
New Acylcarnitine Ratio as a Reliable Indicator of Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Sample Preparation
2.3. Biomarker Analysis
2.4. Genotyping
2.5. Statistical Analysis
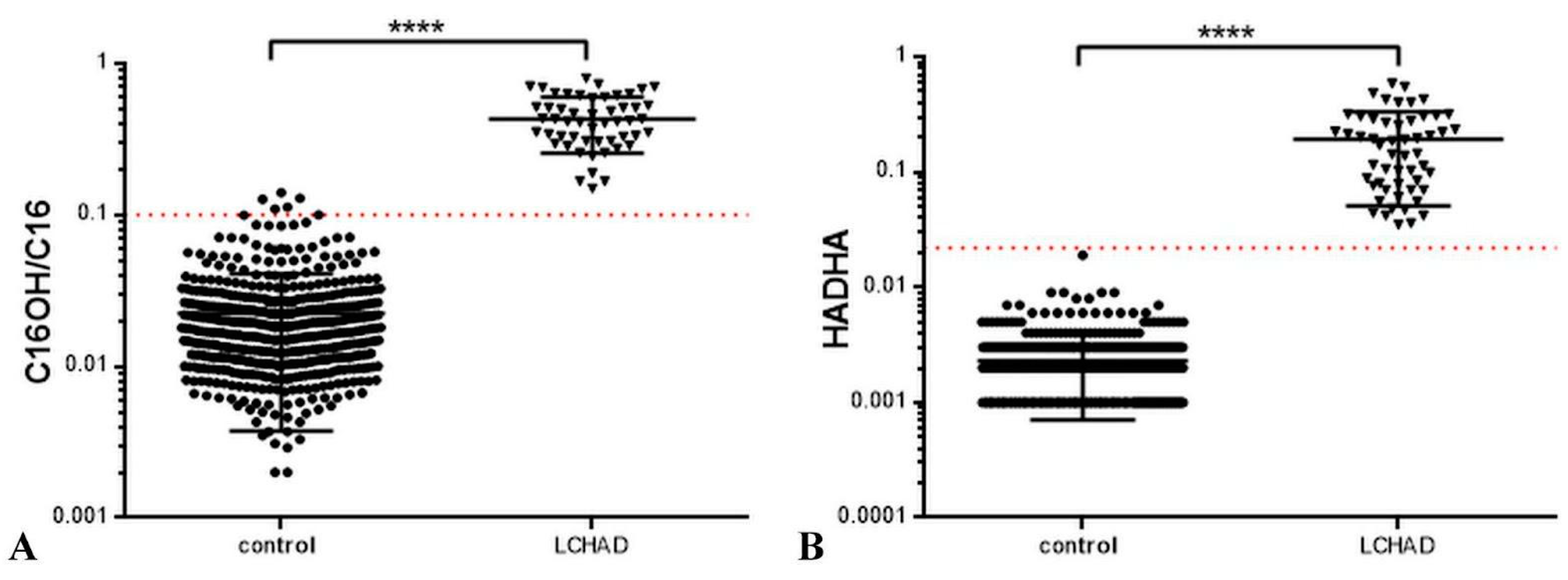
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sykut-Cegielska, J.; Gradowska, W.; Piekutowska-Abramczuk, D.; Andresen, B.S.; Olsen, R.K.J.; Ołtarzewski, M.; Pronicki, M.; Pajdowska, M.; Bogdańska, A.; Jabłońska, E.; et al. Urgent metabolic service improves survival in long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency detected by symptomatic identification and pilot newborn screening. J. Inherit. Metab. Dis. 2011, 34, 185–195. [Google Scholar] [CrossRef] [PubMed]
- Dagher, R.; Massie, R.; Gentil, B.J. MTP deficiency caused by HADHB mutations: Pathophysiology and clinical manifestations. Mol. Genet. Metab. 2021, 133, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Marsden, D.; Bedrosian, C.L.; Vockley, J. Impact of newborn screening on the reported incidence and clinical outcomes associated with medium- and long-chain fatty acid oxidation disorders. Genet. Med. 2021, 23, 816–829. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.; Duran, M.; Ijlst, L.; de Jager, J.P.; van Gennip, A.H.; Jakobs, C.; Dorland, L.; van Sprang, F.J. Sudden infant death and long-chain 3-hydroxyacyl-CoA dehydrogenase. Lancet 1989, 2, 52–53. [Google Scholar] [CrossRef]
- Olpin, S.E.; Clark, S.; Andresen, B.S.; Bischoff, C.; Olsen, R.K.J.; Gregersen, N.; Chakrapani, A.; Downing, M.; Manning, N.J.; Sharrard, M.; et al. Biochemical, clinical and molecular findings in LCHAD and general mitochondrial trifunctional protein deficiency. J. Inherit. Metab. Dis. 2005, 28, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Bross, P.; Andresen, B.S. Genetic defects in fatty acid β-oxidation and acyl-CoA dehydrogenases: Molecular pathogenesis and genotype–phenotype relationships. Eur. J. Biochem. 2004, 271, 470–482. [Google Scholar] [CrossRef]
- Haglind, C.B.; Nordenström, A.; Ask, S.; von Döbeln, U.; Gustafsson, J.; Stenlid, M.H. Increased and early lipolysis in children with long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency during fast. J. Inherit. Metab. Dis. 2015, 38, 315–322. [Google Scholar] [CrossRef]
- Stinton, C.; Fraser, H.; Geppert, J.; Johnson, R.; Connock, M.; Johnson, S.; Clarke, A.; Taylor-Phillips, S. Newborn screening for long-chain 3-hydroxyacyl-CoA dehydrogenase and mitochondrial trifunctional protein deficiencies using acylcarnitines measurement in dried blood spots—A systematic review of test accuracy. Front. Pediatr. 2021, 9, 606194. [Google Scholar] [CrossRef]
- Spiekerkoetter, U.; Khuchua, Z.; Yue, Z.; Bennett, M.J.; Strauss, A.W. General mitochondrial trifunctional protein (TFP) deficiency as a result of either alpha- or beta-subunit mutations exhibits similar phenotypes because mutations in either subunit alter TFP complex expression and subunit turnover. Pediatr. Res. 2004, 55, 190–196. [Google Scholar] [CrossRef]
- Dessein, A.F.; Hebbar, E.; Vamecq, J.; Lebredonchel, E.; Devos, A.; Ghoumid, J.; Mention, K.; Dobbelaere, D.; Chevalier-Curt, M.J.; Fontaine, M.; et al. A novel HADHA variant associated with an atypical moderate and late-onset LCHAD deficiency. Mol. Genet. Metab. Rep. 2022, 31, 100860. [Google Scholar] [CrossRef]
- Hickmann, F.H.; Cecatto, C.; Kleemann, D.; Monteiro, W.O.; Castilho, R.F.; Amaral, A.U.; Wajner, M. Uncoupling, metabolic inhibition and induction of mitochondrial permeability transition in rat liver mitochondria caused by the major long-chain hydroxyl monocarboxylic fatty acids accumulating in LCHAD deficiency. Biochim. Biophys. Acta (BBA)-Bioenerg. 2015, 1847, 620–628. [Google Scholar] [CrossRef]
- Taylor, W.A.; Mejia, E.M.; Mitchell, R.W.; Choy, P.C.; Sparagna, G.C.; Hatch, G.M. Human trifunctional protein alpha links cardiolipin remodeling to beta-oxidation. PLoS ONE 2012, 7, e48628. [Google Scholar] [CrossRef] [PubMed]
- Miklas, J.W.; Clark, E.; Levy, S.; Detraux, D.; Leonard, A.; Beussman, K.; Showalter, M.R.; Smith, A.T.; Hofsteen, P.; Yang, X.; et al. TFPa/HADHA is required for fatty acid beta-oxidation and cardiolipin re-modeling in human cardiomyocytes. Nat. Commun. 2019, 10, 4671. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Palmfeldt, J.; Gregersen, N.; Makhov, A.M.; Conway, J.F.; Wang, M.; McCalley, S.P.; Basu, S.; Alharbi, H.; Croix, C.S.; et al. Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex. J. Biol. Chem. 2019, 294, 12380–12391. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Ibdah, J.A. Mitochondrial dysfunction and acute fatty liver of pregnancy. Int. J. Mol. Sci. 2022, 23, 3595. [Google Scholar] [CrossRef]
- den Boer, M.E.; Wanders, R.J.; Morris, A.A.; IJlst, L.; Heymans, H.S.A.; Wijburg, F.A. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Clinical presentation and follow-up of 50 patients. Pediatrics 2002, 109, 99–104. [Google Scholar] [CrossRef]
- Weiss, R.E.; Refetoff, S. (Eds.) Genetic Diagnosis of Endocrine Disorders; Academic Press: Cambridge, MA, USA, 2015; pp. 62–63. [Google Scholar]
- Lindner, M.; Hoffmann, G.F.; Matern, D. Newborn screening for disorders of fatty-acid oxidation: Experience and recommendations from an expert meeting. J. Inherit. Metab. Dis. 2010, 33, 521–526. [Google Scholar] [CrossRef]
- Zytkovicz, T.H.; Fitzgerald, E.F.; Marsden, D.; Larson, C.A.; Shih, V.E.; Johnson, D.M.; Strauss, A.W.; Comeau, A.M.; Eaton, R.B.; Grady, G.F. Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: A two-year summary from the New England Newborn Screening Program. Clin. Chem. 2001, 47, 1945–1955. [Google Scholar] [CrossRef]
- McCann, M.R.; George De la Rosa, M.V.; Rosania, G.R.; Stringer, K.A. L-carnitine and acylcarnitines: Mitochondrial biomarkers for precision medicine. Metabolites 2021, 11, 51. [Google Scholar] [CrossRef]
- Lotz-Havla, A.S.; Röschinger, W.; Schiergens, K.; Singer, K.; Karall, D.; Konstantopoulou, V.; Wortmann, S.B.; Maier, E.M. Fatal pitfalls in newborn screening for mitochondrial trifunctional protein (MTP)/long-chain 3-Hydroxyacyl-CoA dehydrogenase (LCHAD) deficiency. Orphanet J. Rare Dis. 2018, 13, 122. [Google Scholar] [CrossRef]
- Spiekerkoetter, U.; Haussmann, U.; Mueller, M.; ter Veld, F.; Stehn, M.; Santer, R.; Lukacs, Z. Tandem mass spectrometry screening for very long-chain acylCoA dehydrogenase deficiency: The value of second-tier enzyme testing. J. Pediatr. 2010, 157, 668–673. [Google Scholar] [CrossRef] [PubMed]
- Okun, J.G.; Kölker, S.; Schulze, A.; Kohlmüller, D.; Olgemöller, K.; Lindner, M.; Hoffmann, G.F.; Wanders, R.J.A.; Mayatepek, E. A method for quantitative acylcarnitine profiling in human skin fibroblasts using unlabelled palmitic acid: Diagnosis of fatty acid oxidation disorders and differentiation between biochemical phenotypes of MCAD deficiency. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2002, 1584, 91–98. [Google Scholar] [CrossRef]
- Diekman, E.; de Sain-van der Velden, M.; Waterham, H.; Kluijtmans, L.; Schielen, P.; van Veen, E.B.; Ferdinandusse, S.; Wijburg, F.; Visser, G. The newborn screening paradox: Sensitivity vs. Overdiagnosis in VLCAD deficiency. JIMD Rep. 2016, 27, 101–106. [Google Scholar] [PubMed]
- Fingerhut, R.; Roschinger, W.; Muntau, A.C.; Röschinger, W.; Muntau, A.C.; Dame, T.; Kreischer, J.; Arnecke, R.; Superti-Furga, A.; Troxler, H.; et al. Hepatic carnitine palmitoyltransferase I deficiency: Acylcarnitine profiles in blood spots are highly specific. Clin. Chem. 2001, 47, 1763–1768. [Google Scholar] [CrossRef]
- McHugh, D.; Cameron, C.A.; Abdenur, J.E.; Abdulrahman, M.; Adair, O.; Al Nuaimi, S.A.; Åhlman, H.; Allen, J.J.; Antonozzi, I.; Archer, S.; et al. Clinical validation of cutoff target ranges in newborn screening of metabolic disorders by tandem mass spectrometry: A worldwide collaborative project. Genet. Med. 2011, 13, 230–254. [Google Scholar] [CrossRef]

{kind=link}
| C0 | C14:1 | C16:1OH | C16OH | C18:1OH | C18OH | “HADHA Ratio” | C16OH/C16 | |
|---|---|---|---|---|---|---|---|---|
| 1 | 4.4 ± 1.8 | 0.21 ± 0.11 | 0.14 ± 0.09 | 0.32 ± 0.20 | 0.49 ± 0.25 | 0.37 ± 0.15 | 0.29 ± 0.14 | 0.43 ± 0.19 |
| 2 | 17.5 ± 8.5 | 0.22 ± 0.09 | 0.12 ± 0.11 | 0.45 ± 0.3 | 0.64 ± 0.77 | 0.5 ± 0.31 | 0.1 ± 0.1 | 0.39 ± 0.15 |
| 3 | 24.0 ± 18.4 | 0.68 ± 0.3 | 0.31 ± 0.14 | 1.03 ± 0.51 | 1.05 ± 0.57 | 0.99 ± 0.62 | 0.21 ± 0.13 | 0.48 ± 0.16 |
| 4 | 5.3 ± 2.2 | 0.08 ± 0.05 | 0.03 ± 0.03 | 0.08 ± 0.01 | 0.09 ± 0.03 | 0.1 ± 0.05 | 0.06 ± 0.01 | 0.21 ± 0.07 |
| RI | 8–90 | <0.32 | <0.22 | <0.18 | <0.16 | <0.15 | <0.027 | <0.1 * |
| AC or Ratio | AUC |
|---|---|
| C16OH | 0.981 |
| C16:1OH | 0.851 |
| C18OH | 0.999 |
| C18:1OH | 0.999 |
| C16OH/C16 | 0.982 |
| “HADHA Ratio” | 1 |
| Cut-Off | Sensitivity (%) | 95% CI | Specificity (%) | 95% CI |
|---|---|---|---|---|
| >0.0085 | 100 | 93.4% to 100% | 99.0 | 97.68% to 99.67% |
| >0.014 | 100 | 93.4% to 100% | 99.8 | 98.89% to 99.99% |
| >0.027 | 100 | 93.4% to 100% | 100 | 99.26% to 100% |
| >0.036 | 98.2 | 90.11% to 99.95% | 100 | 99.26% to 100% |
| >0.039 | 96.3 | 87.25% to 99.55% | 100 | 99.26% to 100% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baydakova, G.V.; Tsygankova, P.G.; Pechatnikova, N.L.; Bazhanova, O.A.; Nazarenko, Y.D.; Zakharova, E.Y. New Acylcarnitine Ratio as a Reliable Indicator of Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency. Int. J. Neonatal Screen. 2023, 9, 48. https://doi.org/10.3390/ijns9030048
Baydakova GV, Tsygankova PG, Pechatnikova NL, Bazhanova OA, Nazarenko YD, Zakharova EY. New Acylcarnitine Ratio as a Reliable Indicator of Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency. International Journal of Neonatal Screening. 2023; 9(3):48. https://doi.org/10.3390/ijns9030048
Chicago/Turabian StyleBaydakova, Galina V., Polina G. Tsygankova, Natalia L. Pechatnikova, Olga A. Bazhanova, Yana D. Nazarenko, and Ekaterina Y. Zakharova. 2023. "New Acylcarnitine Ratio as a Reliable Indicator of Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency" International Journal of Neonatal Screening 9, no. 3: 48. https://doi.org/10.3390/ijns9030048
APA StyleBaydakova, G. V., Tsygankova, P. G., Pechatnikova, N. L., Bazhanova, O. A., Nazarenko, Y. D., & Zakharova, E. Y. (2023). New Acylcarnitine Ratio as a Reliable Indicator of Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency. International Journal of Neonatal Screening, 9(3), 48. https://doi.org/10.3390/ijns9030048