Cell-Type-Specific Quantification of a Scaffold-Based 3D Liver Co-Culture

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Cell Seeding
2.2. Cell Quantification by Optical Methods
2.2.1. Resazurin Conversion
2.2.2. DNA Isolation in 2D and 3D Scaffold Cultures
2.2.3. Absorption Measurement by Using LVIS Micro Drop Plate
2.2.4. Fluorescence-Based CyQuant Measurement
2.2.5. Fluorescence-Based Hoechst 33342 Measurement
2.3. Cell-Type-Specific DNA Quantification
2.3.1. Test of Different Primers for the Usability in a Species-Specific DNA Quantification Method
2.3.2. Conventional Semi-Quantitative PCR
2.3.3. Quantitative Real-Time PCR
2.4. Cell-Type-Specific Cell Labeling
Statistics
3. Results
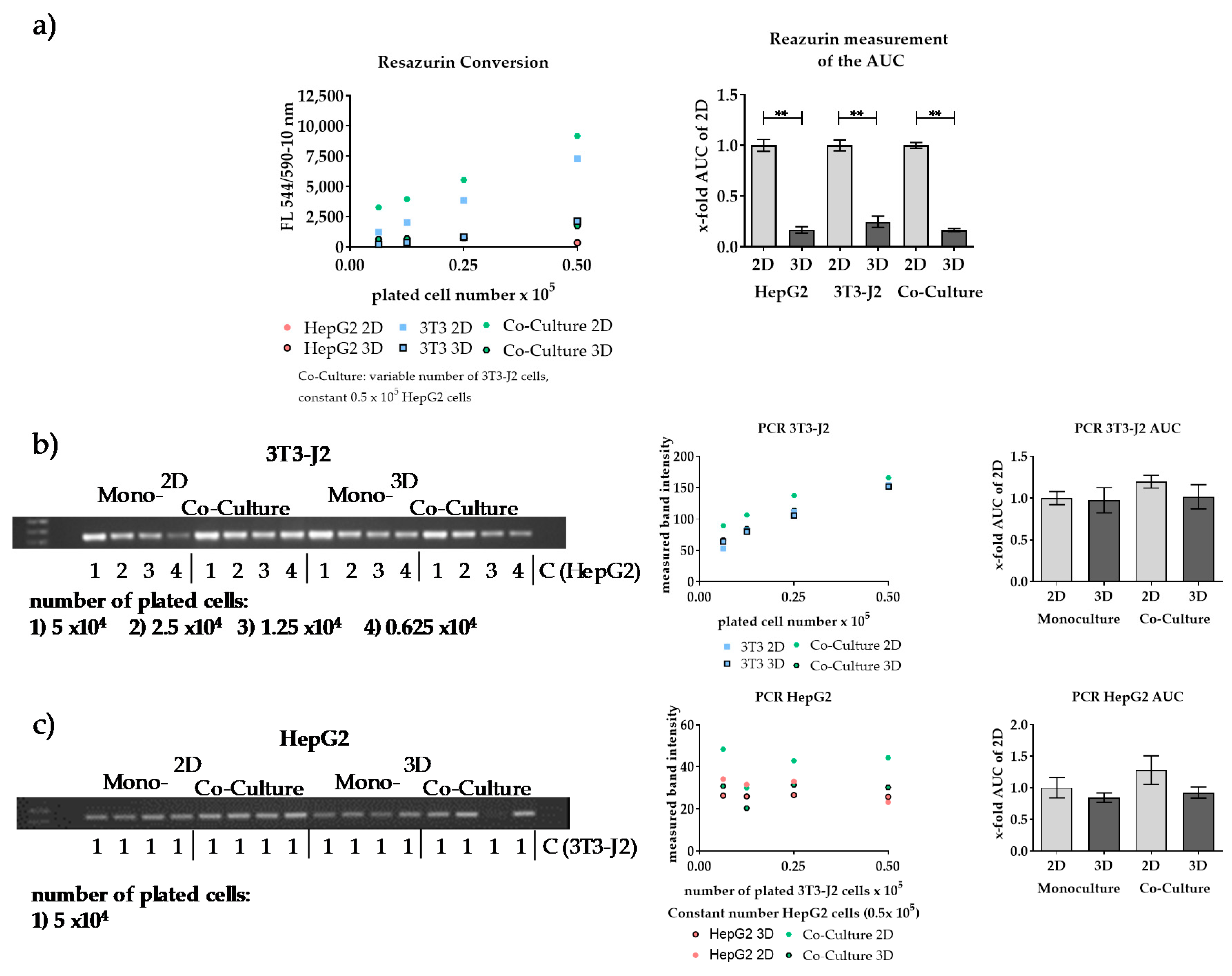
3.1. Morphological Differences of HepG2 and 3T3-J2 Cells Can Be Used with Restrictions for Quantification in 2D Co-Culture but Not in 3D Co-Culture
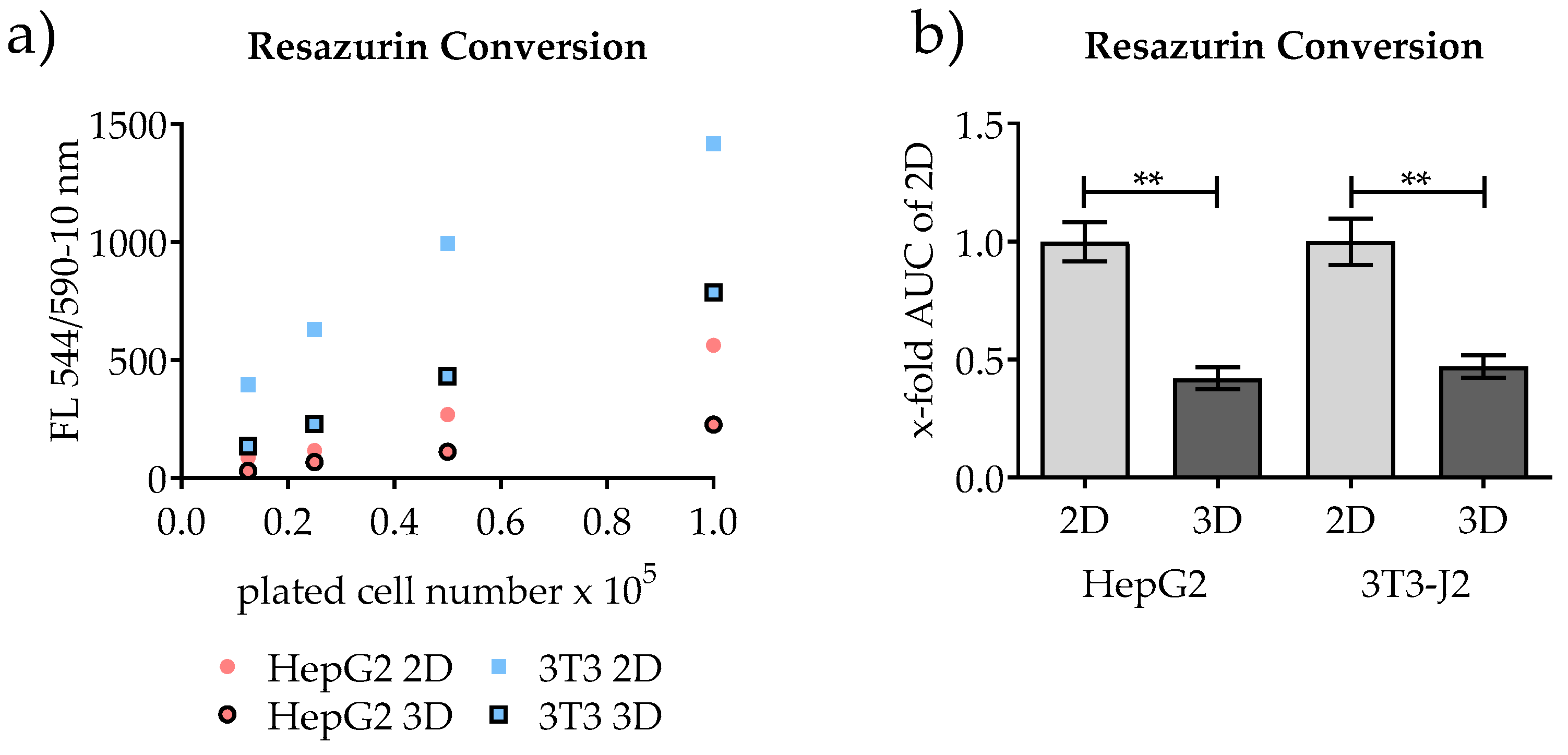
3.2. Quantification of Conventional 2D Culture and 3D Scaffold Culture by Measuring the Mitochondrial Activity
3.3. Comparison of Alternatives to Resazurin Conversion for the Quantification of Conventional 2D Culture and 3D Scaffold Culture
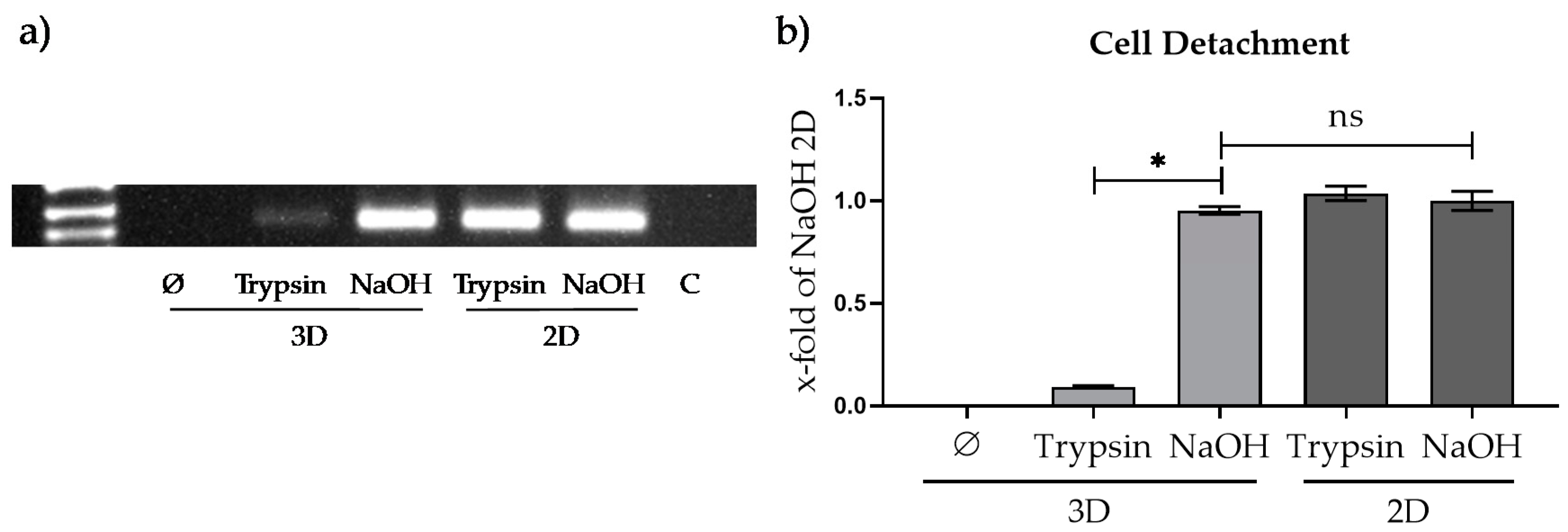
3.3.1. Comparison of Different Approaches for Cell Detachment in 2D and from the Optimaix-3D Scaffold
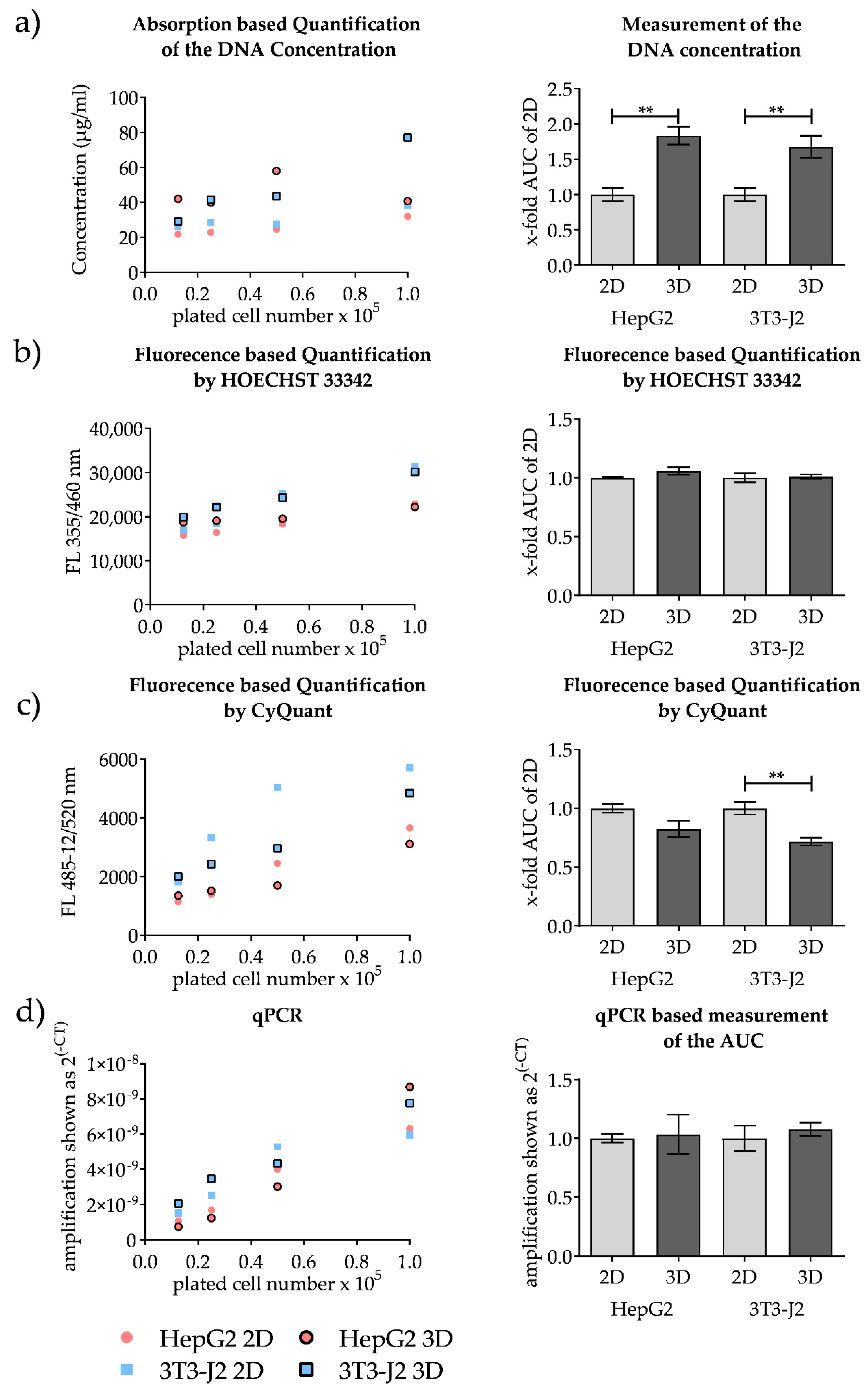
3.3.2. Comparison of Methods for the Quantification of Conventional 2D Culture and 3D Scaffold Culture
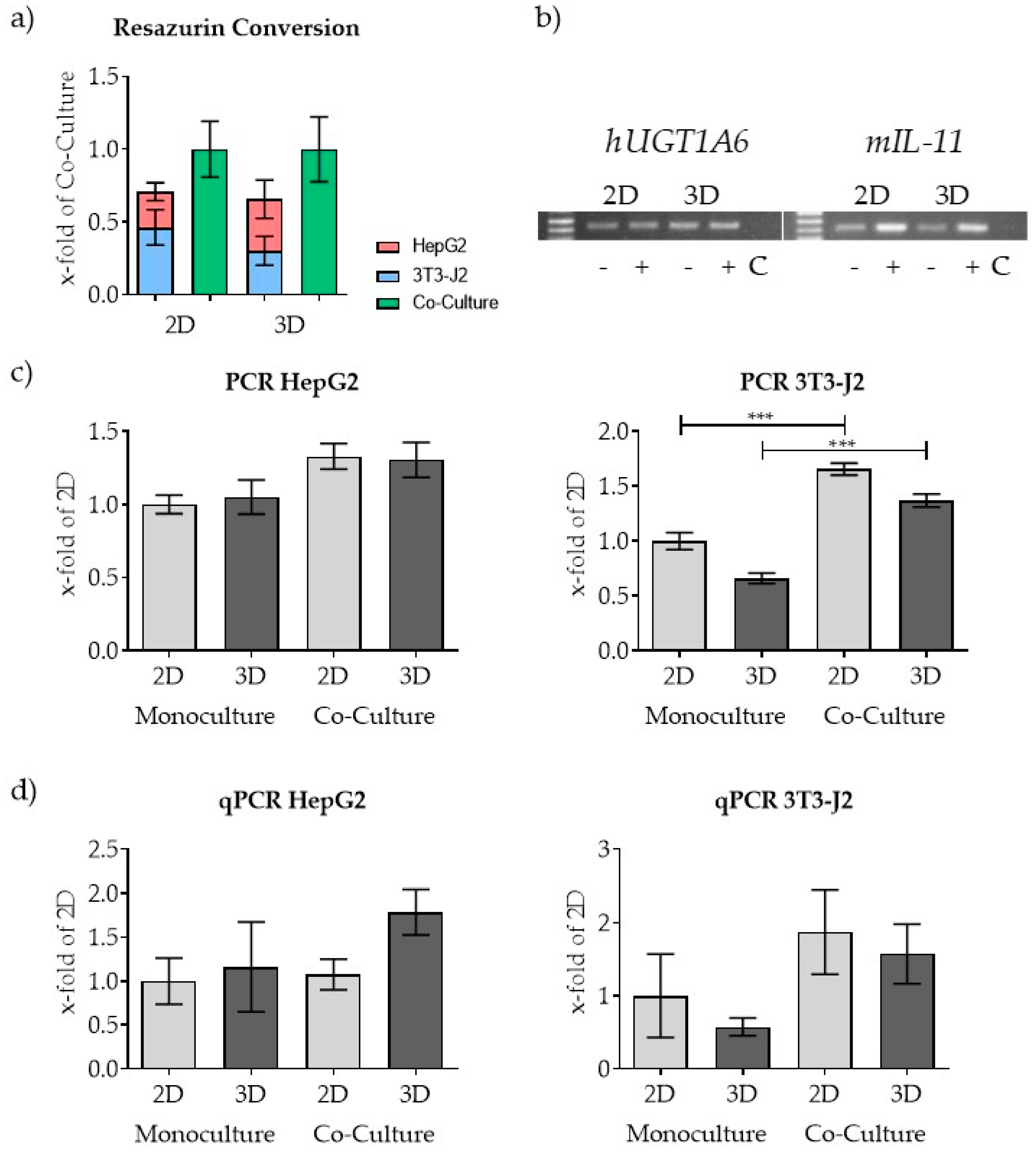
3.4. PCR-Based Co-Culture Quantification
4. Discussions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AUC | Area under the curve |
| BCS | Bovine calf serum |
| Ct | Cycle threshold |
| FCS | Fetal calf serum |
| HPV | Human papillomavirus |
| LOD | Limits of Detection |
| LOQ | Limits of Quantitation |
| MSCs | Mesenchymal stem cells |
| P/S | Penicillin-streptomycin |
| SRB | Sulforhodamine B |
References
- Godoy, P.; Hewitt, N.J.; Albrecht, U.; Andersen, M.E.; Ansari, N.; Bhattacharya, S.; Bode, J.G.; Bolleyn, J.; Borner, C.; Bottger, J.; et al. Recent advances in 2d and 3d in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and adme. Arch. Toxicol. 2013, 87, 1315–1530. [Google Scholar]
- Bachmann, A.; Moll, M.; Gottwald, E.; Nies, C.; Zantl, R.; Wagner, H.; Burkhardt, B.; Sánchez, J.; Ladurner, R.; Thasler, W.; et al. 3d cultivation techniques for primary human hepatocytes. Microarrays 2015, 4, 64–83. [Google Scholar] [CrossRef]
- Luckert, C.; Schulz, C.; Lehmann, N.; Thomas, M.; Hofmann, U.; Hammad, S.; Hengstler, J.G.; Braeuning, A.; Lampen, A.; Hessel, S. Comparative analysis of 3d culture methods on human hepg2 cells. Arch. Toxicol. 2017, 91, 393–406. [Google Scholar] [CrossRef]
- Ruoss, M.; Haussling, V.; Schugner, F.; Olde Damink, L.H.H.; Lee, S.M.L.; Ge, L.; Ehnert, S.; Nussler, A.K. A standardized collagen-based scaffold improves human hepatocyte shipment and allows metabolic studies over 10 days. Bioengineering 2018, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Schyschka, L.; Sanchez, J.J.; Wang, Z.; Burkhardt, B.; Muller-Vieira, U.; Zeilinger, K.; Bachmann, A.; Nadalin, S.; Damm, G.; Nussler, A.K. Hepatic 3d cultures but not 2d cultures preserve specific transporter activity for acetaminophen-induced hepatotoxicity. Arch. Toxicol. 2013, 87, 1581–1593. [Google Scholar] [CrossRef]
- Kumari, J.; Karande, A.A.; Kumar, A. Combined effect of cryogel matrix and temperature-reversible soluble–insoluble polymer for the development of in vitro human liver tissue. ACS Appl. Mater. Interfaces 2015, 8, 264–277. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.J.; Gray, M.; Yue, S.T.; Haugland, R.P.; Singer, V.L. Sensitive determination of cell number using the cyquant® cell proliferation assay. J. Immunol. Methods 2001, 254, 85–98. [Google Scholar] [CrossRef]
- Bell, C.C.; Hendriks, D.F.G.; Moro, S.M.L.; Ellis, E.; Walsh, J.; Renblom, A.; Puigvert, L.F.; Dankers, A.C.A.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspera-Werz, R.H.; Ehnert, S.; Heid, D.; Zhu, S.; Chen, T.; Braun, B.; Sreekumar, V.; Arnscheidt, C.; Nussler, A.K. Nicotine and cotinine inhibit catalase and glutathione reductase activity contributing to the impaired osteogenesis of scp-1 cells exposed to cigarette smoke. Oxidative Med. Cell. Longev. 2018, 2018, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, C.J.; Harman, M.W.; Centuori, S.M.; Wolgemuth, C.W.; Martinez, J.D. Measuring DNA content in live cells by fluorescence microscopy. Cell Div. 2018, 13, 6. [Google Scholar] [CrossRef]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar] [PubMed]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Van Tonder, A.; Joubert, A.M.; Cromarty, A.D. Limitations of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2h-tetrazolium bromide (mtt) assay when compared to three commonly used cell enumeration assays. BMC Res. Notes 2015, 8, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreekumar, V.; Aspera-Werz, R.; Ehnert, S.; Strobel, J.; Tendulkar, G.; Heid, D.; Schreiner, A.; Arnscheidt, C.; Nussler, A.K. Resveratrol protects primary cilia integrity of human mesenchymal stem cells from cigarette smoke to improve osteogenic differentiation in vitro. Arch. Toxicol. 2018, 92, 1525–1538. [Google Scholar] [CrossRef] [PubMed]
- Aslantürk, Ö.S. In vitro cytotoxicity and cell viability assays: Principles, advantages, and disadvantages. In Genotoxicity-A Predictable Risk to Our Actual World; IntechOpen: Rijeka, Croatia, 2017. [Google Scholar]
- Huyck, L.; Ampe, C.; Van Troys, M. The xtt cell proliferation assay applied to cell layers embedded in three-dimensional matrix. Assay Drug Dev. Technol. 2012, 10, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampersad, S.N. Multiple applications of alamar blue as an indicator of metabolic function and cellular health in cell viability bioassays. Sensors 2012, 12, 12347–12360. [Google Scholar] [CrossRef]
- Allen, M.; Millett, P.; Dawes, E.; Rushton, N. Lactate dehydrogenase activity as a rapid and sensitive test for the quantification of cell numbers in vitro. Clin. Mater. 1994, 16, 189–194. [Google Scholar] [CrossRef]
- Ruoß, M.; Damm, G.; Vosough, M.; Ehret, L.; Grom-Baumgarten, C.; Petkov, M.; Naddalin, S.; Ladurner, R.; Seehofer, D.; Nussler, A.; et al. Epigenetic modifications of the liver tumor cell line hepg2 increase their drug metabolic capacity. Int. J. Mol. Sci. 2019, 20, 347. [Google Scholar] [CrossRef] [Green Version]
- Uzarski, J.S.; DiVito, M.D.; Wertheim, J.A.; Miller, W.M. Essential design considerations for the resazurin reduction assay to noninvasively quantify cell expansion within perfused extracellular matrix scaffolds. Biomaterials 2017, 129, 163–175. [Google Scholar] [CrossRef]
- Rai, Y.; Pathak, R.; Kumari, N.; Sah, D.K.; Pandey, S.; Kalra, N.; Soni, R.; Dwarakanath, B.S.; Bhatt, A.N. Mitochondrial biogenesis and metabolic hyperactivation limits the application of mtt assay in the estimation of radiation induced growth inhibition. Sci. Rep. 2018, 8, 1531. [Google Scholar] [CrossRef] [Green Version]
- Surmaitis, R.L.; Arias, C.J.; Schlenoff, J.B. Stressful surfaces: Cell metabolism on a poorly adhesive substrate. Langmuir 2018, 34, 3119–3125. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Sandrin, L. Liver stiffness: A novel parameter for the diagnosis of liver disease. Hepatic Med. 2010, 2, 49–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skardal, A.; Mack, D.; Atala, A.; Soker, S. Substrate elasticity controls cell proliferation, surface marker expression and motile phenotype in amniotic fluid-derived stem cells. J. Mech. Behav. Biomed. Mater. 2013, 17, 307–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, T.S.; Yu, H.; Moore, A.; Khetani, S.R.; Tweedie, D. Meeting the challenge of predicting hepatic clearance of compounds slowly metabolized by cytochrome p450 using a novel hepatocyte model, hepatopac. Drug Metab. Dispos. 2013, 41, 2024–2032. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, D.; Hussain, A.; Yip, D.; Parekh, A.; Shrirao, A.; Cho, C.H. Long-term liver-specific functions of hepatocytes in electrospun chitosan nanofiber scaffolds coated with fibronectin. J. Biomed. Mater. Res. Part A 2017, 105, 2119–2128. [Google Scholar] [CrossRef]
- Cho, C.H.; Berthiaume, F.; Tilles, A.W.; Yarmush, M.L. A new technique for primary hepatocyte expansion in vitro. Biotechnol. Bioeng. 2008, 101, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Schyschka, L.; Muhl-Benninghaus, R.; Neumann, J.; Hao, L.; Nussler, N.; Dooley, S.; Liu, L.; Stockle, U.; Nussler, A.K.; et al. Comparative analysis of phase i and ii enzyme activities in 5 hepatic cell lines identifies huh-7 and hcc-t cells with the highest potential to study drug metabolism. Arch. Toxicol. 2012, 86, 87–95. [Google Scholar] [CrossRef]
- Meeker, N.D.; Hutchinson, S.A.; Ho, L.; Trede, N.S. Method for isolation of pcr-ready genomic DNA from zebrafish tissues. Biotechniques 2007, 43, 610–614. [Google Scholar] [CrossRef]
- Richards, W.L.; Song, M.K.; Krutzsch, H.; Evarts, R.P.; Marsden, E.; Thorgeirsson, S.S. Measurement of cell-proliferation in microculture using hoechst-33342 for the rapid semiautomated microfluorimetric determination of chromatin DNA. Exp. Cell Res. 1985, 159, 235–246. [Google Scholar] [CrossRef]
- Logan, D.J.; Shan, J.; Bhatia, S.N.; Carpenter, A.E. Quantifying co-cultured cell phenotypes in high-throughput using pixel-based classification. Methods 2016, 96, 6–11. [Google Scholar] [CrossRef] [Green Version]
- Krtolica, A.; Ortiz de Solorzano, C.; Lockett, S.; Campisi, J. Quantification of epithelial cells in coculture with fibroblasts by fluorescence image analysis. Cytometry 2002, 49, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Asthana, V.; Tang, Y.; Ferguson, A.; Bugga, P.; Asthana, A.; Evans, E.R.; Chen, A.L.; Stern, B.S.; Drezek, R.A. An inexpensive, customizable microscopy system for the automated quantification and characterization of multiple adherent cell types. PeerJ 2018, 6, e4937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, G.; Wang, J.; Lv, Q.; Liu, M.; Xu, H.; Zhang, H.; Jin, L.; Yu, J.; Wang, X. Three-dimensional coculture of primary hepatocytes and stellate cells in silk scaffold improves hepatic morphology and functionality in vitro. J. Biomed. Mater. Res. Part A 2018, 106, 2171–2180. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.F.; Chua, K.N.; Zhang, P.C.; Lim, W.S.; Ramakrishna, S.; Leong, K.W.; Mao, H.Q. Three-dimensional co-culture of rat hepatocyte spheroids and nih/3t3 fibroblasts enhances hepatocyte functional maintenance. Acta Biomater. 2005, 1, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Glicklis, R.; Shapiro, L.; Agbaria, R.; Merchuk, J.C.; Cohen, S. Hepatocyte behavior within three-dimensional porous alginate scaffolds. Biotechnol. Bioeng. 2000, 67, 344–353. [Google Scholar] [CrossRef]
- Antoni, D.; Burckel, H.; Josset, E.; Noel, G. Three-dimensional cell culture: A breakthrough in vivo. Int. J. Mol. Sci. 2015, 16, 5517–5527. [Google Scholar] [CrossRef]
- Bonnier, F.; Keating, M.E.; Wrobel, T.P.; Majzner, K.; Baranska, M.; Garcia-Munoz, A.; Blanco, A.; Byrne, H.J. Cell viability assessment using the alamar blue assay: A comparison of 2d and 3d cell culture models. Toxicol. In Vitro 2015, 29, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Kepp, O.; Galluzzi, L.; Lipinski, M.; Yuan, J.Y.; Kroemer, G. Cell death assays for drug discovery. Nat. Rev. Drug Discov. 2011, 10, 221–237. [Google Scholar] [CrossRef]
- Koh, B.; Jeon, H.; Kim, D.; Kang, D.; Kim, K.R. Effect of fibroblast co-culture on the proliferation, viability and drug response of colon cancer cells. Oncol. Lett. 2019, 17, 2409–2417. [Google Scholar] [CrossRef] [Green Version]
- Breslin, S.; O’Driscoll, L. The relevance of using 3d cell cultures, in addition to 2d monolayer cultures, when evaluating breast cancer drug sensitivity and resistance. Oncotarget 2016, 7, 45745–45756. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.X.; Xiao, G.Y.; Wang, X.; Dong, Z.G.; Ma, Z.Y.; Li, L.; Li, Y.H.; Pan, X.; Nie, L. Biocompatibility and osteogenesis of calcium phosphate composite scaffolds containing simvastatin-loaded plga microspheres for bone tissue engineering. J. Biomed. Mater. Res. Part A 2015, 103, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.X.; Wang, B.; Zhao, Y.N.; Xiao, Z.F.; Li, J.; Cui, Y.; Han, S.F.; Wei, J.S.; Chen, B.; Han, J.; et al. Collagen scaffold microenvironments modulate cell lineage commitment for differentiation of bone marrow cells into regulatory dendritic cells. Sci. Rep. 2017, 7, 42049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, D.; Yu, S.C.; Ping, Y.F.; Wu, H.; Zhao, X.; Zhang, H.; Cui, Y.; Chen, B.; Zhang, X.; Dai, J.; et al. A three-dimensional collagen scaffold cell culture system for screening anti-glioma therapeutics. Oncotarget 2016, 7, 56904–56914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaibani, M.; Luni, C.; Sbalchiero, E.; Elvassore, N. Flow cytometric cell cycle analysis of muscle precursor cells cultured within 3d scaffolds in a perfusion bioreactor. Biotechnol. Prog. 2009, 25, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Bigdeli, N.; Karlsson, C.; Strehl, R.; Concaro, S.; Hyllner, J.; Lindahl, A. Coculture of human embryonic stem cells and human articular chondrocytes results in significantly altered phenotype and improved chondrogenic differentiation. Stem Cells 2009, 27, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Prins, H.J.; Helder, M.N.; van Blitterswijk, C.A.; Karperien, M. Trophic effects of mesenchymal stem cells in chondrocyte co-cultures are independent of culture conditions and cell sources. Tissue Eng. Part A 2012, 18, 1542–1551. [Google Scholar] [CrossRef]
- Ren, D.; Madsen, J.S.; de la Cruz-Perera, C.I.; Bergmark, L.; Sorensen, S.J.; Burmolle, M. High-throughput screening of multispecies biofilm formation and quantitative pcr-based assessment of individual species proportions, useful for exploring interspecific bacterial interactions. Microb. Ecol. 2014, 68, 146–154. [Google Scholar] [CrossRef]
- Simmons, J.G.; Pucilowska, J.B.; Lund, P.K. Autocrine and paracrine actions of intestinal fibroblast-derived insulin-like growth factors. Am. J. Physiol. 1999, 276, G817–G827. [Google Scholar] [CrossRef]
- Fasolino, I.; Guarino, V.; Marrese, M.; Cirillo, V.; Vallifuoco, M.; Tamma, M.L.; Vassallo, V.; Bracco, A.; Calise, F.; Ambrosio, L. Hepg2 and human healthy hepatocyte in vitro culture and co-culture in pcl electrospun platforms. Biomed. Mater. 2017, 13, 015017. [Google Scholar] [CrossRef] [Green Version]
- O’Callaghan, N.J.; Fenech, M. A quantitative pcr method for measuring absolute telomere length. Biol. Proced. Online 2011, 13, 3. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Pan, X.; Kalmbach, K.; Seth-Smith, M.L.; Ye, X.; Antumes, D.M.; Yin, Y.; Liu, L.; Keefe, D.L.; Weissman, S.M. Robust measurement of telomere length in single cells. Proc. Natl. Acad. Sci. USA 2013, 110, E1906–E1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.V.; Cox, C.L.; Eirew, P.; Knapp, D.J.; Pellacani, D.; Kannan, N.; Carles, A.; Moksa, M.; Balani, S.; Shah, S.; et al. DNA barcoding reveals diverse growth kinetics of human breast tumour subclones in serially passaged xenografts. Nat. Commun. 2014, 5, 5871. [Google Scholar] [CrossRef] [Green Version]
- Nolan-Stevaux, O.; Tedesco, D.; Ragan, S.; Makhanov, M.; Chenchik, A.; Ruefli-Brasse, A.; Quon, K.; Kassner, P.D. Measurement of cancer cell growth heterogeneity through lentiviral barcoding identifies clonal dominance as a characteristic of in vivo tumor engraftment. PLoS ONE 2013, 8, e67316. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.V.; Makarem, M.; Carles, A.; Moksa, M.; Kannan, N.; Pandoh, P.; Eirew, P.; Osako, T.; Kardel, M.; Cheung, A.M.; et al. Clonal analysis via barcoding reveals diverse growth and differentiation of transplanted mouse and human mammary stem cells. Cell Stem Cell 2014, 14, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuruga, Y.; Kiyono, T.; Matsushita, M.; Takahashi, T.; Kasai, H.; Matsumoto, S.; Todo, S. Establishment of immortalized human hepatocytes by introduction of hpv16 e6/e7 and htert as cell sources for liver cell-based therapy. Cell Transplant. 2008, 17, 1083–1094. [Google Scholar] [CrossRef]
- Truong, A.S.; Lochbaum, C.A.; Boyce, M.W.; Lockett, M.R. Tracking the invasion of small numbers of cells in paper-based assays with quantitative pcr. Anal. Chem. 2015, 87, 11263–11270. [Google Scholar] [CrossRef]
- Gaskell, H.; Sharma, P.; Colley, H.E.; Murdoch, C.; Williams, D.P.; Webb, S.D. Characterization of a functional c3a liver spheroid model. Toxicol. Res. 2016, 5, 1053–1065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stecklum, M.; Wulf-Goldenberg, A.; Purfürst, B.; Siegert, A.; Keil, M.; Eckert, K.; Fichtner, I. Cell differentiation mediated by co-culture of human umbilical cord blood stem cells with murine hepatic cells. In Vitro Cell. Dev. Biol. Anim. 2015, 51, 183–191. [Google Scholar] [CrossRef]
- Alajbeg, I.; Alic, I.; Andabak-Rogulj, A.; Brailo, V.; Mitrecic, D. Human- and mouse-derived neurons can be simultaneously obtained by co-cultures of human oral mucosal stem cells and mouse neural stem cells. Oral Dis. 2018, 24, 5–10. [Google Scholar] [CrossRef]
- Schröder, H.C.; Wang, X.H.; Wiens, M.; Diehl-Seifert, B.; Kropf, K.; Schloßmacher, U.; Müller, W.E.G. Silicate modulates the cross-talk between osteoblasts (saos-2) and osteoclasts (raw 264.7 cells): Inhibition of osteoclast growth and differentiation. J. Cell. Biochem. 2012, 113, 3197–3206. [Google Scholar] [CrossRef]
- Hendriks, J.A.A.; Miclea, R.L.; Schotel, R.; de Bruijn, E.; Moroni, L.; Karperien, M.; Riesle, J.; van Blitterswijk, C.A. Primary chondrocytes enhance cartilage tissue formation upon co-culture with a range of cell types. Soft Matter 2010, 6, 5080–5088. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondrial Activity (Resazurin, MTT, or XTT) | ATP Measurement | LDH Measurement | DNA Staining (CyQuant) | Protein Staining (SRB) | Protein Quantification (Lowry) | |
|---|---|---|---|---|---|---|
| Assay principle | Measurement of mitochondrial dehydrogenase activity | Measurement of total ATP levels | Measurement of released LDH | Staining of total DNA | Staining of total protein | Measurement of soluble protein |
| Advantages | Wide range of applications, distinction of dead and living cells is possible | Very sensitive assay, distinction of dead and living cells possible, not affected by stress level of the cells | Very sensitive assay, not affected by stress level of the cells | Very sensitive assay, not affected by stress level of the cells | Favorable and stable assay for quantification of adherent cells, not affected by stress level of the cells | Used for adherent and suspension cells |
| Disadvantages | Affected by stress level of the cells | Lysis of cells necessary | Lysis of cells necessary for normalization, assay is susceptible to interference (e.g., by FCS) | Lysis of cells necessary, assay is susceptible to interference (e.g., by phenol red of the medium) | Cannot be used for quantification in 3D culture | Lysis of cells necessary, assay is susceptible to interference (e.g., by FCS or scaffold ingredients) |
| No distinction between the different cell types in co-culture possible | ||||||
| References | [4,6,16,17] | [8] | [18] | [7] | [12,13,19] | [11] |
| Gene | mIL-11 | hUGT1A6 |
|---|---|---|
| Forward-Sequence 5′-3′ | TGCTGACAAGGCTTCGAGTAG | TGGTGCCTGAAGTTAATTTGCT |
| Reverse-Sequence 5′-3′ | ACATCAAGAGCTGTAAACGGC | GCTCTGGCAGTTGATGAAGTA |
| Amplicon in bp | 156 | 209 |
| Annealing Temperature in °C | 62 | 62 |
| Cycle Number | 30 | 30 |
| Reference Sequence | NC_000073.6 | NC_000002.12 |
| Method | Number of Cells | |||
|---|---|---|---|---|
| Cell Line | LOD | LOQ | Sensitivity (%) | |
| Absorption-based quantification | HepG2 | 2183 | 7277 | 95 |
| 3T3-J2 | 2557 | 8523 | 98 | |
| Fluorescence-based quantification (HOECHST 33342) | HepG2 | 5291 | 17,635 | 88 |
| 3T3-J2 | 3400 | 11,334 | 98 | |
| Fluorescence-based quantification (CyQuant) | HepG2 | 1506 | 5018 | 104 |
| 3T3-J2 | 471 | 1571 | 101 | |
| qPCR-based quantification | HepG2 | 1742 | 5808 | 99 |
| 3T3-J2 | 1447 | 4824 | 99 | |
| Organ System | Cell Type I | Cell Type II | Ref |
|---|---|---|---|
| Liver (HEPATOPAC®) | Primary human hepatocytes | m3T3-J2 | [25] |
| Liver | HepG2 | m3T3-J2 | [26] |
| Liver | primary rat hepatocytes | m3T3-J2 | [27] |
| Liver | human cord blood stem cells | hepatic alpha mouse liver 12 cells | [59] |
| Nervous system | human oral mucosal stem cells | mouse neural stem cells | [60] |
| Bone | SaOS2 | RAW 264.7 cells | [61] |
| Cartilage | bovine primary chondrocytes | Three different cell lines from mouse/human | [62] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruoß, M.; Kieber, V.; Rebholz, S.; Linnemann, C.; Rinderknecht, H.; Häussling, V.; Häcker, M.; Olde Damink, L.H.H.; Ehnert, S.; Nussler, A.K. Cell-Type-Specific Quantification of a Scaffold-Based 3D Liver Co-Culture. Methods Protoc. 2020, 3, 1. https://doi.org/10.3390/mps3010001
Ruoß M, Kieber V, Rebholz S, Linnemann C, Rinderknecht H, Häussling V, Häcker M, Olde Damink LHH, Ehnert S, Nussler AK. Cell-Type-Specific Quantification of a Scaffold-Based 3D Liver Co-Culture. Methods and Protocols. 2020; 3(1):1. https://doi.org/10.3390/mps3010001
Chicago/Turabian StyleRuoß, Marc, Vanessa Kieber, Silas Rebholz, Caren Linnemann, Helen Rinderknecht, Victor Häussling, Marina Häcker, Leon H. H. Olde Damink, Sabrina Ehnert, and Andreas K. Nussler. 2020. "Cell-Type-Specific Quantification of a Scaffold-Based 3D Liver Co-Culture" Methods and Protocols 3, no. 1: 1. https://doi.org/10.3390/mps3010001
APA StyleRuoß, M., Kieber, V., Rebholz, S., Linnemann, C., Rinderknecht, H., Häussling, V., Häcker, M., Olde Damink, L. H. H., Ehnert, S., & Nussler, A. K. (2020). Cell-Type-Specific Quantification of a Scaffold-Based 3D Liver Co-Culture. Methods and Protocols, 3(1), 1. https://doi.org/10.3390/mps3010001