An Assay to Study Intra-Chromosomal Deletions in Yeast

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Design, Methods and Materials
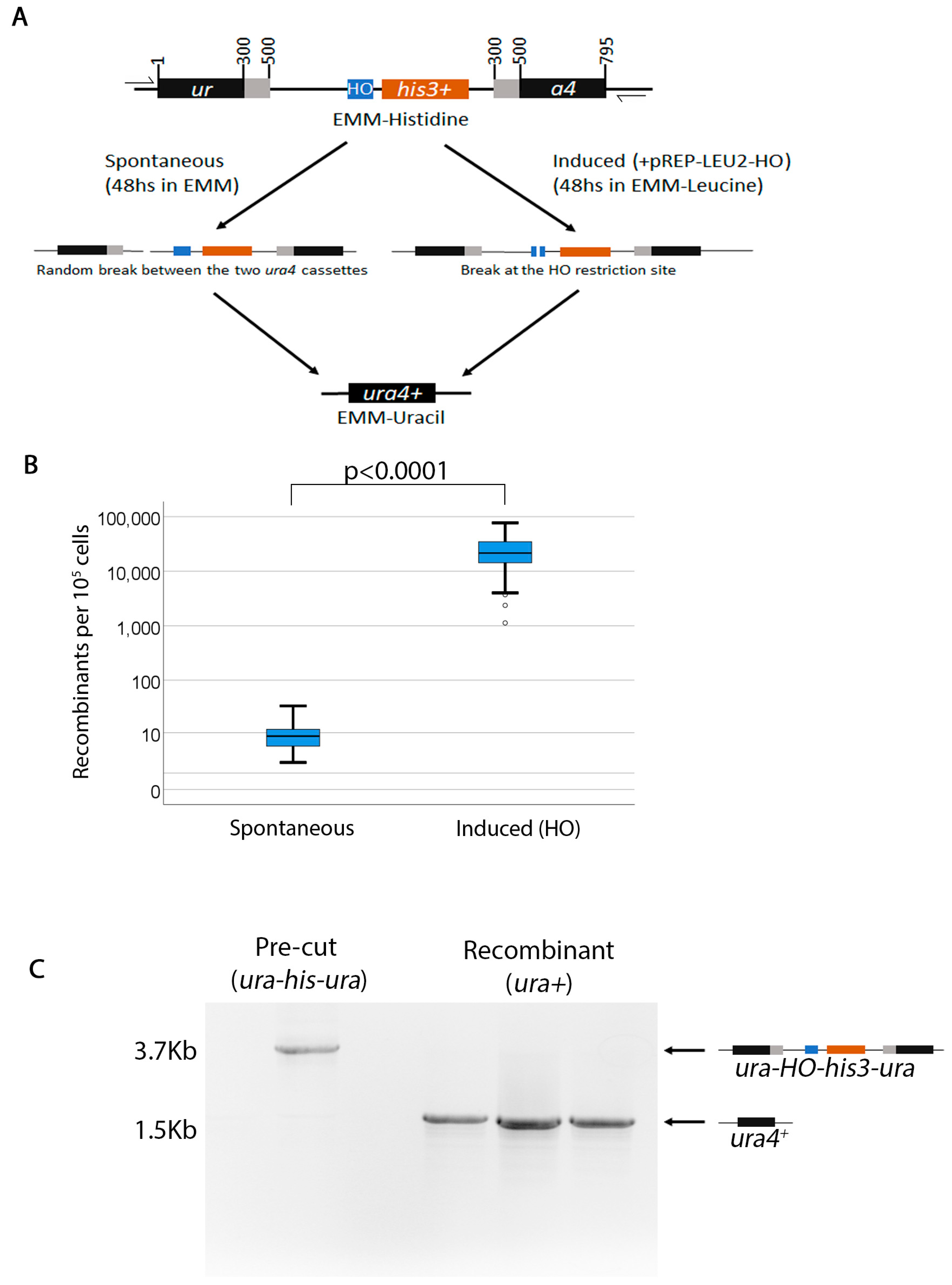
2.1. Spontaneous Break Recombination Protocol
- Streak cells onto EMM-Histidine plates from the −70 °C freezer. Grow at 32 °C for 3–4 days until colonies appear. Although the cassette is quite stable, to ensure that starting cells are ura−his+, it may be necessary to replica plate the EMM-His onto 5-FOA and choose only those colonies that grow on both plates.
- Resuspend 10 colonies each in 100 μL water in microtubes, count cells and release in 4 mL liquid EMM+UraHisLeuAde at 100 cells/microliter. Incubate tubes at 32 °C in the rotator for approximately 48 h.
- Determine the concentration of the cells in the tubes by counting cells using a hemocytometer and plate onto EMM-Uracil+Phloxin B at 105–106 cells per plate. Because ura− cells tend to cannibalize themselves, sometimes false positives appear. The addition of Phloxin B makes it easier to identify false positive because it stains ura− cells bright red. Phloxin B does not have an effect on recombination rate (Supplementary Tables S2 and S3). Furthermore, we recommend using large 150 mm × 15 mm plates particularly when plating at higher density. Plate a YES control as well for each colony plated on EMM-Uracil at 1000 cells per plate. This control is important to check for cell viability and accuracy in counting. Although we used YES for this control, a better control may be EMM+Uracil+Histidine. This maintains consistency with the experimental plate which is EMM not YES.
- Incubate all plates at 32 °C until colonies appear—usually 3%#x2013;5 days for WT and longer for mutants.
- Count colonies on both the YES control and EMM-Uracil plates and record the numbers. Although Phloxin B allows for easier differentiation of ura4+ prototrophic colonies, to ensure that all colonies on the EMM-Uracil plates are in fact Ura+, this plate can be replica plated onto 5-FOA. All ura4+ colonies that grow on EMM-Uracil should die on 5-FOA.
2.2. Induced Break Recombination Protocol
- Streak cells onto EMM-Leucine+Thiamine plates from the −70°C freezer. Incubate at 32 °C for 3–4 days.
- Resuspend 10 colonies in water, count cells and release in 4 mL liquid EMM-Leucine at 100 cells/microliter. Incubate tubes at 32 °C with rotation for approximately 48 h.
- Determine the concentration of the cells in the tubes and plate onto EMM-Uracil (100 mm × 15 mm plates) at 104 cells per plate. Plate on YES as well at 103 cells per plate.
- Incubate all plates at 32 °C until colonies appear.
- Count colonies on both YES and EMM-Uracil plates and record the numbers.
2.3. Characterization of the Assay
2.4. Strains
2.5. PCR Analysis
2.6. Data Analysis
3. Genetic Validation of the Assay and Discussion
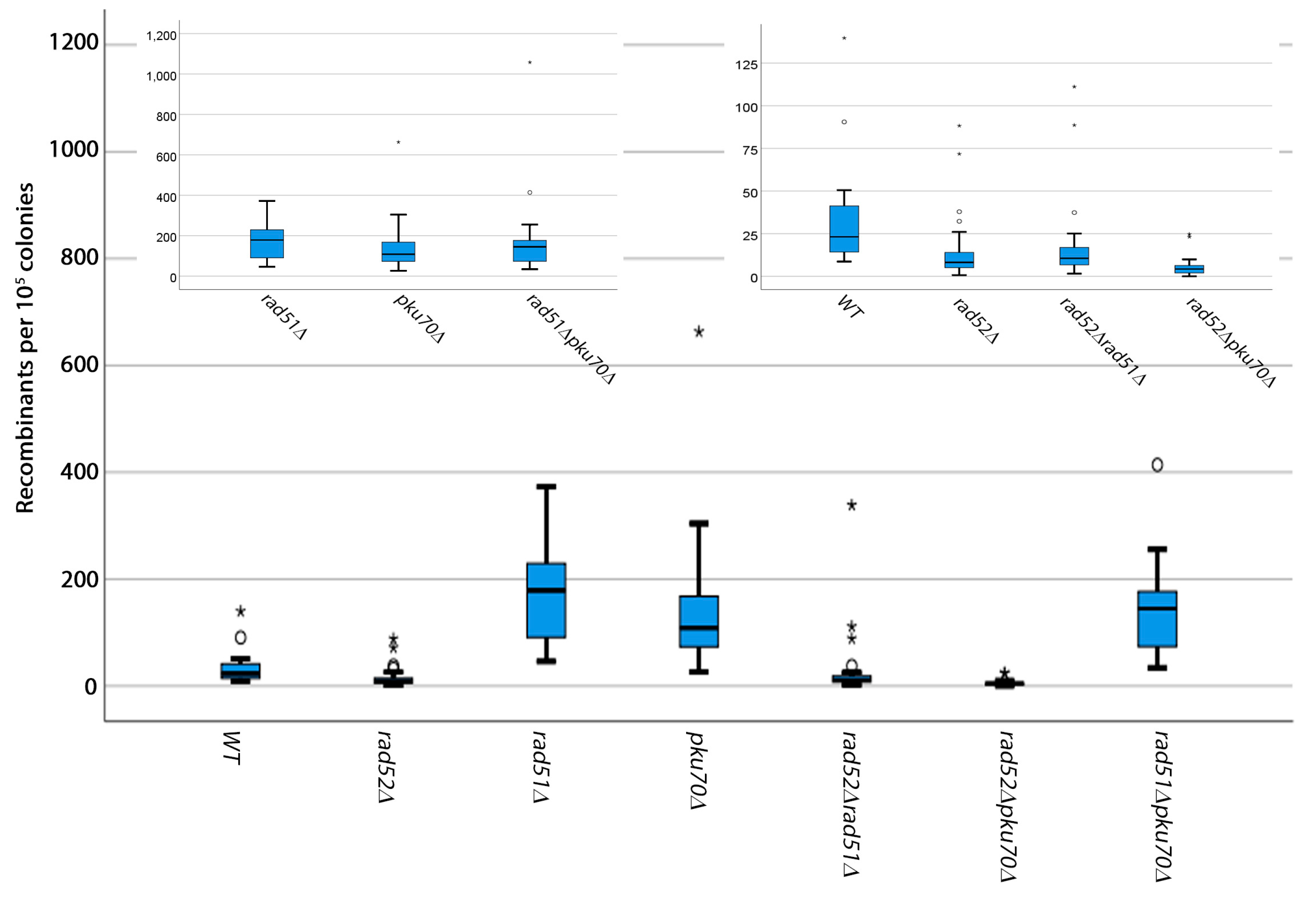
3.1. Analysis of Spontaneous Breaks
3.2. Analysis of Induced Breaks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Janssen, A.; van der Burg, M.; Szuhai, K.; Kops, G.J.; Medema, R.H. Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 2011, 333, 1895–1898. [Google Scholar] [CrossRef] [PubMed]
- Lasko, D.; Cavenee, W.; Nordenskjold, M. Loss of constitutional heterozygosity in human cancer. Annu. Rev. Genet 1991, 25, 281–314. [Google Scholar] [CrossRef] [PubMed]
- Ortega, V.; Chaubey, A.; Mendiola, C.; Ehman, W., Jr.; Vadlamudi, K.; Dupont, B.; Velagaleti, G. Complex Chromosomal Rearrangements in B-Cell Lymphoma: Evidence of Chromoanagenesis? A Case Report. Neoplasia 2016, 18, 223–228. [Google Scholar] [CrossRef][Green Version]
- Mehta, A.; Haber, J.E. Sources of DNA double-strand breaks and models of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [PubMed]
- Nikolov, I.; Taddei, A. Linking replication stress with heterochromatin formation. Chromosoma 2016, 125, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Tabancay, A.P., Jr.; Forsburg, S.L. Eukaryotic DNA replication in a chromatin context. Curr. Top Dev. Biol. 2006, 76, 129–184. [Google Scholar]
- Prado, F.; Aguilera, A. Impairment of replication fork progression mediates RNA polII transcription-associated recombination. EMBO J. 2005, 24, 1267–1276. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. Origins of the machinery of recombination and sex. Heredity (Edinb) 2002, 88, 125–141. [Google Scholar] [CrossRef]
- Mortensen, U.H.; Lisby, M.; Rothstein, R. Rad52. Curr. Biol. 2009, 19, R676–R677. [Google Scholar] [CrossRef]
- Symington, L.S. Role of RAD52 epistasis group genes in homologous recombination and double-strand break repair. Microbiol. Mol. Biol. Rev. 2002, 66, 630–670. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Aylon, Y.; Liefshitz, B.; Kupiec, M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004, 23, 4868–4875. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Lisby, M.; Rothstein, R. Differential regulation of the cellular response to DNA double-strand breaks in G1. Mol. Cell 2008, 30, 73–85. [Google Scholar] [CrossRef][Green Version]
- Clerici, M.; Mantiero, D.; Guerini, I.; Lucchini, G.; Longhese, M.P. The Yku70-Yku80 complex contributes to regulate double-strand break processing and checkpoint activation during the cell cycle. EMBO Rep. 2008, 9, 810–818. [Google Scholar] [CrossRef] [PubMed]
- Ira, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; Haber, J.E.; Foiani, M. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Caspari, T.; Murray, J.M.; Carr, A.M. Cdc2-cyclin B kinase activity links Crb2 and Rqh1-topoisomerase III. Genes Dev. 2002, 16, 1195–1208. [Google Scholar] [CrossRef]
- Fortunato, E.A.; Osman, F.; Subramani, S. Analysis of spontaneous and double-strand break-induced recombination in rad mutants of S. pombe. Mutat. Res. 1996, 364, 14–60. [Google Scholar] [CrossRef]
- Cullen, J.K.; Prudden, J.; Wee, B.Y.; Davé, A.; Findlay, J.S.; Savory, A.P.; Humphrey, T.C. Break-induced loss of heterozygosity in fission yeast: Dual roles for homologous recombination in promoting translocations and preventing de novo telomere addition. Mol. Cell Biol. 2007, 27, 7745–7757. [Google Scholar] [CrossRef]
- Pierce, A.J.; Johnson, R.D.; Thompson, L.H.; Jasin, M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999, 13, 2633–2638. [Google Scholar] [CrossRef]
- Stark, J.M.; Pierce, A.J.; Oh, J.; Pastink, A.; Jasin, M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell Biol. 2004, 24, 9305–9316. [Google Scholar] [CrossRef]
- Sugawara, N.; Haber, J.E. Repair of DNA double strand breaks: In vivo biochemistry. Methods Enzymol. 2006, 408, 416–429. [Google Scholar]
- Gonzalez-Barrera, S.; Cortés-Ledesma, F.; Wellinger, R.E.; Aguilera, A. Equal sister chromatid exchange is a major mechanism of double-strand break repair in yeast. Mol. Cell 2003, 11, 1661–1671. [Google Scholar] [CrossRef]
- Liddell, L.; Manthey, G.; Pannunzio, N.; Bailis, A. Quantitation and analysis of the formation of HO-endonuclease stimulated chromosomal translocations by single-strand annealing in Saccharomyces cerevisiae. J. Vis. Exp. 2011, 55, 3150. [Google Scholar] [CrossRef]
- McDonald, J.P.; Rothstein, R. Unrepaired heteroduplex DNA in Saccharomyces cerevisiae is decreased in RAD1 RAD52-independent recombination. Genetics 1994, 137, 393–405. [Google Scholar]
- Osman, F.; Fortunato, E.A.; Subramani, S. Double-strand break-induced mitotic intrachromosomal recombination in the fission yeast Schizosaccharomyces pombe. Genetics 1996, 142, 341–357. [Google Scholar]
- Li, P.C.; Petreaca, R.C.; Jensen, A.; Yuan, J.P.; Green, M.D.; Forsburg, S.L. Replication fork stability is essential for the maintenance of centromere integrity in the absence of heterochromatin. Cell Rep. 2013, 3, 638–645. [Google Scholar] [CrossRef]
- Du, L.L.; Nakamura, T.M.; Moser, B.A.; Russell, P. Retention but not recruitment of Crb2 at double-strand breaks requires Rad1 and Rad3 complexes. Mol. Cell Biol. 2003, 23, 6150–6158. [Google Scholar] [CrossRef]
- Prudden, J.; Evans, J.S.; Hussey, S.P.; Deans, B.; O’Neill, P.; Thacker, J.; Humphrey, T. Pathway utilization in response to a site-specific DNA double-strand break in fission yeast. EMBO J. 2003, 22, 1419–1430. [Google Scholar] [CrossRef]
- Burke, J.D.; Gould, K.L. Molecular cloning and characterization of the Schizosaccharomyces pombe his3 gene for use as a selectable marker. Mol. Gen Genet. 1994, 242, 169–176. [Google Scholar] [CrossRef]
- Osman, F.; Whitby, M.C. Monitoring homologous recombination following replication fork perturbation in the fission yeast Schizosaccharomyces pombe. Methods Mol. Biol. 2009, 521, 535–552. [Google Scholar]
- Nakamura, K.; Shitanda, T.; Kaweeteerawat, C.; Takahashi, T.S.; Itoh, T.; Shirahige, K.; Masukata, H.; Nakagawa, T. Rad51 suppresses gross chromosomal rearrangement at centromere in Schizosaccharomyces pombe. EMBO J. 2008, 27, 3036–3046. [Google Scholar] [CrossRef]
- Doe, C.L.; Osman, F.; Dixon, J.; Matthew, C. WhitbyDNA repair by a Rad22-Mus81-dependent pathway that is independent of Rhp51. Nucleic Acids Res. 2004, 32, 5570–5581. [Google Scholar] [CrossRef]
- Onaka, A.T. Rad51 and Rad54 promote noncrossover recombination between centromere repeats on the same chromatid to prevent isochromosome formation. Nucleic Acids Res. 2016, 44, 10744–10757. [Google Scholar] [CrossRef]
- Teixeira-Silva, A. The end-joining factor Ku acts in the end-resection of double strand break-free arrested replication forks. Nat. Commun. 2017, 8, 1982. [Google Scholar] [CrossRef]
- Osman, F.; Adriance, M.; McCready, S. The genetic control of spontaneous and UV-induced mitotic intrachromosomal recombination in the fission yeast Schizosaccharomyces pombe. Curr. Genet. 2000, 38, 113–125. [Google Scholar] [CrossRef]
- Yamaguchi-Iwai, Y. Homologous recombination, but not DNA repair, is reduced in vertebrate cells deficient in RAD52. Mol. Cell Biol. 1998, 18, 6430–6435. [Google Scholar] [CrossRef]
- Tomita, K.; Matsuura, A.; Caspari, T.; Carr, A.M.; Akamatsu, Y.; Iwasaki, H.; Mizuno, K.; Ohta, K.; Uritani, M.; Ushimaru, T. Competition between the Rad50 complex and the Ku heterodimer reveals a role for Exo1 in processing double-strand breaks but not telomeres. Mol. Cell Biol. 2003, 23, 5186–5197. [Google Scholar] [CrossRef]
- Ferreira, M.G.; Cooper, J.P. Two modes of DNA double-strand break repair are reciprocally regulated through the fission yeast cell cycle. Genes Dev. 2004, 18, 2249–2254. [Google Scholar] [CrossRef]
- Florl, A.R.; Schulz, W.A. Peculiar structure and location of 9p21 homozygous deletion breakpoints in human cancer cells. Genes Chromosomes Cancer 2003, 37, 141–148. [Google Scholar] [CrossRef]
- Frazao, L.; Carmo, M.; Mafra, M. BRAF V600E mutation and 9p21: CDKN2A/B and MTAP co-deletions Markers in the clinical stratification of pediatric gliomas. BMC Cancer 2018, 18, 1259. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lucas, B.E.; McPherson, M.T.; Hawk, T.M.; Wilson, L.N.; Kroh, J.M.; Hickman, K.G.; Fitzgerald, S.R.; Disbennett, W.M.; Rollins, P.D.; Hylton, H.M.; et al. An Assay to Study Intra-Chromosomal Deletions in Yeast. Methods Protoc. 2019, 2, 74. https://doi.org/10.3390/mps2030074
Lucas BE, McPherson MT, Hawk TM, Wilson LN, Kroh JM, Hickman KG, Fitzgerald SR, Disbennett WM, Rollins PD, Hylton HM, et al. An Assay to Study Intra-Chromosomal Deletions in Yeast. Methods and Protocols. 2019; 2(3):74. https://doi.org/10.3390/mps2030074
Chicago/Turabian StyleLucas, Bailey E., Matthew T. McPherson, Tila M. Hawk, Lexia N. Wilson, Jacob M. Kroh, Kyle G. Hickman, Sean R. Fitzgerald, W. Miguel Disbennett, P. Daniel Rollins, Hannah M. Hylton, and et al. 2019. "An Assay to Study Intra-Chromosomal Deletions in Yeast" Methods and Protocols 2, no. 3: 74. https://doi.org/10.3390/mps2030074
APA StyleLucas, B. E., McPherson, M. T., Hawk, T. M., Wilson, L. N., Kroh, J. M., Hickman, K. G., Fitzgerald, S. R., Disbennett, W. M., Rollins, P. D., Hylton, H. M., Baseer, M. A., Montgomery, P. N., Wu, J.-Q., & Petreaca, R. C. (2019). An Assay to Study Intra-Chromosomal Deletions in Yeast. Methods and Protocols, 2(3), 74. https://doi.org/10.3390/mps2030074