Influence of Metabolite Extraction Methods on 1H-NMR-Based Metabolomic Profiling of Enteropathogenic Yersinia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Culture Conditions
2.2. Sampling and Metabolite Extraction
2.3. NMR Data Acquisition
2.4. Pre-Processing
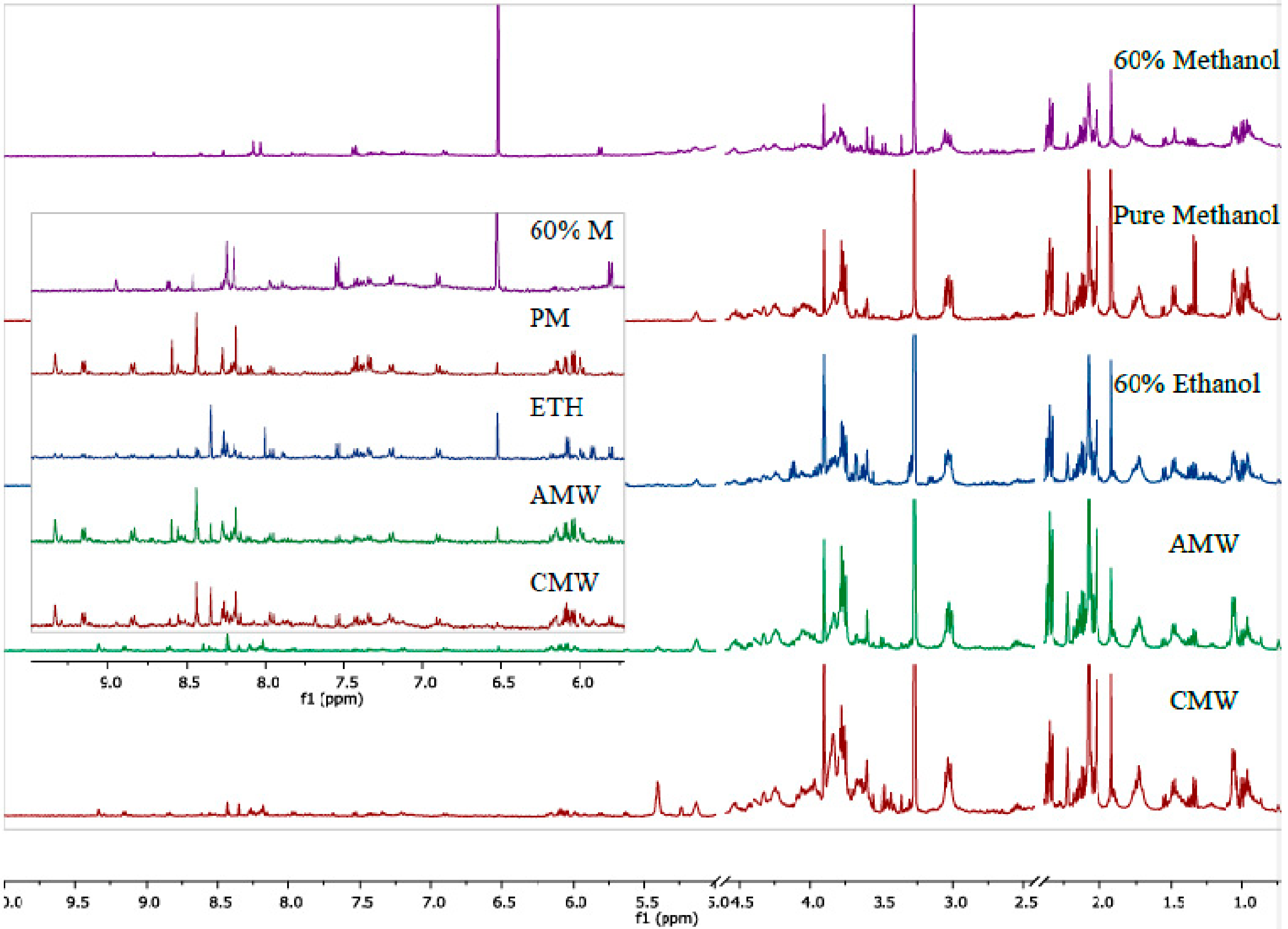
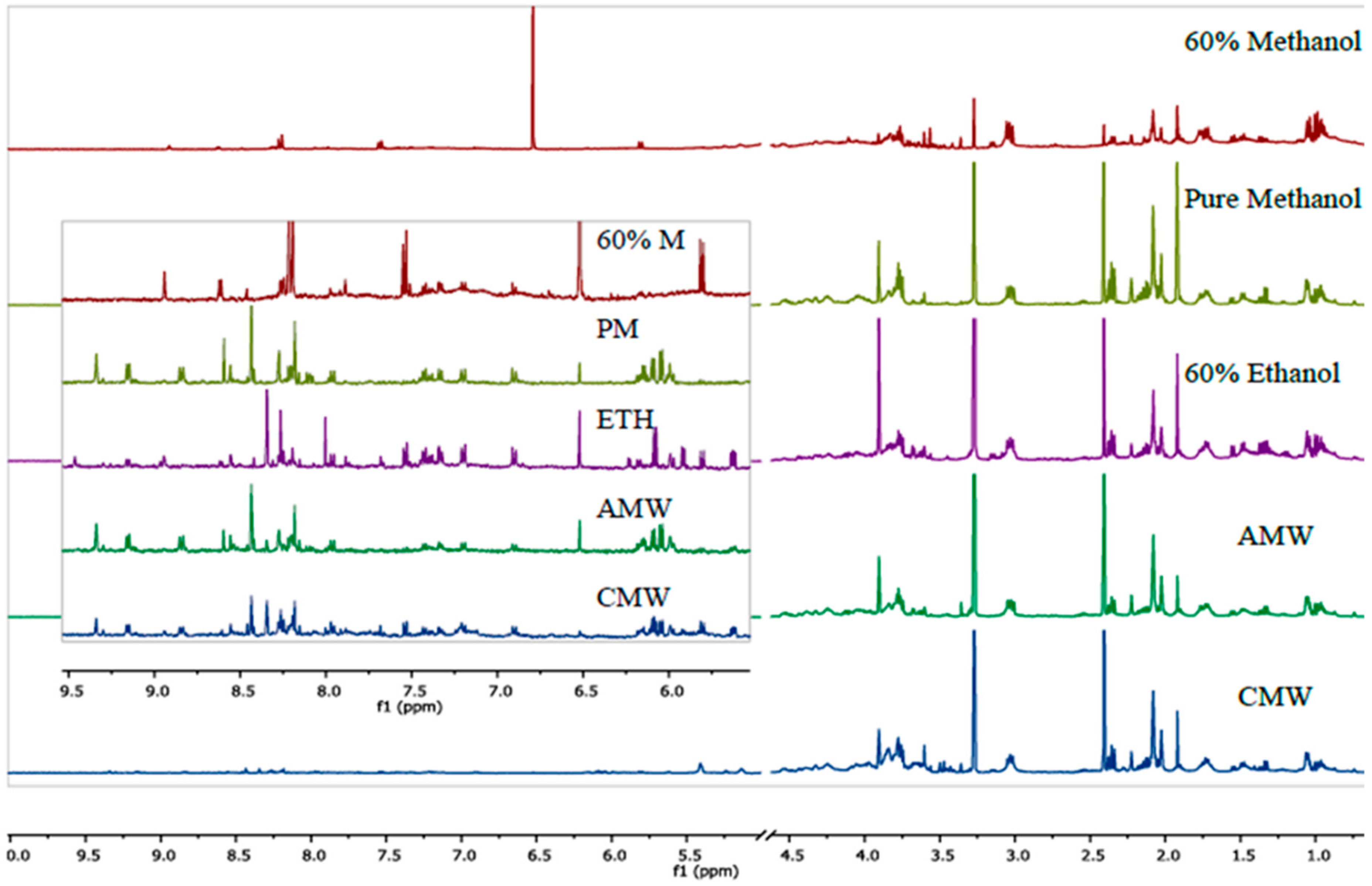
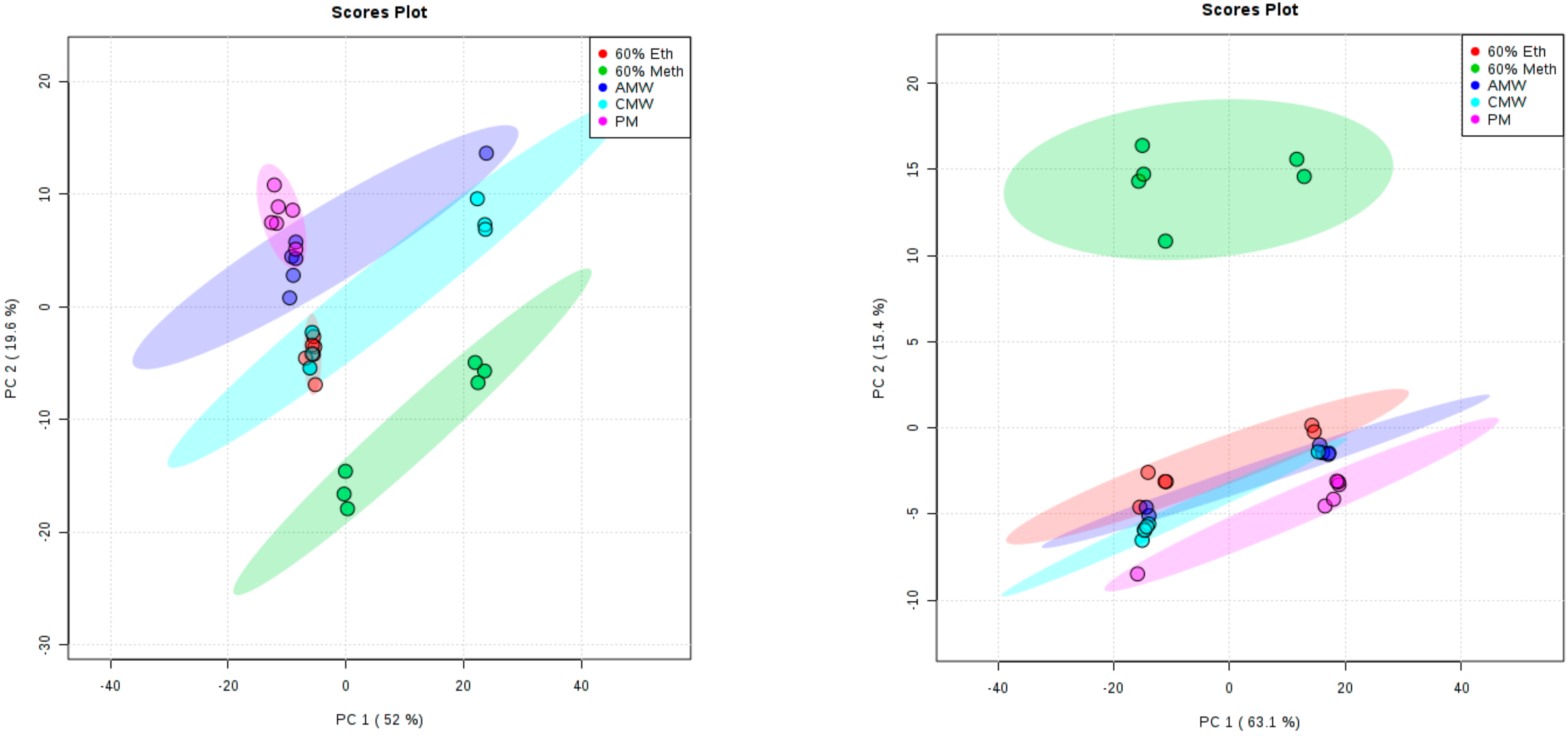
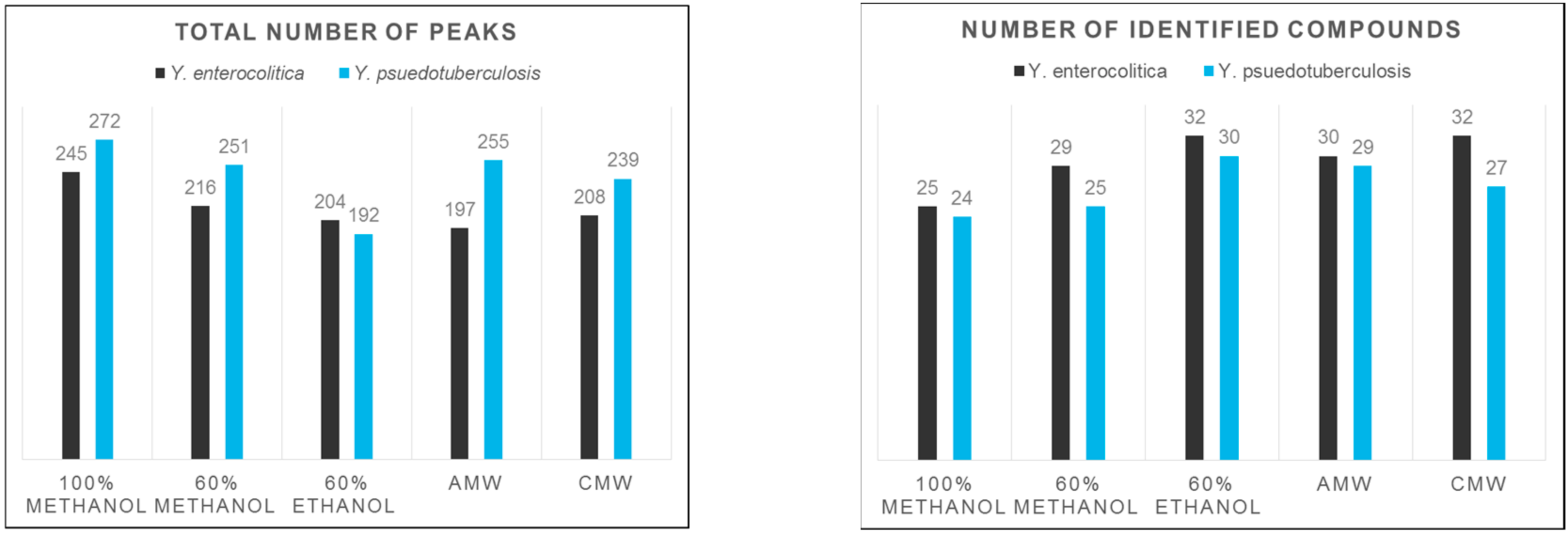
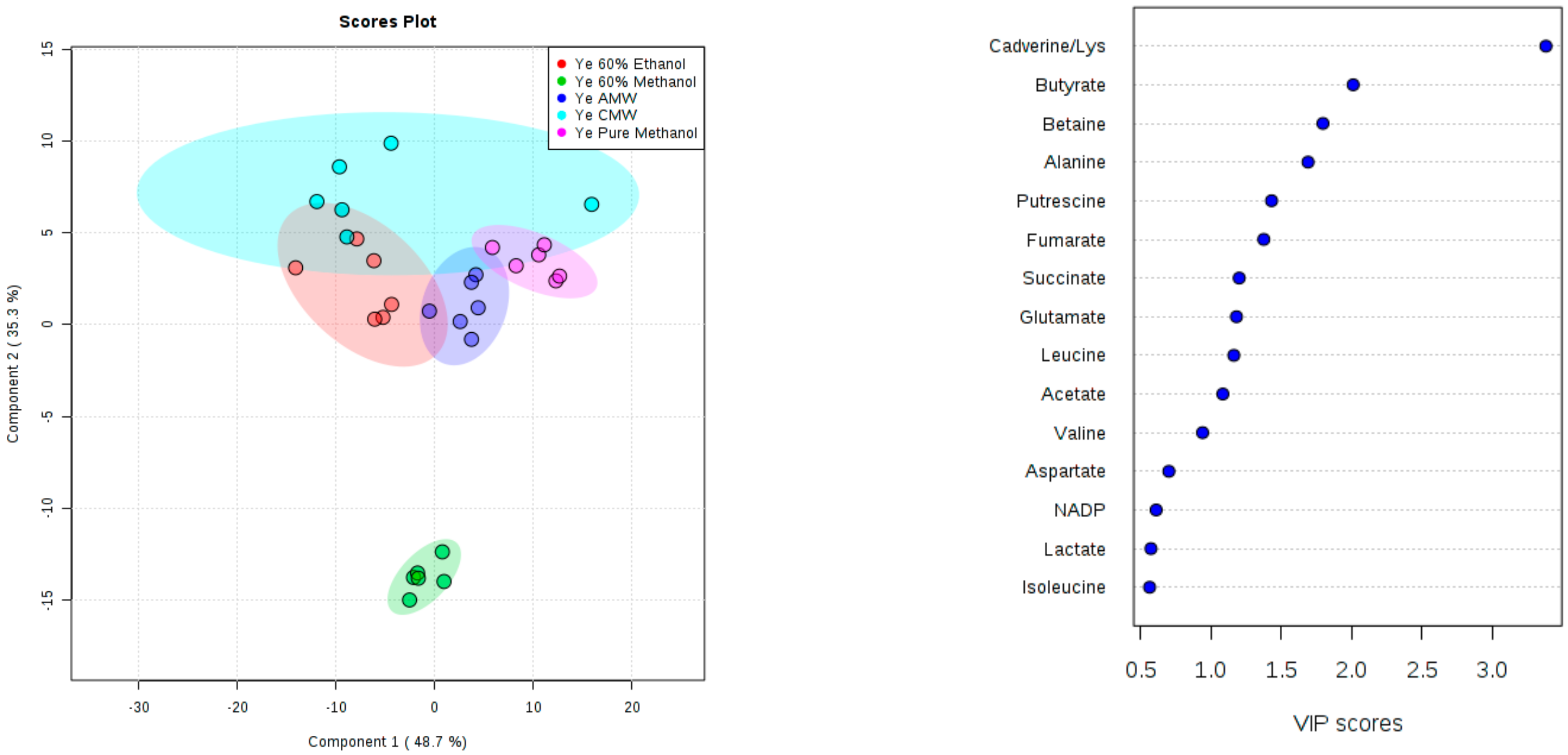
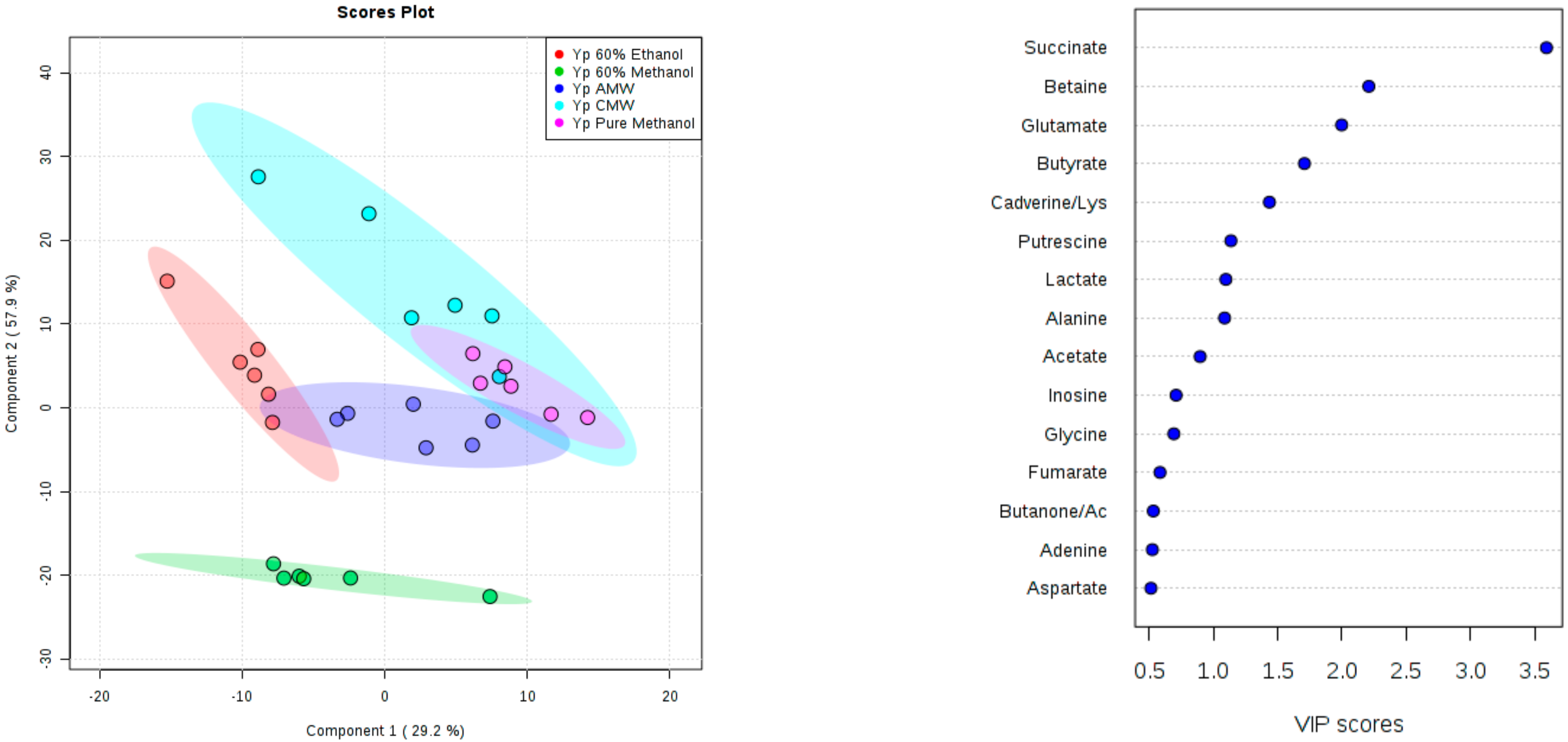
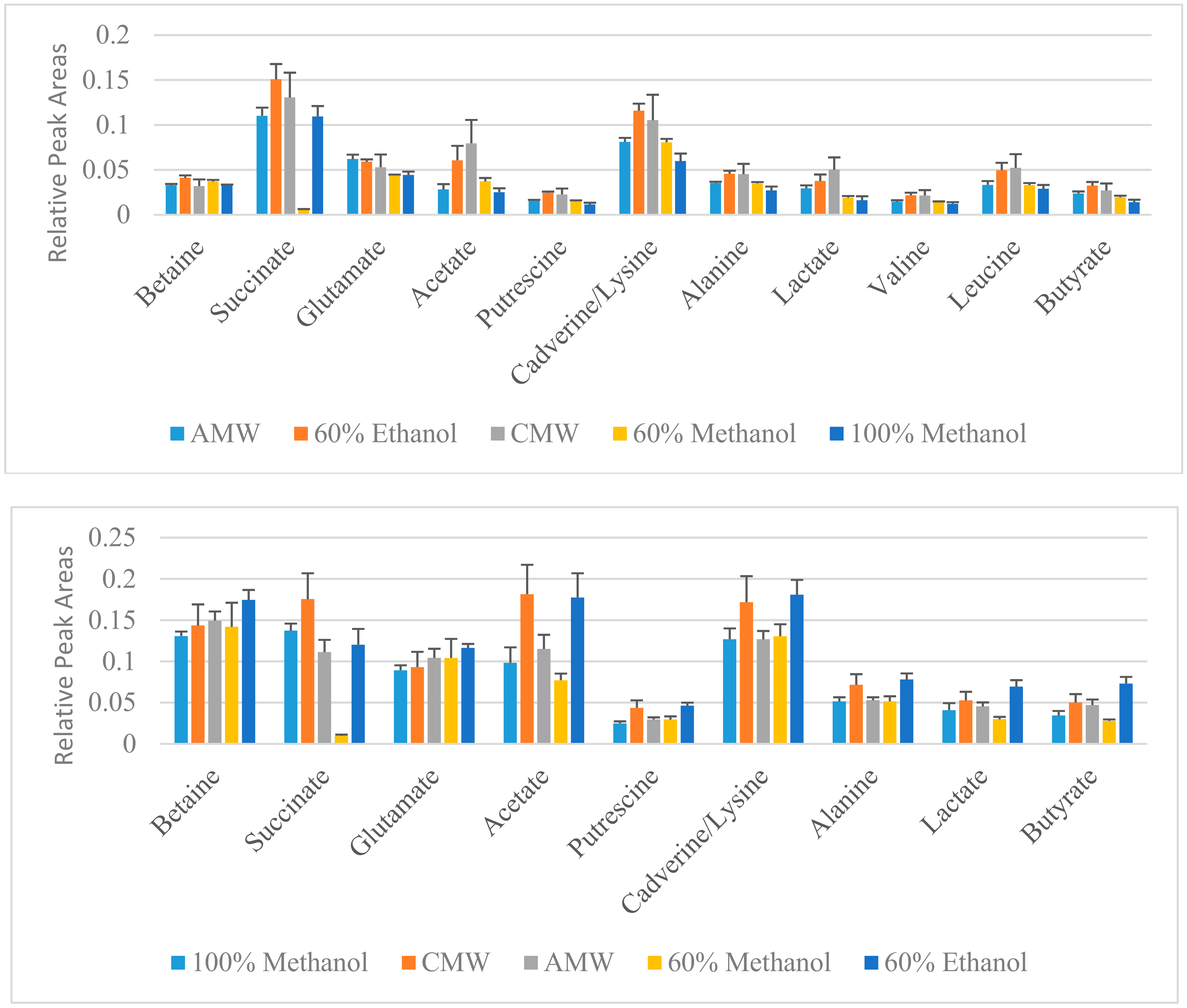
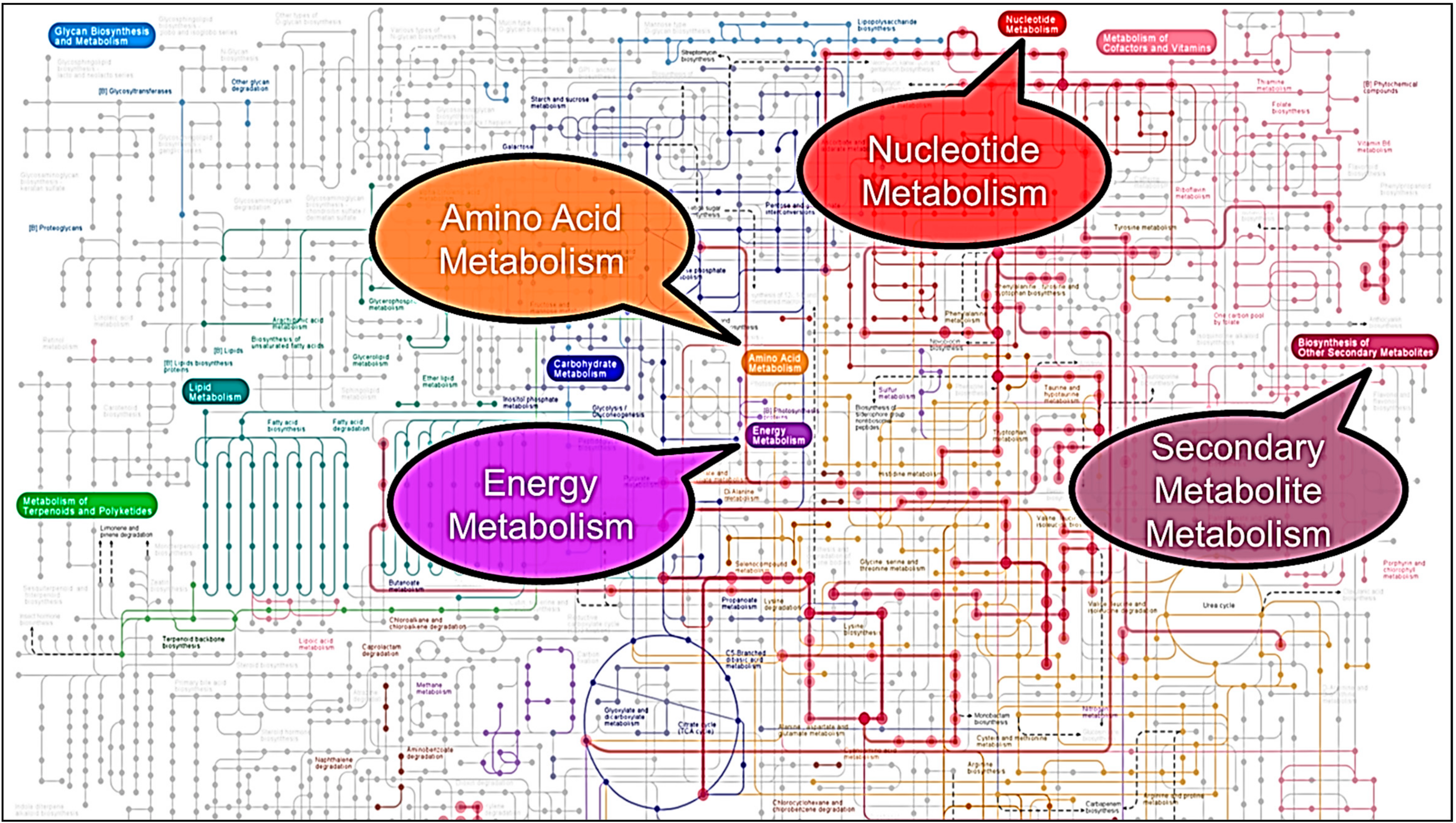
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Feijo Delgado, F.; Cermak, N.; Hecht, V.C.; Son, S.; Li, Y.; Knudsen, S.M.; Olcum, S.; Higgins, J.M.; Chen, J.; Grover, W.H.; et al. Intracellular water exchange for measuring the dry mass, water mass and changes in chemical composition of living cells. PloS ONE 2013, 8, e67590. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.C.; Jewison, T.; Wilson, M.; Liu, Y.; Knox, C.; Djoumbou, Y.; Lo, P.; Mandal, R.; Krishnamurthy, R.; Wishart, D.S. ECMDB: The E. coli metabolome database. Nucleic Acids Res. 2013, 41, D625–D630. [Google Scholar] [CrossRef] [PubMed]
- National Academies of Sciences Engineering Medicine. Use of metabolomics to advance research on environmental exposures and the human exposome. In Proceedings of the The National Academies Press 2016, Workshop in Brief, Washington, DC, USA, 28–29 May 2015. [Google Scholar]
- Weckwerth, W.; Morgenthal, K. Metabolomics: From pattern recognition to biological interpretation. Drug Discov. Today 2005, 10, 1551–1558. [Google Scholar] [CrossRef]
- Kim, S.; Lee, D.Y.; Wohlgemuth, G.; Park, H.S.; Fiehn, O.; Kim, K.H. Evaluation and optimization of metabolome sample preparation methods for Saccharomyces cerevisiae. Anal. Chem. 2013, 85, 2169–2176. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.; Weidmann, H.; Lalk, M. Methodological approaches to help unravel the intracellular metabolome of Bacillus subtilis. Microb. Cell Factor. 2013, 12, 69. [Google Scholar] [CrossRef] [PubMed]
- Steven, H.; Bo, Z.; Rosmarie, G.; Shulei, L.; Emily, S.; Fenton, R.; Barletta, R.G.; Somerville, G.; Powers, R. Revisiting protocols for the NMR analysis of bacterial metabolomes. J. Integr. OMICS 2013, 3, 120–137. [Google Scholar]
- Liu, H.; Garrett, T.J.; Tayyari, F.; Gu, L. Profiling the metabolome changes caused by cranberry procyanidins in plasma of female rats using 1H NMR and UHPLC-Q-orbitrap-HRMS global metabolomics approaches. Mol. Nutr. Food Res. 2015, 59, 2107–2118. [Google Scholar] [CrossRef] [PubMed]
- Li, A.-P.; Li, Z.-Y.; Sun, H.-F.; Li, K.; Qin, X.-M.; Du, G.-H. Comparison of two different astragali radix by a 1H NMR-based metabolomic approach. J. Proteome Res. 2015, 14, 2005–2016. [Google Scholar] [CrossRef] [PubMed]
- Worley, B.; Powers, R. PCA as a practical indicator of OPLS-DA model reliability. Curr. Metabolomics 2016, 4, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Zennaro, L.; Vanzani, P.; Nicolè, L.; Cappellesso, R.; Fassina, A. Metabonomics by proton nuclear magnetic resonance in human pleural effusions: A route to discriminate between benign and malignant pleural effusions and to target small molecules as potential cancer biomarkers. Cancer Cytopathol. 2017, 125, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Smolinska, A.; Blanchet, L.; Buydens, L.M.C.; Wijmenga, S.S. NMR and pattern recognition methods in metabolomics: From data acquisition to biomarker discovery: A review. Anal. Chimica Acta 2012, 750, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Miele, M.M.; Irving, B.A.; Wenrich, B.R.; Martin, P.L.; Rovnyak, D. Reproducibility and stability of aqueous metabolite levels in extracted serum by NMR spectroscopy. Curr. Metabolomics 2017, 5, 45–54. [Google Scholar] [CrossRef]
- Galindo, C.L.; Rosenzweig, J.A.; Kirtley, M.L.; Chopra, A.K. Pathogenesis of Y. enterocolitica and Y. pseudotuberculosis in human yersiniosis. J. Pathog. 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson-Ahomaa, M. Enteropathogenic Yersinia spp. In Zoonoses—Infections Affecting Humans and Animals: Focus on Public Health Aspects; Sing, A, Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 213–234. [Google Scholar]
- Gupta, V.; Gulati, P.; Bhagat, N.; Dhar, M.; Virdi, J. Detection of Yersinia enterocolitica in food: An overview. Eur. J. Clin. Microbiol. Infec. Dis. 2015, 34, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Laporte, J.; Savin, C.; Lamourette, P.; Devilliers, K.; Volland, H.; Carniel, E.; Creminon, C.; Simon, S. Fast and sensitive detection of enteropathogenic Yersinia by immunoassays. J. Clin. Microbiol. 2015, 53, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Abdela, W.; Graham, M.; Habtemariam, T.; Samuel, T.; Yehualaeshet, T. Effects of orange juice pH on survival, urease activity and DNA profiles of Yersinia enterocolitica and Yersinia pseudotuberculosis stored at 4 °C. J. Food Saf. 2011, 31, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Nie, W.; Lv, H. Metabolic phenotyping of the Yersinia high-pathogenicity island that regulates central carbon metabolism. Analyst 2015, 140, 3356–3361. [Google Scholar] [CrossRef] [PubMed]
- Palama, T.L.; Canard, I.; Rautureau, G.J.; Mirande, C.; Chatellier, S.; Elena-Herrmann, B. Identification of bacterial species by untargeted NMR spectroscopy of the exo-metabolome. Analyst 2016, 141, 4558–4561. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Using MetaboAnalyst 3.0 for comprehensive metabolomics data analysis. Curr. Protoc. Bioinform. 2016, 55, 14.10.1–14.10.91. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vazquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Lewis, I.A.; Hegeman, A.D.; Anderson, M.E.; Li, J.; Schulte, C.F.; Westler, W.M.; Eghbalnia, H.R.; Sussman, M.R.; Markley, J.L. Metabolite identification via the Madison Metabolomics Consortium Database. Nat. Biotechnol. 2008, 26, 162–164. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Dutta, T.; Persson, X.-M.T.; Mielke, M.M.; Petersen, R.C. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PloS ONE 2013, 8, e63644. [Google Scholar] [CrossRef] [PubMed]
- Beltran, A.; Suarez, M.; Rodriguez, M.A.; Vinaixa, M.; Samino, S.; Arola, L.; Correig, X.; Yanes, O. Assessment of compatibility between extraction methods for NMR- and LC/MS-based metabolomics. Anal. Chem. 2012, 84, 5838–5844. [Google Scholar] [CrossRef] [PubMed]
- Vinaixa, M.; Rodriguez, M.A.; Samino, S.; Díaz, M.; Beltran, A.; Mallol, R.; Bladé, C.; Ibañez, L.; Correig, X.; Yanes, O. Metabolomics reveals reduction of metabolic oxidation in women with polycystic ovary syndrome after pioglitazone-flutamide-metformin polytherapy. PloS ONE 2011, 6, e29052. [Google Scholar] [CrossRef] [PubMed]
- Büscher, J.M.; Czernik, D.; Ewald, J.C.; Sauer, U.; Zamboni, N. Cross-platform comparison of methods for quantitative metabolomics of primary metabolism. Anal. Chem. 2009, 81, 2135–2143. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Bruschweiler, R.; Edison, A.S.; Eghbalnia, H.R.; Powers, R.; Raftery, D.; Wishart, D.S. The future of NMR-based metabolomics. Curr. Opin. Biotechnol. 2017, 43, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Wilson, I.D.; Gika, H.; Theodoridis, G.; Plumb, R.S.; Shockcor, J.; Holmes, E.; Nicholson, J.K. Global metabolic profiling procedures for urine using UPLC-MS. Nat. Protoc. 2010, 5, 1005–1018. [Google Scholar] [CrossRef] [PubMed]
- Lanza, I.R.; Zhang, S.; Ward, L.E.; Karakelides, H.; Raftery, D.; Nair, K.S. Quantitative metabolomics by 1H-NMR and LC-MS/MS confirms altered metabolic pathways in diabetes. PloS ONE 2010, 5, e10538. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gines, B.R.; Collier, W.E.; Abdalla, M.A.; Yehualaeshet, T. Influence of Metabolite Extraction Methods on 1H-NMR-Based Metabolomic Profiling of Enteropathogenic Yersinia. Methods Protoc. 2018, 1, 45. https://doi.org/10.3390/mps1040045
Gines BR, Collier WE, Abdalla MA, Yehualaeshet T. Influence of Metabolite Extraction Methods on 1H-NMR-Based Metabolomic Profiling of Enteropathogenic Yersinia. Methods and Protocols. 2018; 1(4):45. https://doi.org/10.3390/mps1040045
Chicago/Turabian StyleGines, Brandon R., Willard E. Collier, Mohamed A. Abdalla, and Teshome Yehualaeshet. 2018. "Influence of Metabolite Extraction Methods on 1H-NMR-Based Metabolomic Profiling of Enteropathogenic Yersinia" Methods and Protocols 1, no. 4: 45. https://doi.org/10.3390/mps1040045
APA StyleGines, B. R., Collier, W. E., Abdalla, M. A., & Yehualaeshet, T. (2018). Influence of Metabolite Extraction Methods on 1H-NMR-Based Metabolomic Profiling of Enteropathogenic Yersinia. Methods and Protocols, 1(4), 45. https://doi.org/10.3390/mps1040045