
A New Approach to Objectively Evaluate Inherited Metabolic Diseases for Inclusion on Newborn Screening Programmes

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
Analysis of the 10 Classic Screening Principles
3. Results
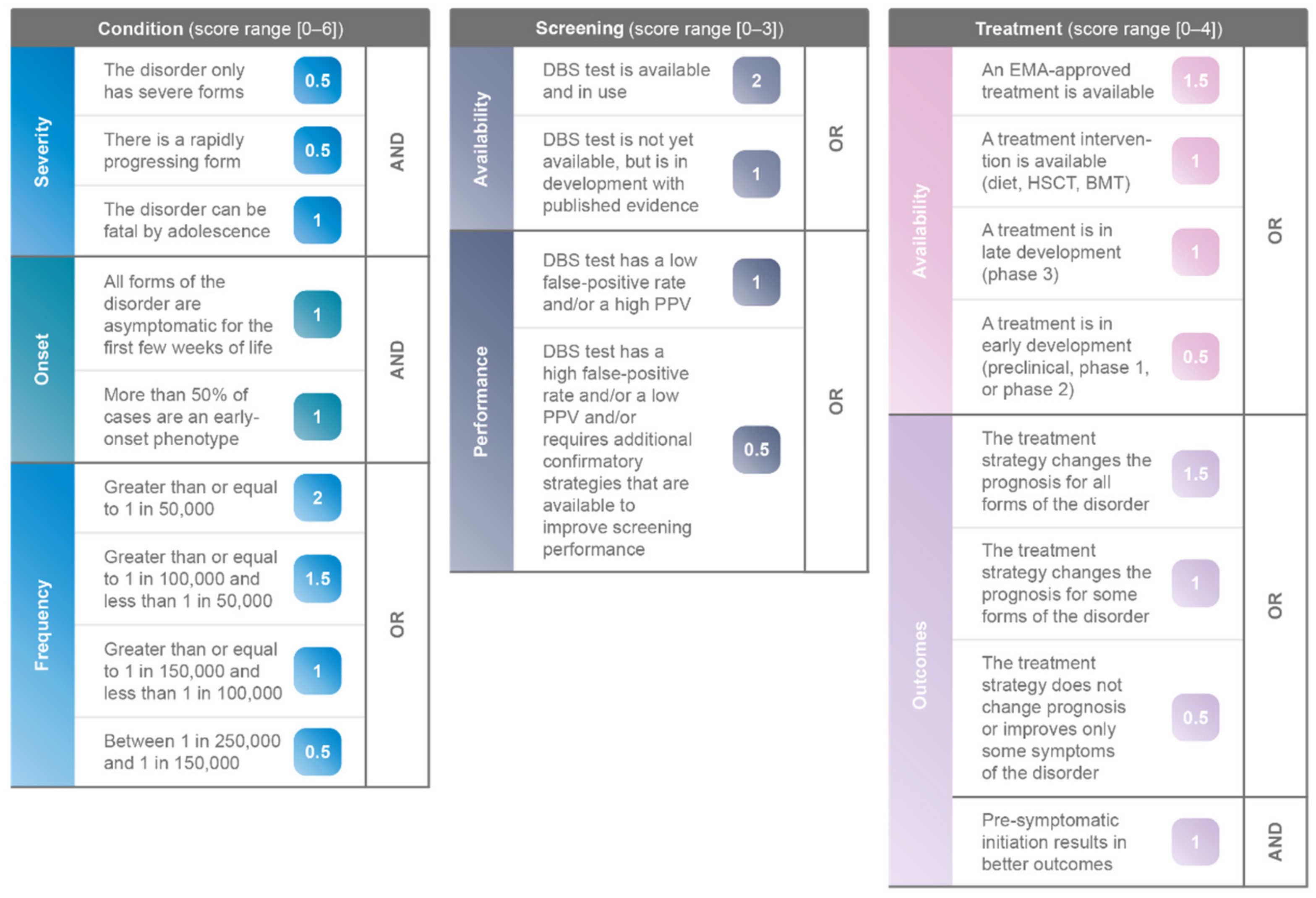
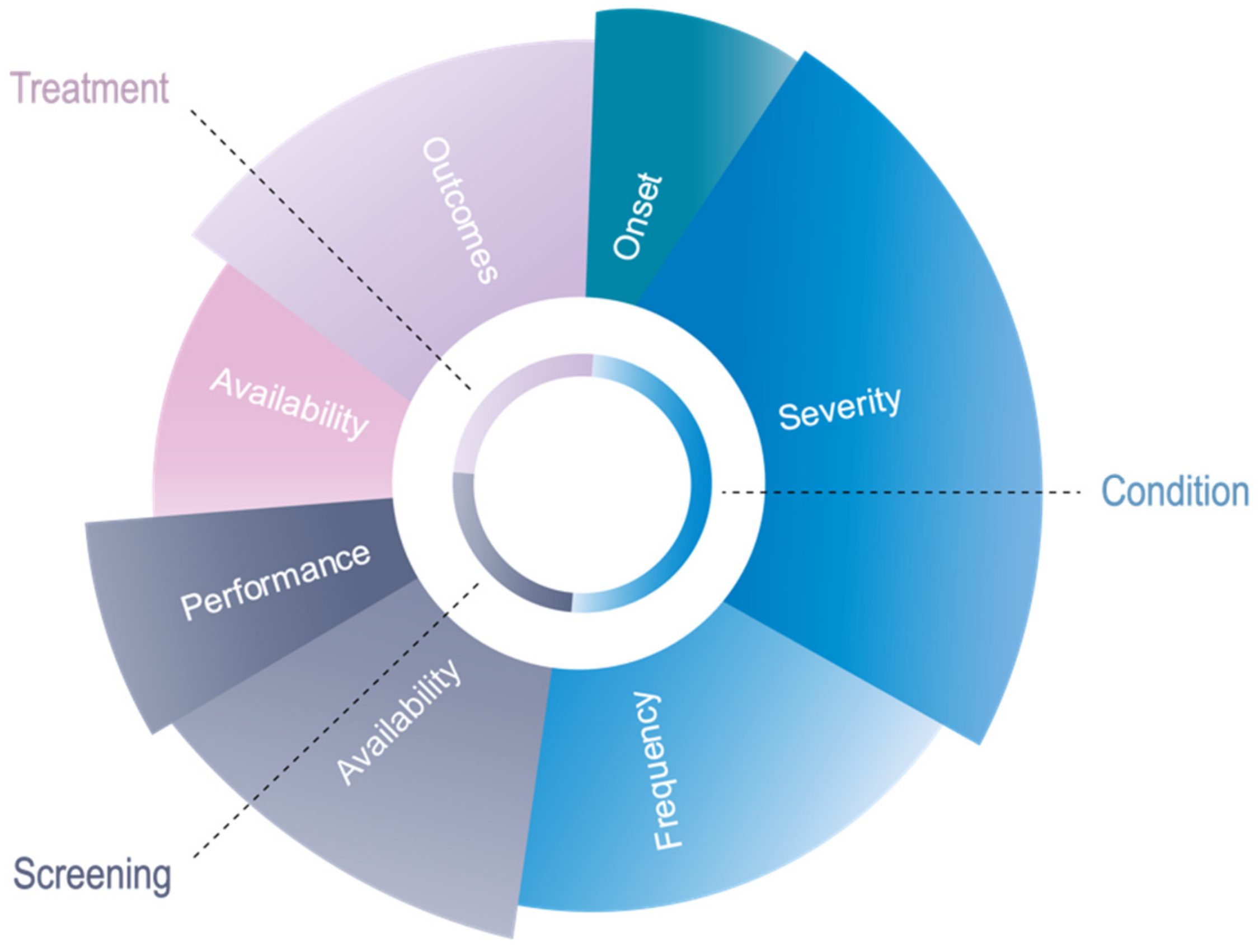
3.1. Components of Novel Algorithm
3.1.1. Pillar 1: Condition
3.1.2. Pillar 2: Screening
3.1.3. Pillar 3: Treatment
3.2. Weighting of Novel Algorithm
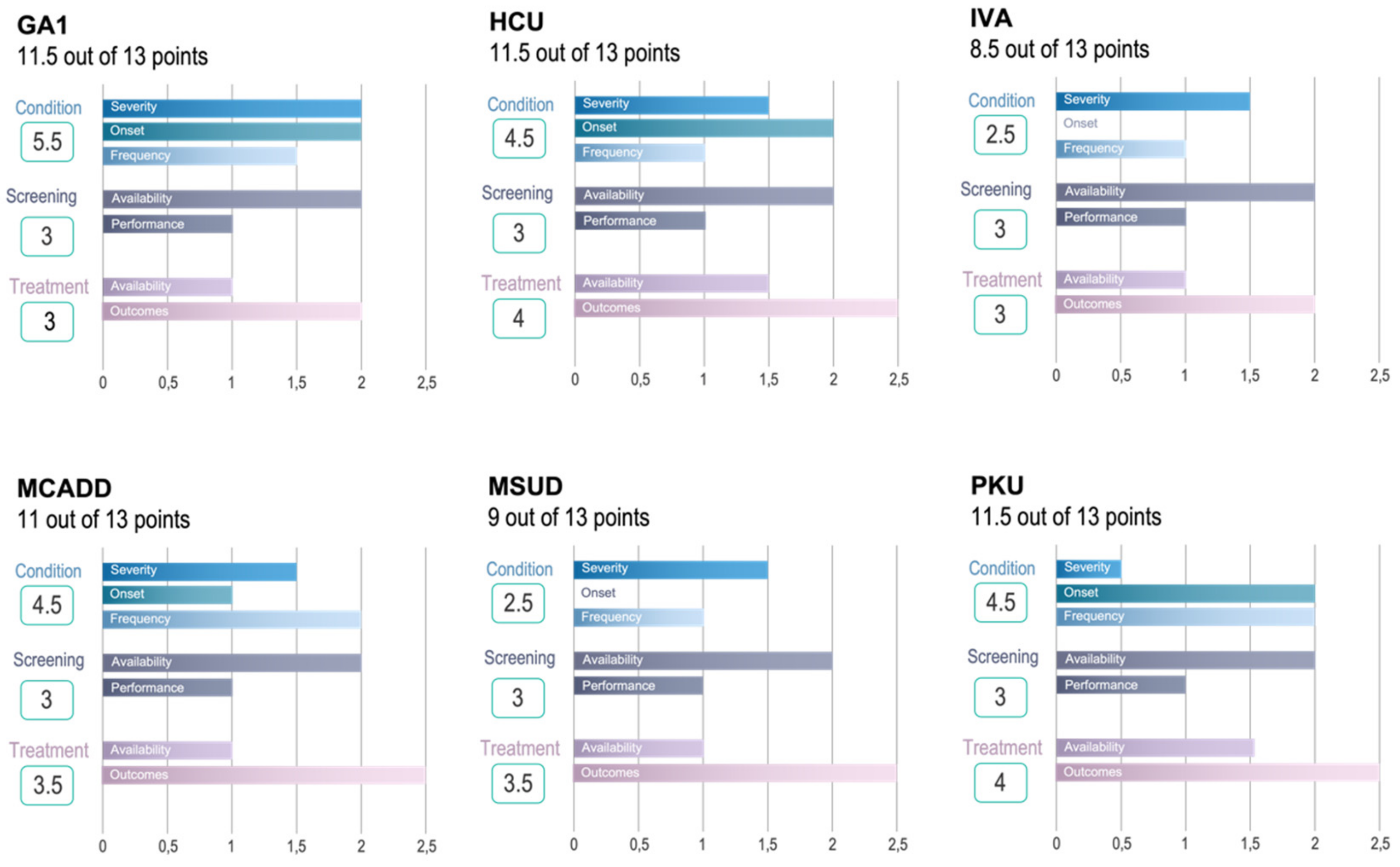
3.3. Validation of Novel Algorithm
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A. Detailed Scoring for the Six IMDs on the UK Newborn Screening Panel are Shown in the Following Tables

{kind=link}
{kind=link}
{kind=link}
| Condition (5.5 out of 6 Points) | Screening (3 out of 3 Points) | Treatment (3 out of 4 Points) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Severity | The disorder only has severe forms | 0.5 | AND | Availability | DBS test is available and in use | 2 | OR | Availability | An EMA-approved treatment is available | 0 | OR |
| There is a rapidly progressing form | 0.5 | DBS test is not yet available, but is in development with published evidence | 0 | A treatment intervention is available (diet, HSCT, BMT) | 1 | ||||||
| The disorder can be fatal by adolescence | 1 | Performance | DBS test has a low false-positive rate and/or a high PPV | 1 | OR | A treatment is in late development (phase 3) | 0 | ||||
| Onset | All forms of the disorder are asymptomatic for the first few weeks of life | 1 | AND | DBS test has a high false-positive rate and/or a low PPV and/or requires additional confirmatory strategies that are available to improve screening performance | 0 | A treatment is in early development (preclinical, phase 1, or phase 2) | 0 | ||||
| More than 50% of cases are an early-onset phenotype | 1 | Outcomes | The treatment strategy changes the prognosis for all forms of the disorder | 0 | OR | ||||||
| Frequency | Greater than or equal to 1 in 50,000 | 0 | OR | The treatment strategy changes the prognosis for some forms of the disorder | 1 | ||||||
| Greater than or equal to 1 in 100,000 and less than 1 in 50,000 | 1.5 | The treatment strategy does not change prognosis or improves only some symptoms of the disorder | 0 | ||||||||
| Greater than or equal to 1 in 150,000 and less than 1 in 100,000 | 0 | Pre-symptomatic initiation results in better outcomes | 1 | AND | |||||||
| Between 1 in 250,000 and 1 in 150,000 | 0 | ||||||||||
| Condition | Screening | Treatment | |||
|---|---|---|---|---|---|
| Severity | There is a rapidly progressing form The condition only has severe forms The condition can be fatal by adolescence - If not promptly and properly treated, GA1 will typically cause serious, irreversible neurologic damage that can permanently affect control of voluntary muscle movement and can severely impact life and shorten life expectancy, especially if damage occurs before the age of six. (Rarediseases.org) - The low excreting patients have the same risk of developing striatal injury as the high excretors and must not be considered to have a “mild” clinical phenotype. [23] | Availability | DBS test is available and in use | Availability | A therapeutic strategy is available (diet, HSCT, BMT) Treatment consists of a low lysine diet and oral carnitine supplementation as well as intermittent emergency treatment during episodes that are likely to induce catabolism. (Rarediseases.org) |
| Onset | All forms of the condition are asymptomatic for the first few weeks of life More than 50% of cases are an early-onset phenotype Neonates are mainly asymptomatic, with onset usually between three months and three years. (Rarediseases.org) | Performance | DBS test has a low false-positive rate or a high PPV [23] Note on sensitivity: Patients with the low excreting phenotype may be missed by newborn screening. (Rarediseases.org; [23]) | Outcomes | The therapeutic strategy changes the prognosis only for some forms of the condition Pre-symptomatic initiation results in better outcomes For 80–90% of people with GA1, motor symptom development is preventable, but this requires early diagnosis by newborn screening and treatment from birth on. (Rarediseases.org) |
| Frequency | Incidence/prevalence is≥1/100,000 and <1/50,000 Birth incidence is 1/100,000 [24] | ||||
| Condition (4.5 out of 6 Points) | Screening (3 out of 3 Points) | Treatment (4 out of 4 Points) | |||||||||
| Severity | The disorder only has severe forms | 0 | AND | Availability | DBS test is available and in use | 2 | OR | Availability | An EMA-approved treatment is available | 1.5 | OR |
| There is a rapidly progressing form | 0.5 | DBS test is not yet available, but is in development with published evidence | 0 | A treatment intervention is available (diet, HSCT, BMT) | 0 | ||||||
| The disorder can be fatal by adolescence | 1 | Performance | DBS test has a low false-positive rate and/or a high PPV | 1 | OR | A treatment is in late development (phase 3) | 0 | ||||
| Onset | All forms of the disorder are asymptomatic for the first few weeks of life | 1 | AND | DBS test has a high false-positive rate and/or a low PPV and/or requires additional confirmatory strategies that are available to improve screening performance | 0 | A treatment is in early development (preclinical, phase 1, or phase 2) | 0 | ||||
| More than 50% of cases are an early-onset phenotype | 1 | Outcomes | The treatment strategy changes the prognosis for all forms of the disorder | 1.5 | OR | ||||||
| Frequency | Greater than or equal to 1 in 50,000 | 0 | OR | The treatment strategy changes the prognosis for some forms of the disorder | 0 | ||||||
| Greater than or equal to 1 in 100,000 and less than 1 in 50,000 | 0 | The treatment strategy does not change prognosis or improves only some symptoms of the disorder | 0 | ||||||||
| Greater than or equal to 1 in 150,000 and less than 1 in 100,000 | 1 | Pre-symptomatic initiation results in better outcomes | 1 | AND | |||||||
| Between 1 in 250,000 and 1 in 150,000 | 0 | ||||||||||
| Condition | Screening | Treatment | |||
|---|---|---|---|---|---|
| Severity | There is a rapidly progressing form The condition can be fatal by adolescence Infants with untreated HCU will slowly develop the various symptoms associated with the disorder. The major cause of early death are blood clots, especially in untreated patients. (Rarediseases.org) | Availability | DBS test is available and in use | Availability | An EMA-approved therapy is available Betaine anhydrous is a methyl donor that may lead to lowering of homocysteine levels in these individuals and can be used as an adjunct to such a diet. It obtained EU marketing authorization as an orphan drug for the treatment of HCU in 2007. (Orphanet) |
| Onset | All forms of the condition are asymptomatic for the first few weeks of life More than 50% of cases are an early-onset phenotype The signs and symptoms typically develop within the first year of life. (Rarediseases.info) Some mildly affected people may not develop features until later in childhood or adulthood. (Rarediseases.info) | Performance | DBS test has a low false-positive rate or a high PPV | Outcomes | The therapeutic strategy changes the prognosis for all forms of the condition Pre-symptomatic initiation results in better outcomes If the disease is diagnosed in the newborn infant, the treatment may ensure the development of normal intelligence and prevent the development of other complications. If treatment starts later, it can prevent life-endangering thromboembolic events and further escalation of the complications. |
| Frequency | Incidence/prevalence is≥1/150,000 and <1/100,000 Prevalence is 1:144,000 in the UK. (expandedscreening.org) | ||||
| Condition (2.5 out of 6 Points) | Screening (3 out of 3 Points) | Treatment (3 out of 4 Points) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Severity | The disorder only has severe forms | 0 | AND | Availability | DBS test is available and in use | 2 | OR | Availability | An EMA-approved treatment is available | 0 | OR |
| There is a rapidly progressing form | 0.5 | DBS test is not yet available, but is in development with published evidence | 0 | A treatment intervention is available (diet, HSCT, BMT) | 1 | ||||||
| The disorder can be fatal by adolescence | 1 | Performance | DBS test has a low false-positive rate and/or a high PPV | 1 | OR | A treatment is in late development (phase 3) | 0 | ||||
| Onset | All forms of the disorder are asymptomatic for the first few weeks of life | 0 | AND | DBS test has a high false-positive rate and/or a low PPV and/or requires additional confirmatory strategies that are available to improve screening performance | 0 | A treatment is in early development (preclinical, phase 1, or phase 2) | 0 | ||||
| More than 50% of cases are an early-onset phenotype | 0 | Outcomes | The treatment strategy changes the prognosis for all forms of the disorder | 0 | OR | ||||||
| Frequency | Greater than or equal to 1 in 50,000 | 0 | OR | The treatment strategy changes the prognosis for some forms of the disorder | 1 | ||||||
| Greater than or equal to 1 in 100,000 and less than 1 in 50,000 | 0 | The treatment strategy does not change prognosis or improves only some symptoms of the disorder | 0 | ||||||||
| Greater than or equal to 1 in 150,000 and less than 1 in 100,000 | 1 | Pre-symptomatic initiation results in better outcomes | 1 | AND | |||||||
| Between 1 in 250,000 and 1 in 150,000 | 0 | ||||||||||
| Condition | Screening | Treatment | |||
|---|---|---|---|---|---|
| Severity | There is a rapidly progressing form About half of babies identified through newborn screening have a very mild deficiency that remains asymptomatic and requires no therapy. (Rarediseases.org) Acute presentation progresses to coma. Two major clinical scenarios are often described, an acute form and a chronic intermittent form, but in reality the disease is best thought of as a continuous spectrum from asymptomatic to life threatening. (Rarediseases.org) | Availability | DBS test is available and in use | Availability | A therapeutic strategy is available (diet, HSCT, BMT) Lifelong management is with a low protein diet. (Orphanet) |
| Onset | All forms of the condition are asymptomatic for the first few weeks of life More than 50% of cases are an early-onset phenotype Acute, neonatal presentation is characterized by onset in the first two weeks of life. Later onset is relatively non-specific with failure to thrive and/or developmental delay. (Rarediseases.org) About half of babies identified through newborn screening have a very mild deficiency that remains asymptomatic and requires no therapy. (Rarediseases.org) The proportion of neonatal onset cases versus later onset cases is unknown. | Performance | DBS test has a low false-positive rate or a high PPV [23] | Outcomes | The therapeutic strategy changes the prognosis for all forms of the condition Pre-symptomatic initiation results in better outcomes If identified prior to the development of symptoms, outcomes are generally better, with normal growth and development. (Rarediseases) Patients who present symptomatically can have significant neurologic sequelae including neurodevelopmental delay, especially if acidosis and hyperammonemia are severe. (Orphanet) |
| Frequency | Incidence/prevalence is≥1/150,000 and <1/100,000 Birth incidence is 0.83/100,000 (~1/120,000) [24] | ||||
| Condition (4.5 out of 6 Points) | Screening (3 out of 3 Points) | Treatment (3.5 out of 4 Points) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Severity | The disorder only has severe forms | 0 | AND | Availability | DBS test is available and in use | 2 | OR | Availability | An EMA-approved treatment is available | 0 | OR | |
| There is a rapidly progressing form | 0.5 | DBS test is not yet available, but is in development with published evidence | 0 | A treatment intervention is available (diet, HSCT, BMT) | 1 | |||||||
| The disorder can be fatal by adolescence | 1 | Performance | DBS test has a low false-positive rate and/or a high PPV | 1 | OR | A treatment is in late development (phase 3) | 0 | |||||
| Onset | All forms of the disorder are asymptomatic for the first few weeks of life | 1 | AND | DBS test has a high false-positive rate and/or a low PPV and/or requires additional confirmatory strategies that are available to improve screening performance | 0 | A treatment is in early development (preclinical, phase 1, or phase 2) | 0 | |||||
| More than 50% of cases are an early-onset phenotype | 0 | Outcomes | The treatment strategy changes the prognosis for all forms of the disorder | 1.5 | OR | |||||||
| Frequency | Greater than or equal to 1 in 50,000 | 2 | OR | The treatment strategy changes the prognosis for some forms of the disorder | 0 | |||||||
| Greater than or equal to 1 in 100,000 and less than 1 in 50,000 | 0 | The treatment strategy does not change prognosis or improves only some symptoms of the disorder | 0 | |||||||||
| Greater than or equal to 1 in 150,000 and less than 1 in 100,000 | 0 | Pre-symptomatic initiation results in better outcomes | 1 | AND | ||||||||
| Between 1 in 250,000 and 1 in 150,000 | 0 | |||||||||||
| Condition | Screening | Treatment | |||
|---|---|---|---|---|---|
| Severity | There is a rapidly progressing form The condition can be fatal by adolescence About 25% of undiagnosed patients die during their first presentation of a crisis. (Orphanet) | Availability | DBS test is available and in use | Availability | A therapeutic strategy is available (diet, HSCT, BMT) Diet: Strict avoidance of fasting is the primary objective. Medium chain triglycerides should be avoided but no other special dietary restrictions are required. (Orphanet) |
| Onset | All forms of the condition are asymptomatic for the first few weeks of life More than 50% of cases are an early-onset phenotype - MCADD usually presents 3threeto 24 months after birth in previously healthy infants. (Orphanet) - Many affected individuals remain asymptomatic throughout life. (Orphanet) | Performance | DBS test has a low false-positive rate or a high PPV [25] | Outcomes | The therapeutic strategy changes the prognosis for all forms of the condition Without treatment MCADD can be fatal. About 25% of undiagnosed patients die during their first presentation of a crisis. (Orphanet) Pre-symptomatic initiation results in better outcomes |
| Frequency | Incidence/prevalence is≥1/50.000 MCADD is estimated to affect up to 1/10,000 babies born in the UK. (www.nhs.uk/conditions/mcadd, accessed on 31 May 2021) | ||||
| Condition (2.5 out of 6 Points) | Screening (3 out of 3 Points) | Treatment (3.5 out of 4 Points) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Severity | The disorder only has severe forms | 0 | AND | Availability | DBS test is available and in use | 2 | OR | Availability | An EMA-approved treatment is available | 0 | OR |
| There is a rapidly progressing form | 0.5 | DBS test is not yet available, but is in development with published evidence | 0 | A treatment intervention is available (diet, HSCT, BMT) | 1 | ||||||
| The disorder can be fatal by adolescence | 1 | Performance | DBS test has a low false-positive rate and/or a high PPV | 1 | OR | A treatment is in late development (phase 3) | 0 | ||||
| Onset | All forms of the disorder are asymptomatic for the first few weeks of life | 0 | AND | DBS test has a high false-positive rate and/or a low PPV and/or requires additional confirmatory strategies that are available to improve screening performance | 0 | A treatment is in early development (preclinical, phase 1, or phase 2) | 0 | ||||
| More than 50% of cases are an early-onset phenotype | 0 | Outcomes | The treatment strategy changes the prognosis for all forms of the disorder | 1.5 | OR | ||||||
| Frequency | Greater than or equal to 1 in 50,000 | 0 | OR | The treatment strategy changes the prognosis for some forms of the disorder | 0 | ||||||
| Greater than or equal to 1 in 100,000 and less than 1 in 50,000 | 1 | The treatment strategy does not change prognosis or improves only some symptoms of the disorder | 0 | ||||||||
| Greater than or equal to 1 in 150,000 and less than 1 in 100,000 | 0 | Pre-symptomatic initiation results in better outcomes | 1 | AND | |||||||
| Between 1 in 250,000 and 1 in 150,000 | 0 | ||||||||||
| Condition | Screening | Treatment | |||
|---|---|---|---|---|---|
| Severity | There is a rapidly progressing form The condition can be fatal by adolescence Classic MSUD: If untreated, progressive brain damage is inevitable and death occurs usually within weeks or months. (Rarediseases.org) | Availability | DBS test is available and in use | Availability | A therapeutic strategy is available (diet, HSCT, BMT) The treatment of classic, intermediate, intermittent, and thiamine-responsive MSUD has three chief components: 1. Lifelong therapy to maintain an acceptable diet; 2. Life-long maintenance of normal metabolic conditions including the levels of the BCAAs in the body; 3. immediate medical intervention for metabolic crises. (Rarediseases) Even if affected individuals follow the specialized diet strictly, the risk of metabolic crisis always remains. (Rarediseases) |
| Onset | Onset of classic MSUD (50–75% of cases) (Orphanet) occurs in the neonatal period, usually within the first 24–48 h of life. (https://www.ncbi.nlm.nih.gov/gtr/conditions/C0024776, accessed on 31 May 2021) | Performance | DBS test has a low false-positive rate or a high PPV [24] | Outcomes | The therapeutic strategy changes the prognosis for all forms of the condition Pre-symptomatic initiation results in better outcomes If identified prior to the development of symptoms, outcomes are generally better, with normal growth and development. (Rarediseases) Patients who present symptomatically can have significant neurologic sequelae including neurodevelopmental delay, especially if acidosis and hyperammonemia are severe. (Orphanet) |
| Frequency | Incidence/prevalence is≥1/150,000 and <1/100,000 Birth incidence is 0.74/100,000 (~1/137,000) [24] | ||||
| Condition (4.5 out of 6 Points) | Screening (3 out of 3 Points) | Treatment (4 out of 4 Points) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Severity | The disorder only has severe forms | 0 | AND | Availability | DBS test is available and in use | 2 | OR | Availability | An EMA-approved treatment is available | 1.5 | OR |
| There is a rapidly progressing form | 0.5 | DBS test is not yet available, but is in development with published evidence | 0 | A treatment intervention is available (diet, HSCT, BMT) | 0 | ||||||
| The disorder can be fatal by adolescence | 0 | Performance | DBS test has a low false-positive rate and/or a high PPV | 1 | OR | A treatment is in late development (phase 3) | 0 | ||||
| Onset | All forms of the disorder are asymptomatic for the first few weeks of life | 1 | AND | DBS test has a high false-positive rate and/or a low PPV and/or requires additional confirmatory strategies that are available to improve screening performance | 0 | A treatment is in early development (preclinical, phase 1, or phase 2) | 0 | ||||
| More than 50% of cases are an early-onset phenotype | 1 | Outcomes | The treatment strategy changes the prognosis for all forms of the disorder | 1.5 | OR | ||||||
| Frequency | Greater than or equal to 1 in 50,000 | 2 | OR | The treatment strategy changes the prognosis for some forms of the disorder | 0 | ||||||
| Greater than or equal to 1 in 100,000 and less than 1 in 50,000 | 0 | The treatment strategy does not change prognosis or improves only some symptoms of the disorder | 0 | ||||||||
| Greater than or equal to 1 in 150,000 and less than 1 in 100,000 | 0 | Pre-symptomatic initiation results in better outcomes | 1 | AND | |||||||
| Between 1 in 250,000 and 1 in 150,000 | 0 | ||||||||||
| Condition | Screening | Treatment | |||
|---|---|---|---|---|---|
| Severity | There is a rapidly progressing form | Availability | DBS test is available and in use | Availability | An EMA-approved therapy is available - Palynziq is indicated for the treatment of patients with PKU aged 16 years and older who have inadequate blood phenylalanine control despite prior management with available treatment options. - Kuvan is indicated for the treatment of hyperphenylalaninaemia in adults and paediatric patients of all ages with PKU who have been shown to be responsive to such treatment. |
| Onset | All forms of the condition are asymptomatic for the first few weeks of life More than 50% of cases are an early-onset phenotype - Infants with PKU typically appear normal at birth. Symptoms develop within a few months of birth. (Orphanet) - Classical phenylketonuria is the most common form | Performance | DBS test has a low false-positive rate or a high PPV | Outcomes | The therapeutic strategy changes the prognosis for all forms of the condition Pre-symptomatic initiation results in better outcomes |
| Frequency | Incidence/prevalence is ≥1/50,000 Prevalence 1/10,000 live births in Europe (Orphanet) | ||||
References
- Kelly, N.; Makarem, D.C.; Wasserstein, M.P. Screening of Newborns for Disorders with High Benefit-Risk Ratios Should Be Mandatory. J. Law Med. Ethics 2016, 44, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Lehotay, D.C.; Hall, P.; Lepage, J.; Eichhorst, J.C.; Etter, M.L.; Greenberg, C.R. LC-MS/MS Progress in Newborn Screening. Clin. Biochem. 2011, 44, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Jungner, Y.G. Principles and practice of mass screening for disease. Bol. Oficina Sanit. Panam. 1968, 65, 281–393. [Google Scholar] [PubMed]
- Pitt, J.J. Newborn Screening. Clin. Biochem. Rev. 2010, 31, 57–68. [Google Scholar] [PubMed]
- Loeber, J.G.; Platis, D.; Zetterström, R.H.; Almashanu, S.; Boemer, F.; Bonham, J.R.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal Screening in Europe Revisited: An ISNS Perspective on the Current State and Developments Since 2010. Int. J. Neonatal Screen. 2021, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Andermann, A.; Blancquaert, I.; Beauchamp, S.; Costea, I. Guiding Policy Decisions for Genetic Screening: Developing a Systematic and Transparent Approach. Public Health Genom. 2011, 14, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Orphanet. Classic Maple Syrup Urine Disease. Page Last Updated in April 2014. Available online: https://www.orpha.net/consor/cgi-bin/Disease_Search.php?lng=EN&data_id=20168&Disease_Disease_Search_diseaseGroup=MSUD&Disease_Disease_Search_diseaseType=Pat&Disease(s)/group%20of%20diseases=Classic-maple-syrup-urine-disease&title=Classic%20maple%20syrup%20urine%20disease&search=Disease_Search_Simple (accessed on 31 May 2021).
- Knottnerus, S.J.G.; Pras-Raves, M.L.; van der Ham, M.; Ferdinandusse, S.; Houtkooper, R.H.; Schielen, P.C.J.I.; Visser, G.; Wijburg, F.A.; de Sain-van der Velden, M.G.M. Prediction of VLCAD Deficiency Phenotype by a Metabolic Fingerprint in Newborn Screening Bloodspots. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165725. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Morillo, E.; Prieto García, B.; Álvarez Menéndez, F.V. Challenges for Worldwide Harmonization of Newborn Screening Programs. Clin. Chem. 2016, 62, 689–698. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cedars Sinai. Carnitine Palmitoyltransferase Deficiency. Available online: https://www.cedars-sinai.org/health-library/diseases-and-conditions/c/carnitine-palmitoyl-transferase-deficiency.html (accessed on 31 May 2021).
- MedlinePlus. Homocystinuria. Page last updated on 18 August 2020. Available online: https://medlineplus.gov/genetics/condition/homocystinuria/ (accessed on 21 May 2021).
- Lisi, E.C.; Gillespie, S.; Laney, D.; Ali, N. Patients’ Perspectives on Newborn Screening for Later-Onset Lysosomal Storage Diseases. Mol. Genet. Metab. 2016, 119, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Richter, T.; Nestler-Parr, S.; Babela, R.; Khan, Z.M.; Tesoro, T.; Molsen, E.; Hughes, D.A.; International Society for Pharmacoeconomics and Outcomes Research Rare Disease Special Interest Group. Rare Disease Terminology and Definitions-A Systematic Global Review: Report of the ISPOR Rare Disease Special Interest Group. Value Health 2015, 18, 906–914. [Google Scholar] [CrossRef] [PubMed]
- George, R.S.; Moat, S.J. Effect of Dried Blood Spot Quality on Newborn Screening Analyte Concentrations and Recommendations for Minimum Acceptance Criteria for Sample Analysis. Clin. Chem. 2016, 62, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Maxim, L.D.; Niebo, R.; Utell, M.J. Screening Tests: A Review with Examples. Inhal. Toxicol. 2014, 26, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Wiltink, R.C.; Kruijshaar, M.E.; van Minkelen, R.; Onkenhout, W.; Verheijen, F.W.; Kemper, E.A.; van Spronsen, F.J.; van der Ploeg, A.T.; Niezen-Koning, K.E.; Saris, J.J.; et al. Neonatal Screening for Profound Biotinidase Deficiency in the Netherlands: Consequences and Considerations. Eur. J. Hum. Genet. 2016, 24, 1424–1429. [Google Scholar] [CrossRef] [PubMed]
- Donati, M.A.; Pasquini, E.; Spada, M.; Polo, G.; Burlina, A. Newborn Screening in Mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44 (Suppl 2), 126. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. From Laboratory to Patient: The Journey of a Medicine Assessed by EMA. 2019. Available online: https://www.ema.europa.eu/en/documents/other/laboratory-patient-journey-centrally-authorised-medicine_en.pdf (accessed on 21 May 2021).
- Cornel, M.; Rigter, T.; Weinreich, S.; Burgard, P.; Hoffmann, G.F.; Lindner, M.; Loeber, J.G.; Rupp, K.; Taruscio, D.; Vittozzi, L.; et al. Newborn Screening in Europe; Expert Opinion document. EU Network of Experts on Newborn Screening. 2011. Available online: https://isns-neoscreening.org/wp-content/uploads/2016/06/Expert-opinion-document-on-NBS-FINAL.pdf (accessed on 5 June 2021).
- Castiñeras, D.E.; Couce, M.-L.; Marin, J.L.; González-Lamuño, D.; Rocha, H. Newborn screening for metabolic disorders in Spain and worldwide. An. Pediatr. 2019, 91, 128.e1–128.e14. [Google Scholar] [CrossRef]
- Haute Autorité de Santé. Évaluation a Priori de l’Extension du Dépistage Néonatal à Une ou Plusieurs Erreurs Innées du Métabolisme par la Technique de Spectrométrie de Masse en Tandem en Population Générale en France (volet 2). Service Evaluation Economique et Santé Publique. 2020. Available online: https://www.has-sante.fr (accessed on 20 December 2021).
- Kellar-Guenther, Y.; McKasson, S.; Hale, K.; Singh, S.; Sontag, M.K.; Ojodu, J. Implementing Statewide Newborn Screening for New Disorders: U.S. Program Experiences. Int. J. Neonatal Screen. 2020, 6, 35. [Google Scholar] [CrossRef] [PubMed]
- Kölker, S.; Christensen, E.; Leonard, J.V.; Greenberg, C.R.; Boneh, A.; Burlina, A.B.; Burlina, A.P.; Dixon, M.; Duran, M.; García Cazorla, A.; et al. Diagnosis and Management of Glutaric Aciduria Type I—Revised Recommendations. J. Inherit. Metab. Dis. 2011, 34, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Bessey, A.; Chilcott, J.; Pandor, A.; Paisley, S. The Cost-Effectiveness of Expanding the UK Newborn Bloodspot Screening Programme to Include Five Additional Inborn Errors of Metabolism. Int. J. Neonatal Screen. 2020, 6, E93. [Google Scholar] [CrossRef] [PubMed]
- Oerton, J.; Khalid, J.M.; Besley, G.; Dalton, R.N.; Downing, M.; Green, A.; Henderson, M.; Krywawych, S.; Leonard, J.; Andresen, B.S.; et al. Newborn Screening for Medium Chain Acyl-CoA Dehydrogenase Deficiency in England: Prevalence, Predictive Value and Test Validity Based on 1.5 million Screened Babies. J. Med. Screen. 2011, 18, 173–181. [Google Scholar] [CrossRef] [PubMed]



| Wilson and Jungner Classic Screening Principles [3] | Category | |
|---|---|---|
| 1 | The condition sought should be an important health problem | Condition |
| 2 | There should be an accepted treatment for patients with recognised disease | Treatment |
| 3 | Facilities for diagnosis and treatment should be available | Other |
| 4 | There should be a recognisable latent or early symptomatic stage | Condition |
| 5 | There should be a suitable test or examination | Screening |
| 6 | The test should be acceptable to the population | Screening |
| 7 | The natural history of the condition, including development from latent to declared disease, should be adequately understood | Condition |
| 8 | There should be an agreed policy on whom to treat as patients | Other |
| 9 | The cost of case-finding (including diagnosis and treatment of patients diagnosed) should be economically balanced in relation to possible expenditures on medical care as a whole | Other |
| 10 | Case-finding should be a continual process and not a “once and for all” project | Other |
| Pillar | Wilson and Jungner Classic Screening Principles |
|---|---|
| Pillar 1: Condition | Principle 1: The condition sought should be an important health problem |
| Principle 4: There should be a recognisable latent or early symptomatic stage | |
| Principle 7: The natural history of the condition, including development from latent to declared disease, should be adequately understood | |
| Pillar 2: Screening | Principle 5: There should be a suitable test or examination |
| Principle 6: The test should be acceptable to the population | |
| Pillar 3: Treatment | Principle 2: There should be an accepted treatment for patients with recognised disease |
| Parameter. | Description | Score | Interaction |
|---|---|---|---|
| Severity | The disorder only has severe forms | 0.5 | AND |
| There is a rapidly progressing form | 0.5 | ||
| The disorder can be fatal by adolescence | 1 | ||
| Onset | All forms of the disorder are asymptomatic for the first few weeks of life | 1 | AND |
| More than 50% of cases are an early-onset phenotype | 1 | ||
| Frequency | Greater than or equal to 1 in 50,000 | 2 | OR |
| Greater than or equal to 1 in 100,000 and less than 1 in 50,000 | 1.5 | ||
| Greater than or equal to 1 in 150,000 and less than 1 in 100,000 | 1 | ||
| Between 1 in 250,000 and 1 in 150,000 | 0.5 |
| Parameter | Description | Score | Interaction |
|---|---|---|---|
| Availability | DBS test is available and in use | 2 | OR |
| DBS test is not yet available, but is in development with published evidence | 1 | ||
| Performance | DBS test has a low false-positive rate and/or a high positive predictive value (PPV) | 1 | OR |
| DBS test has a high false-positive rate and/or a low PPV and/or requires additional confirmatory strategies that are available to improve screening performance | 0.5 |
| Parameter | Description | Score | Interaction |
|---|---|---|---|
| Availability | An EMA-approved treatment is available | 1.5 | AND |
| A treatment intervention is available (diet, HSCT, BMT) | 1 | ||
| A treatment is in late development (phase 3) | 1 | ||
| A treatment is in early development (preclinical, phase 1, or phase 2) | 0.5 | ||
| Outcomes | The treatment strategy changes the prognosis for all forms of the disorder | 1.5 | OR |
| The treatment strategy changes the prognosis for some forms of the disorder | 1 | ||
| The treatment strategy does not change prognosis or improves only some symptoms of the disorder | 0.5 | ||
| Pre-symptomatic initiation results in better outcomes | 1 | AND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burlina, A.; Jones, S.A.; Chakrapani, A.; Church, H.J.; Heales, S.; Wu, T.H.Y.; Morton, G.; Roberts, P.; Sluys, E.F.; Cheillan, D. A New Approach to Objectively Evaluate Inherited Metabolic Diseases for Inclusion on Newborn Screening Programmes. Int. J. Neonatal Screen. 2022, 8, 25. https://doi.org/10.3390/ijns8020025
Burlina A, Jones SA, Chakrapani A, Church HJ, Heales S, Wu THY, Morton G, Roberts P, Sluys EF, Cheillan D. A New Approach to Objectively Evaluate Inherited Metabolic Diseases for Inclusion on Newborn Screening Programmes. International Journal of Neonatal Screening. 2022; 8(2):25. https://doi.org/10.3390/ijns8020025
Chicago/Turabian StyleBurlina, Alberto, Simon A. Jones, Anupam Chakrapani, Heather J. Church, Simon Heales, Teresa H. Y. Wu, Georgina Morton, Patricia Roberts, Erica F. Sluys, and David Cheillan. 2022. "A New Approach to Objectively Evaluate Inherited Metabolic Diseases for Inclusion on Newborn Screening Programmes" International Journal of Neonatal Screening 8, no. 2: 25. https://doi.org/10.3390/ijns8020025
APA StyleBurlina, A., Jones, S. A., Chakrapani, A., Church, H. J., Heales, S., Wu, T. H. Y., Morton, G., Roberts, P., Sluys, E. F., & Cheillan, D. (2022). A New Approach to Objectively Evaluate Inherited Metabolic Diseases for Inclusion on Newborn Screening Programmes. International Journal of Neonatal Screening, 8(2), 25. https://doi.org/10.3390/ijns8020025