
Use of Molecular Genetic Analyses in Danish Routine Newborn Screening

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
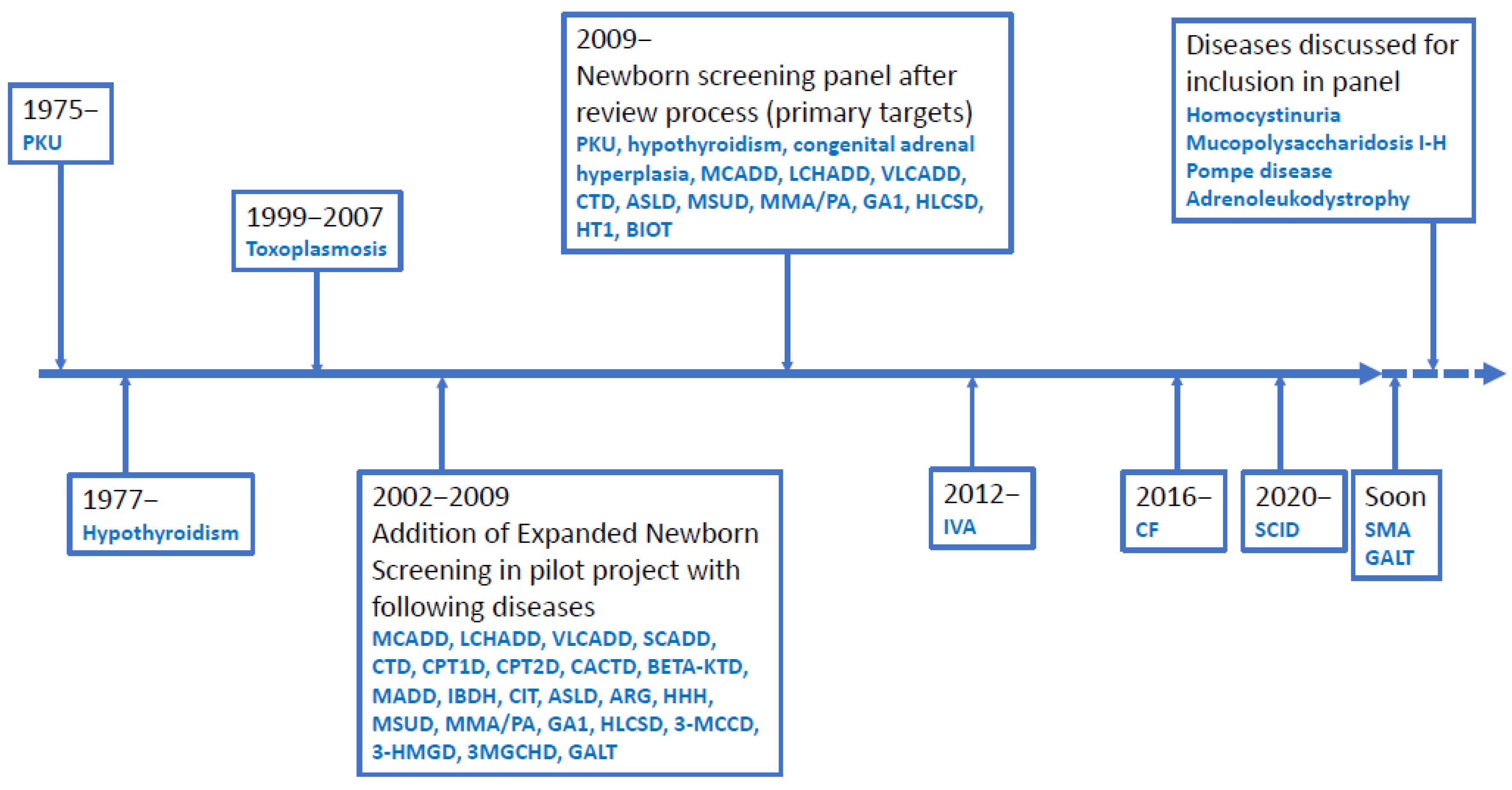
2.1. General Set-Up of Danish Newborn Screening
2.2. Molecular Genetic Analyses
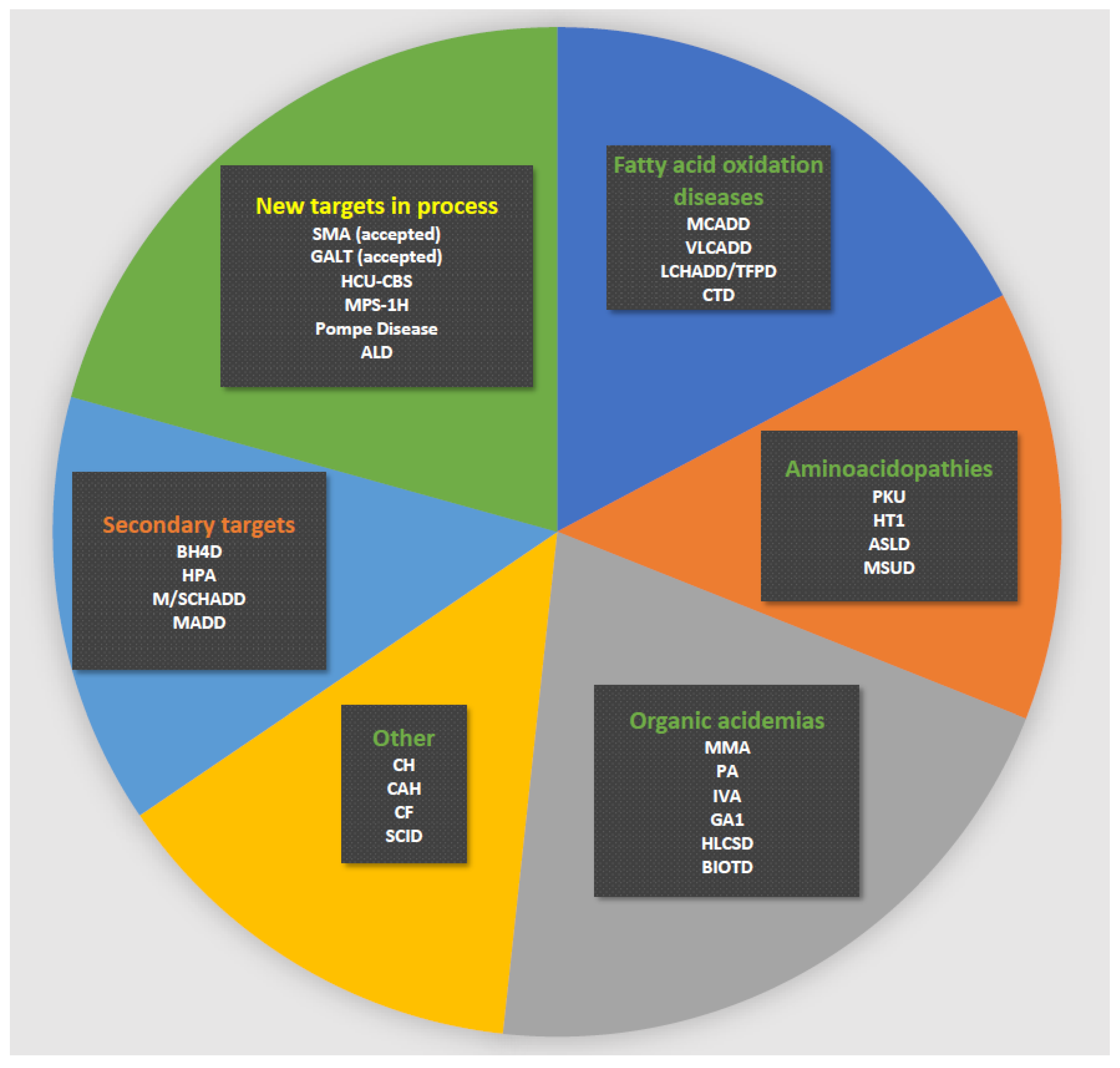
3. Results
3.1. Medium-Chain Acyl-CoA Dehydrogenase Deficiency (OMIM ID: 201450) (MCADD)
3.2. Very Long-Chain Acyl-CoA Dehydrogenase Deficiency (OMIM ID: 201475) (VLCADD)
3.3. Long-Chain 3-Hydroxy Acyl-CoA Dehydrogenase Deficiency (OMIM ID: 609016) (LCHADD)
3.4. Multiple Acyl-CoA Dehydrogenase Deficiency (OMIM ID: 231680) (MADD)
3.5. Carnitine Palmitoyl Transferase 1 Deficiency (OMIM ID: 600528) (CPT1D)
3.6. Biotinidase Deficiency (OMIM ID: 253260) (BIOTD)
3.7. Holocarboxylase Synthase Deficiency (OMIM ID: 253270) (HLCSD)
3.8. Isovaleric Acidemia (OMIM ID: 243500) (IVA)
3.9. Cystic Fibrosis (OMIM ID: 219700) (CF)
3.10. Severe Combined Immunodeficiency (OMIM ID: 300400) (SCID)
3.11. q-Spinal Muscular Atrophy (OMIM ID: 253300) (SMA)
3.12. Other Diagnoses
4. Discussion
4.1. Molecular Genetic Studies May Decrease the False Positive Rate
4.2. Molecular Genetic Studies May Filter Diseases and Disease Subtypes
4.3. Use of Molecular Genetic Studies: Possibilities and Drawbacks
4.3.1. Reporting of Carriers
4.3.2. Use as First-Tier Test
4.3.3. Secondary/Incidental/VUS Findings
4.3.4. Turnaround Time
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-HMGD | 3-hydroxy-3-methyl-glutaryl-CoA lyase deficiency (OMIM ID: 246450) |
| 3-MCCD | 3-methylcrotonyl-CoA-carboxylase deficiency (OMIM IDs: 210200, 210210) |
| 3-MGCHD | 3-methylglutaconyl-CoA hydratase deficiency (OMIM ID: 250950) |
| ALD | adrenoleucodystrophy (OMIM ID: 300100) |
| ARG | hyperargininemia due to arginase deficiency (OMIM ID: 207800) |
| ASLD | argininosuccinate lyase deficiency (OMIM ID: 207900) |
| BETA-KTD | beta-ketothiolase deficiency (OMIM ID: 203750) |
| BH4D | biopterin cofactor deficiencies (OMIM IDs 233910, 261530, 261640) |
| BIOTD | biotinidase deficiency (OMIM ID: 253260) |
| CACTD | carnitine/acylcarnitine translocase deficiency (OMIM ID: 212138) |
| CAH | congenital adrenal hyperplasia (OMIM ID: 201910 |
| CF | cystic fibrosis (OMIM ID: 219700) |
| CFTR | cystic fibrosis transmembrane conductance regulator |
| CH | congenital hypothyroidism |
| CIT | citrullinemia type 1 due to arginosuccinate synthase deficiency (OMIM ID: 215700) |
| CPT1D | carnitine palmitoyl transferase 1 deficiency (OMIM ID: 600528) |
| CPT2D | carnitine palmitoyl transferase 2 deficiency (OMIM IDs: 255110, 600649, 608836) |
| CTD | carnitine transporter deficiency (OMIM ID: 600528) |
| eNBS | expanded newborn screening |
| FAOD | fatty acid oxidation disease |
| GA1 | glutaric aciduria type 1 due to glutaryl-CoA dehydrogenase deficiency (OMIM ID: 231670) |
| GALT | galactosemia due to galactose 1-phosphate uridyl transferase deficiency (OMIM ID: 230400) |
| IEM | inborn error of metabolism |
| HCU-CBS | homocystinuria due to cystathionine-beta-synthase deficiency (OMIM ID: 236200) |
| HHH | hyperornithinemia, hyperammonemia, homocitrullinuria due to ornithine translocase deficiency (OMIM ID: 238970) |
| HLCSD | holocarboxylase synthase deficiency (OMIM ID: 253270) |
| HPA | non-PKU mild hyperphenylalaninemia (OMIM ID: 261660) |
| HT1 | hepatorenal tyrosinemia (type 1) due to fumarylacetoacetase deficiency (OMIM ID: 276700) |
| IVA | isovaleric acidemia due to isovaleryl-CoA dehydrogenase deficiency (OMIM ID: 243500) |
| LCHADD | long-chain 3-hydroxy acyl-CoA dehydrogenase deficiency (OMIM ID 609016) |
| MADD | multiple acyl-CoA dehydrogenase deficiency (OMIM ID: 231680) |
| MCADD | medium-chain acyl-CoA dehydrogenase deficiency (OMIM ID: 201450) |
| MMA | methylmalonic aciduria (many OMIM IDs, most relevant here are: 251000, 251100, 251110); |
| MPS-1H | mucopolysaccharidosis type 1H (OMIM ID: 607014) |
| MS/MS | tandem mass spectrometry |
| M/SCHADD | medium/-short-chain L-3-hydroxy acyl-CoA dehydrogenase deficiency (OMIM ID: 231530) |
| MSUD | classic maple syrup urine disease due to branched-chain alfa-keto acid dehydrogenase deficiency (OMIM ID: 248600) |
| NBS | newborn screening |
| NGS | next generation sequencing |
| PA | propionic acidemia due to propionyl-CoA carboxylase deficiency (OMIM ID: 606054) |
| PKU | classical phenylketonuria due to phenylalanine hydroxylase deficiency (OMIM ID: 261660) |
| PPV | positive predictive value |
| SCADD | short-chain acyl-CoA dehydrogenase deficiency (OMIM ID: 201470) |
| SCID | severe combined immunodeficiency |
| SSI | Statens Serum Institute |
| TPD | trifunctional protein deficiency (OMIM ID: 609015); |
| VLCADD | very long-chain acyl-CoA dehydrogenase deficiency (OMIM ID: 201475) |
| WGS | whole genome sequencing |
References
- Almannai, M.; Marom, R.; Sutton, V.R. Newborn screening: A review of history, recent advancements, and future perspectives in the era of next generation sequencing. Curr. Opin. Pediatr. 2016, 28, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Andresen, B.S.; Lund, A.; Hougaard, D.M.; Christensen, E.; Gahrn, B.; Christensen, M.; Bross, P.; Vested, A.; Simonsen, H.; Skogstrand, K.; et al. MCAD deficiency in Denmark. Mol. Genet. Metab. 2012, 106, 175–188. [Google Scholar] [CrossRef]
- Boy, N.; Mengler, K.; Thimm, E.; Schiergens, K.A.; Marquardt, T.; Weinhold, N.; Marquardt, I.; Das, A.M.; Freisinger, P.; Grünert, S.C.; et al. Newborn screening: A disease-changing intervention for glutaric aciduria type 1. Ann. Neurol. 2018, 83, 970–979. [Google Scholar] [CrossRef]
- Furnier, S.; Durkin, M.; Baker, M. Translating Molecular Technologies into Routine Newborn Screening Practice. Int. J. Neonatal Screen. 2020, 6, 80. [Google Scholar] [CrossRef]
- Tangeraas, T.; Sæves, I.; Klingenberg, C.; Jørgensen, J.; Kristensen, E.; Gunnarsdottir, G.; Hansen, E.V.; Strand, J.; Lundman, E.; Ferdinandusse, S.; et al. Performance of Expanded Newborn Screening in Norway Supported by Post-Analytical Bioinformatics Tools and Rapid Second-Tier DNA Analyses. Int. J. Neonatal Screen. 2020, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Loeber, J.; Platis, D.; Zetterström, R.; Almashanu, S.; Boemer, F.; Bonham, J.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal Screening in Europe Revisited: An ISNS Perspective on the Current State and Developments Since 2010. Int. J. Neonatal Screen. 2021, 7, 15. [Google Scholar] [CrossRef] [PubMed]
- Remec, Z.I.; Podkrajsek, K.T.; Lampret, B.R.; Kovac, J.; Groselj, U.; Tesovnik, T.; Battelino, T.; Debeljak, M. Next-Generation Sequencing in Newborn Screening: A Review of Current State. Front. Genet. 2021, 12, 662254. [Google Scholar] [CrossRef]
- Bhattacharjee, A.; Sokolsky, T.; Wyman, S.K.; Reese, M.G.; Puffenberger, E.; Strauss, K.; Morton, H.; Parad, R.B.; Naylor, E.W. Development of DNA Confirmatory and High-Risk Diagnostic Testing for Newborns Using Targeted Next-Generation DNA Sequencing. Genet. Med. 2015, 17, 337–347. [Google Scholar] [CrossRef]
- Poulsen, J.B.; Lescai, F.; Grove, J.; Bækvad-Hansen, M.; Christiansen, M.; Hagen, C.M.; Maller, J.; Stevens, C.; Li, S.; Li, Q.; et al. High-Quality Exome Sequencing of Whole-Genome Amplified Neonatal Dried Blood Spot DNA. PLoS ONE 2016, 11, e0153253. [Google Scholar] [CrossRef]
- Van Campen, J.C.; Sollars, E.S.A.; Thomas, R.C.; Bartlett, C.M.; Milano, A.; Parker, M.D.; Dawe, J.; Winship, P.R.; Peck, G.; Grafham, D.; et al. Next Generation Sequencing in Newborn Screening in the United Kingdom National Health Service. Int. J. Neonatal Screen. 2019, 5, 40. [Google Scholar] [CrossRef]
- Malvagia, S.; Forni, G.; Ombrone, D.; La Marca, G. Development of Strategies to Decrease False Positive Results in Newborn Screening. Int. J. Neonatal Screen. 2020, 6, 84. [Google Scholar] [CrossRef]
- Mütze, U.; Henze, L.; Gleich, F.; Lindner, M.; Grünert, S.C.; Spiekerkoetter, U.; Santer, R.; Blessing, H.; Thimm, E.; Ensenauer, R.; et al. Newborn screening and disease variants predict neurological outcome in isovaleric aciduria. J. Inherit. Metab. Dis. 2021. [Google Scholar] [CrossRef]
- Vasquez-Loarte, T.; Thompson, J.D.; Merritt, J.L. Considering Proximal Urea Cycle Disorders in Expanded Newborn Screening. Int. J. Neonatal Screen. 2020, 6, 77. [Google Scholar] [CrossRef]
- Lund, A.M.; Hougaard, D.M.; Simonsen, H.; Andresen, B.S.; Christensen, M.; Dunø, M.; Skogstrand, K.; Olsen, R.; Jensen, U.G.; Cohen, A.; et al. Biochemical screening of 504,049 newborns in Denmark, the Faroe Islands and Greenland—Experience and development of a routine program for expanded newborn screening. Mol. Genet. Metab. 2012, 107, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Lund, A.; Wibrand, F.; Skogstrand, K.; Cohen, A.; Christensen, M.; Jäpelt, R.B.; Dunø, M.; Skovby, F.; Nørgaard-Pedersen, B.; Gregersen, N.; et al. Danish expanded newborn screening is a successful preventive public health programme. Dan. Med. J. 2020, 67, 1–8. [Google Scholar]
- Statistics Denmark. 2019. Available online: https://www.dst.dk/en (accessed on 3 May 2021).
- Skov, M.; Baekvad-Hansen, M.; Hougaard, D.M.; Skogstrand, K.; Lund, A.; Pressler, T.; Olesen, H.V.; Duno, M. Cystic fibrosis newborn screening in Denmark: Experience from the first 2 years. Pediatr. Pulmonol. 2019, 55, 549–555. [Google Scholar] [CrossRef]
- Andresen, B.S.; Dobrowolski, S.F.; O’Reilly, L.; Muenzer, J.; McCandless, S.E.; Frazier, D.M.; Udvari, S.; Bross, P.; Knudsen, I.; Banas, R.; et al. Medium-chain acyl-CoA dehydrogenase (MCAD) mutations identified by MS/MS-based prospective screening of newborns differ from those observed in patients with clinical symptoms: Identification and characterization of a new, prevalent mutation that results in mild MCAD deficiency. Am. J. Hum. Genet. 2001, 68, 1408–1418. [Google Scholar]
- Aksglæde, L.; Christensen, M.; Olesen, J.H.; Dunø, M.; Olsen, R.K.; Andresen, B.S.; Hougaard, D.M.; Lund, A.M. Abnormal Newborn Screening in a Healthy Infant of a Mother with Undiagnosed Medium-Chain Acyl-CoA Dehydrogenase Deficiency. J. Inherit Metab. Dis. Rep. 2015, 23, 67–70. [Google Scholar]
- Andresen, B.S.; Olpin, S.; Poorthuis, B.J.; Scholte, H.R.; Vianey-Saban, C.; Wanders, R.; Ijlst, L.; Morris, A.; Pourfarzam, M.; Bartlett, K.; et al. Clear correlation of genotype with disease phenotype in very-long-chain acyl-CoA dehydrogenase deficiency. Am. J. Hum. Genet. 1999, 64, 479–494. [Google Scholar] [CrossRef]
- Rajakumar, C.; Ban, M.R.; Cao, H.; Young, T.K.; Bjerregaard, P.; Hegele, R.A. Carnitine palmitoyltransferase IA polymorphism P479L is common in Greenland Inuit and is associated with elevated plasma apolipoprotein A-I. J. Lipid Res. 2009, 50, 1223–1228. [Google Scholar] [CrossRef] [PubMed]
- Borch, L.; Lund, A.M.; Wibrand, F.; Christensen, E.; Søndergaard, C.; Gahrn, B.; Hougaard, D.M.; Andresen, B.S.; Gregersen, N.; Olsen, R.K.J. Normal Levels of Plasma Free Carnitine and Acylcarnitines in Follow-Up Samples from a Presymptomatic Case of Carnitine Palmitoyl Transferase 1 (CPT1) Deficiency Detected Through Newborn Screening in Denmark. JIMD Rep. 2011, 3, 11–15. [Google Scholar] [CrossRef]
- Karaceper, M.D.; Chakraborty, P.; Coyle, D.; Wilson, K.; Kronick, J.B.; Hawken, S.; Davies, C.; Brownell, M.; Dodds, L.; Feigenbaum, A.; et al. Canadian Inherited Metabolic Diseases Research, N. The health system impact of false positive newborn screening results for medium-chain acyl-CoA dehydrogenase deficiency: A cohort study. Orphanet. J. Rare Dis. 2016, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Gurian, E.A.; Kinnamon, D.; Henry, J.J.; Waisbren, S.E. Expanded Newborn Screening for Biochemical Disorders: The Effect of a False-Positive Result. Pediatrics 2006, 117, 1915–1921. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.-J.; He, J.; Chen, H.; Shi, X.-D.; Li, Y. Psychological Effects of False-Positive Results in Expanded Newborn Screening in China. PLoS ONE 2012, 7, e36235. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Albers, S.; Amato, S.; Ampola, M.; Brewster, T.G.; Demmer, L.; Eaton, R.B.; Greenstein, R.; Korson, M.; Larson, C.; et al. Effect of Expanded Newborn Screening for Biochemical Genetic Disorders on Child Outcomes and Parental Stress. JAMA 2003, 290, 2564–2572. [Google Scholar] [CrossRef] [PubMed]
- Parens, E.; Appelbaum, P.S. On What We Have Learned and Still Need to Learn about the Psychosocial Impacts of Genetic Testing. Häst. Cent. Rep. 2019, 49, S2–S9. [Google Scholar] [CrossRef]
- Stadler, S.C.; Polanetz, R.; Maier, E.M.; Heidenreich, S.C.; Niederer, B.; Mayerhofer, P.U.; Lagler, F.; Koch, H.G.; Santer, R.; Fletcher, J.M.; et al. Newborn screening for 3-methylcrotonyl-CoA carboxylase deficiency: Population heterogeneity of MCCA and MCCB mutations and impact on risk assessment. Hum. Mutat. 2006, 27, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Waddell, L.; Wiley, V.; Carpenter, K.; Bennetts, B.; Angel, L.; Andresen, B.S.; Wilcken, B. Medium-chain acyl-CoA dehydrogenase deficiency: Genotype-biochemical phenotype correlations. Mol. Genet. Metab. 2006, 87, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Mütze, U.; Walter, M.; Keller, M.; Gramer, G.; Garbade, S.F.; Gleich, F.; Haas, D.; Posset, R.; Grünert, S.C.; Hennermann, J.B.; et al. Health Outcomes of Infants with Vitamin B12 Deficiency Identified by Newborn Screening and Early Treated. J. Pediatr. 2021. [Google Scholar] [CrossRef]
- Marsden, D.; Bedrosian, C.L.; Vockley, J. Impact of newborn screening on the reported incidence and clinical outcomes associated with medium- and long-chain fatty acid oxidation disorders. Genet. Med. 2021, 23, 816–829. [Google Scholar] [CrossRef]
- Clarke, L.A.; Giugliani, R.; Guffon, N.; Jones, S.A.; Keenan, H.A.; Munoz-Rojas, M.V.; Okuyama, T.; Viskochil, D.; Whitley, C.B.; Wijburg, F.A.; et al. Genotype-phenotype relationships in mucopolysaccharidosis type I (MPS I): Insights from the International MPS I Registry. Clin. Genet. 2019, 96, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Peck, D.S.; Lacey, J.M.; White, A.L.; Pino, G.; Studinski, A.L.; Fisher, R.; Ahmad, A.; Spencer, L.; Viall, S.; Shallow, N.; et al. Incorporation of Second-Tier Biomarker Testing Improves the Specificity of Newborn Screening for Mucopolysaccharidosis Type I. Int. J. Neonatal Screen. 2020, 6, 10. [Google Scholar] [CrossRef]
- Lundman, E.; Gaup, H.J.; Bakkeheim, E.; Olafsdottir, E.J.; Rootwelt, T.; Storrøsten, O.T.; Pettersen, R.D. Implementation of newborn screening for cystic fibrosis in Norway. Results from the first three years. J. Cyst. Fibros. 2016, 15, 318–324. [Google Scholar] [CrossRef]
- Mosegaard, S.; DiPace, G.; Bross, P.; Carlsen, J.; Gregersen, N.; Olsen, R.K.J. Riboflavin Deficiency—Implications for General Human Health and Inborn Errors of Metabolism. Int. J. Mol. Sci. 2020, 21, 3847. [Google Scholar] [CrossRef]
- Vissing, C.R.; Duno, M.; Olesen, J.H.; Rafiq, J.; Risom, L.; Christensen, E.; Wibrand, F.; Vissing, C. Recurrent myoglobinuria and deranged acylcarnitines due to a mutation in the mtDNA MT-CO2 gene. Neurology 2013, 80, 1908–1910. [Google Scholar] [CrossRef]
- Roos, S.; Sofou, K.; Hedberg-Oldfors, C.; Kollberg, G.; Lindgren, U.; Thomsen, C.; Tulinius, M.; Oldfors, A. Mitochondrial complex IV deficiency caused by a novel frameshift variant in MT-CO2 associated with myopathy and perturbed acylcarnitine profile. Eur. J. Hum. Genet. 2019, 27, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Smed, V.M.; Petersen, O.B.B.; Gerdes, A.-M.A.; Diness, B.R.; Roos, L.S. Genetic screening of prospective parents. Ugeskr Laeger 2021, 183, 13. [Google Scholar]
- Strauss, K.A.; Puffenberger, E.G.; Carson, V.J. Maple Syrup Urine Disease. In Genereviews(R); Adam, M.P., Ardinger, H.H., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Estrella, J.; Wilcken, B.; Carpenter, K.; Bhattacharya, K.; Tchan, M.; Wiley, V. Expanded newborn screening in New South Wales: Missed cases. J. Inherit. Metab. Dis. 2014, 37, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Puckett, R.; Lorey, F.; Rinaldo, P.; Lipson, M.; Matern, D.; Sowa, M.; Levine, S.; Chang, R.; Wang, R.; Abdenur, J. Maple syrup urine disease: Further evidence that newborn screening may fail to identify variant forms. Mol. Genet. Metab. 2010, 100, 136–142. [Google Scholar] [CrossRef]
- Boemer, F.; Caberg, J.-H.; Dideberg, V.; Dardenne, D.; Bours, V.; Hiligsmann, M.; Dangouloff, T.; Servais, L. Newborn screening for SMA in Southern Belgium. Neuromuscul. Disord. 2019, 29, 343–349. [Google Scholar] [CrossRef]
- Adhikari, A.N.; Gallagher, R.C.; Wang, Y.; Currier, R.J.; Amatuni, G.; Bassaganyas, L.; Chen, F.; Kundu, K.; Kvale, M.; Mooney, S.D.; et al. The role of exome sequencing in newborn screening for inborn errors of metabolism. Nat. Med. 2020, 26, 1–6. [Google Scholar] [CrossRef]
- Johnston, J.; Lantos, J.D.; Goldenberg, A.; Chen, F.; Parens, E.; Koenig, B.A.; Members of the NSIGHT Ethics and Policy Advisory Board. Sequencing Newborns:A Call for Nuanced Use of Genomic Technologies. Häst. Cent. Rep. 2018, 48, S2–S6. [Google Scholar] [CrossRef]
- Narravula, A.; Garber, K.B.; Askree, S.H.; Hegde, M.; Hall, P.L. Variants of uncertain significance in newborn screening disorders: Implications for large-scale genomic sequencing. Genet. Med. 2017, 19, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Amendola, L.M.; Dorschner, M.O.; Robertson, P.D.; Salama, J.S.; Hart, R.; Shirts, B.H.; Murray, M.L.; Tokita, M.J.; Gallego, C.J.; Kim, D.S.; et al. Actionable exomic incidental findings in 6503 participants: Challenges of variant classification. Genome Res. 2015, 25, 305–315. [Google Scholar] [CrossRef]
- Amendola, L.M.; Muenzen, K.; Biesecker, L.G.; Bowling, K.M.; Cooper, G.M.; Dorschner, M.O.; Driscoll, C.; Foreman, A.K.M.; Golden-Grant, K.; Greally, J.M.; et al. Variant Classification Concordance using the ACMG-AMP Variant Interpretation Guidelines across Nine Genomic Implementation Research Studies. Am. J. Hum. Genet. 2020, 107, 932–941. [Google Scholar] [CrossRef]
- Levy, H. Ethical and Psychosocial Implications of Genomic Newborn Screening. Int. J. Neonatal Screen. 2021, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.M.; Hildreth, A.; Batalov, S.; Ding, Y.; Chowdhury, S.; Watkins, K.; Ellsworth, K.; Camp, B.; Kint, C.I.; Yacoubian, C.; et al. Diagnosis of genetic diseases in seriously ill children by rapid whole-genome sequencing and automated phenotyping and interpretation. Sci. Transl. Med. 2019, 11, eaat6177. [Google Scholar] [CrossRef]
- Kingsmore, S.F.; Cakici, J.A.; Clark, M.M.; Gaughran, M.; Feddock, M.; Batalov, S.; Bainbridge, M.N.; Carroll, J.; Caylor, S.A.; Clarke, C.; et al. A Randomized, Controlled Trial of the Analytic and Diagnostic Performance of Singleton and Trio, Rapid Genome and Exome Sequencing in Ill Infants. Am. J. Hum. Genet. 2019, 105, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Walter, J.H.; Patterson, A.; Till, J.; Besley, G.T.N.; Fleming, G.; Henderson, M.J. Bloodspot acylcarnitine and amino acid analysis in cord blood samples: Efficacy and reference data from a large cohort study. J. Inherit. Metab. Dis. 2009, 32, 95–101. [Google Scholar] [CrossRef]
- Yoon, H.R. Screening newborns for metabolic disorders based on targeted metabolomics using tandem mass spectrometry. Ann. Pediatr. Endocrinol. Metab. 2015, 20, 119–124. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gavrilov, D.K.; Piazza, A.L.; Pino, G.; Turgeon, C.; Matern, D.; Oglesbee, D.; Raymond, K.; Tortorelli, S.; Rinaldo, P. The Combined Impact of CLIR Post-Analytical Tools and Second Tier Testing on the Performance of Newborn Screening for Disorders of Propionate, Methionine, and Cobalamin Metabolism. Int. J. Neonatal Screen. 2020, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Sörensen, L.; Von Döbeln, U.; Åhlman, H.; Ohlsson, A.; Engvall, M.; Naess, K.; Backman-Johansson, C.; Nordqvist, Y.; Wedell, A.; Zetterström, R.H. Expanded Screening of One Million Swedish Babies with R4S and CLIR for Post-Analytical Evaluation of Data. Int. J. Neonatal Screen. 2020, 6, 42. [Google Scholar] [CrossRef] [PubMed]
- Hall, P.; Marquardt, G.; McHugh, D.M.; Currier, R.; Tang, H.; Stoway, S.D.; Rinaldo, P. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet. Med. 2014, 16, 889–895. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Screen Positive | True Positive Classic (C) Mild (M) VUS (V) | False Positive Heterozygous (H) | False Negative | Not Reported | |
|---|---|---|---|---|---|
| MCADD | 124 | 109 80 C 18 M 11 V | 15 11 H | 4 | NA |
| VLCADD | 25 | 6 3 C 3 V | 19 11 H | 0 | NA |
| LCHADD | 5 | 5 3 C 2 V | 0 | 0 | NA |
| MADD | 5 | 3 2 C * 1 V | 2 | NA | NA |
| CPT1D | 48 | 27 1 C 26 V | 21 | NA | NA |
| IVA | 10 | 6 6 M | 4 2 H | 0 | NA |
| MSUD | 57 | 3 3 C | 54 | 2 (intermittent) | NA |
| BIOTD | 79 | 47 | 18 pre-2018 0 post-2018 | 0 | 14 post-2018 |
| Raised C5OH | 117 | 5 HLCSD 21 other diagnoses | 9 | 1 | 82 post-2009 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lund, A.M.; Wibrand, F.; Skogstrand, K.; Bækvad-Hansen, M.; Gregersen, N.; Andresen, B.S.; Hougaard, D.M.; Dunø, M.; Olsen, R.K.J. Use of Molecular Genetic Analyses in Danish Routine Newborn Screening. Int. J. Neonatal Screen. 2021, 7, 50. https://doi.org/10.3390/ijns7030050
Lund AM, Wibrand F, Skogstrand K, Bækvad-Hansen M, Gregersen N, Andresen BS, Hougaard DM, Dunø M, Olsen RKJ. Use of Molecular Genetic Analyses in Danish Routine Newborn Screening. International Journal of Neonatal Screening. 2021; 7(3):50. https://doi.org/10.3390/ijns7030050
Chicago/Turabian StyleLund, Allan Meldgaard, Flemming Wibrand, Kristin Skogstrand, Marie Bækvad-Hansen, Niels Gregersen, Brage Storstein Andresen, David M. Hougaard, Morten Dunø, and Rikke Katrine Jentoft Olsen. 2021. "Use of Molecular Genetic Analyses in Danish Routine Newborn Screening" International Journal of Neonatal Screening 7, no. 3: 50. https://doi.org/10.3390/ijns7030050
APA StyleLund, A. M., Wibrand, F., Skogstrand, K., Bækvad-Hansen, M., Gregersen, N., Andresen, B. S., Hougaard, D. M., Dunø, M., & Olsen, R. K. J. (2021). Use of Molecular Genetic Analyses in Danish Routine Newborn Screening. International Journal of Neonatal Screening, 7(3), 50. https://doi.org/10.3390/ijns7030050