yeast Using CRISPR-Cas9" Methods and Protocols 5, no. 5: 79. https://doi.org/10.3390/mps5050079
APA StyleAguilar, R. R., Shen, Z.-J., & Tyler, J. K. (2022). A Simple, Improved Method for Scarless Genome Editing of Budding Yeast Using CRISPR-Cas9. Methods and Protocols, 5(5), 79. https://doi.org/10.3390/mps5050079