
Novel Modification of a Confirmatory SMA Sequencing Assay that Can Be Used to Determine SMN2 Copy Number

Abstract
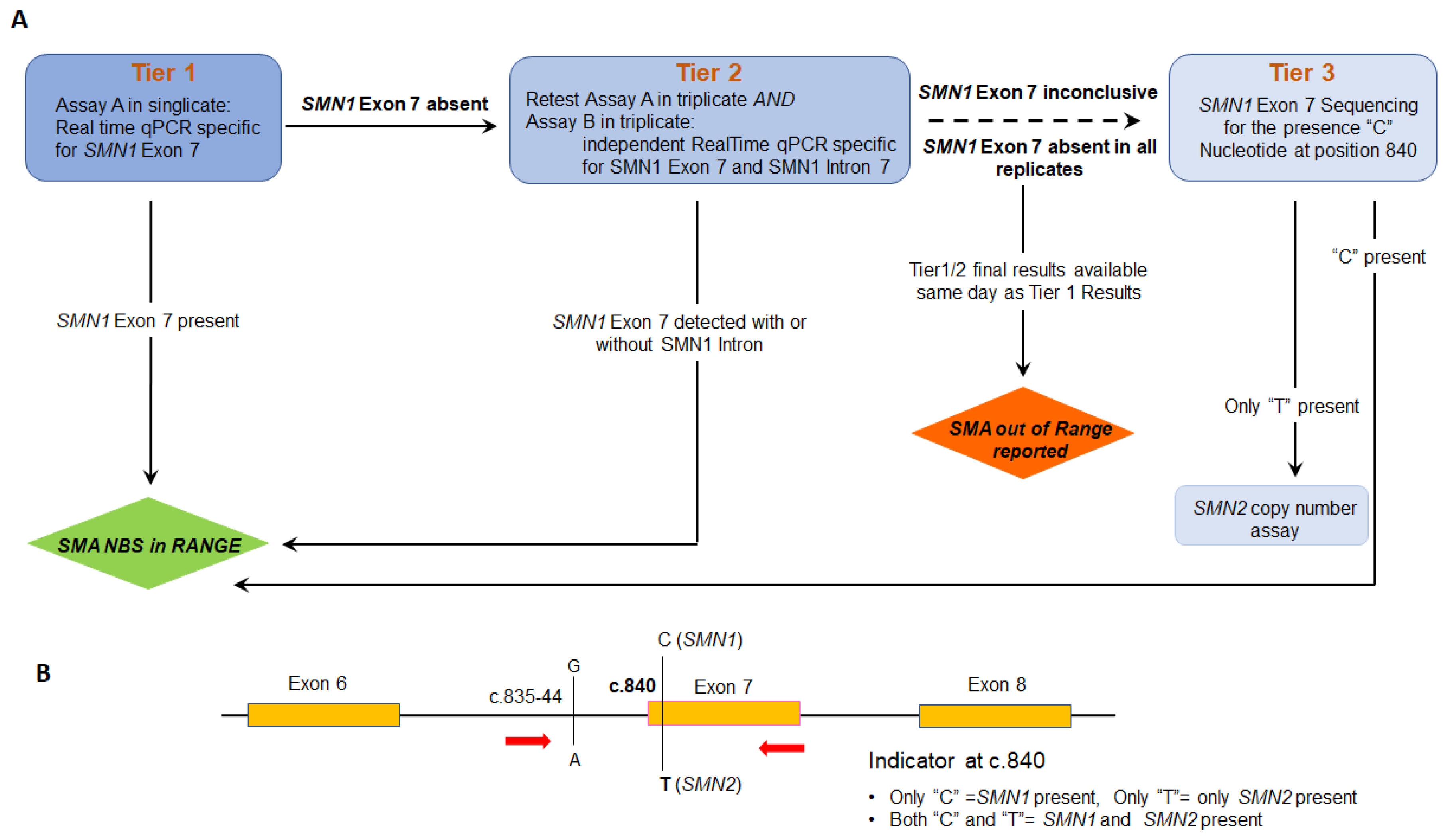
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. DNA Isolation from Dry Blood Spot
2.3. DNA Primers and Sequencing
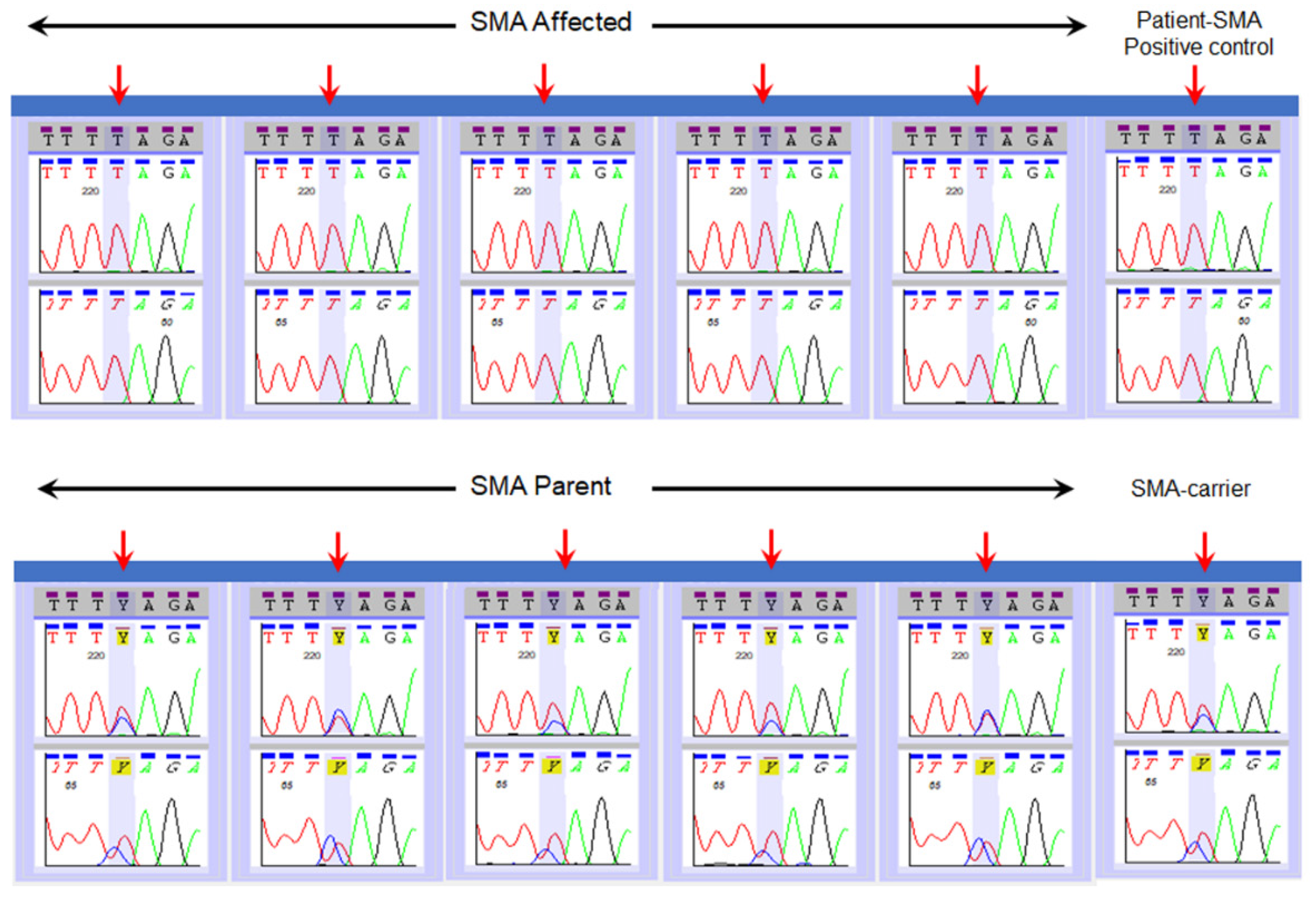
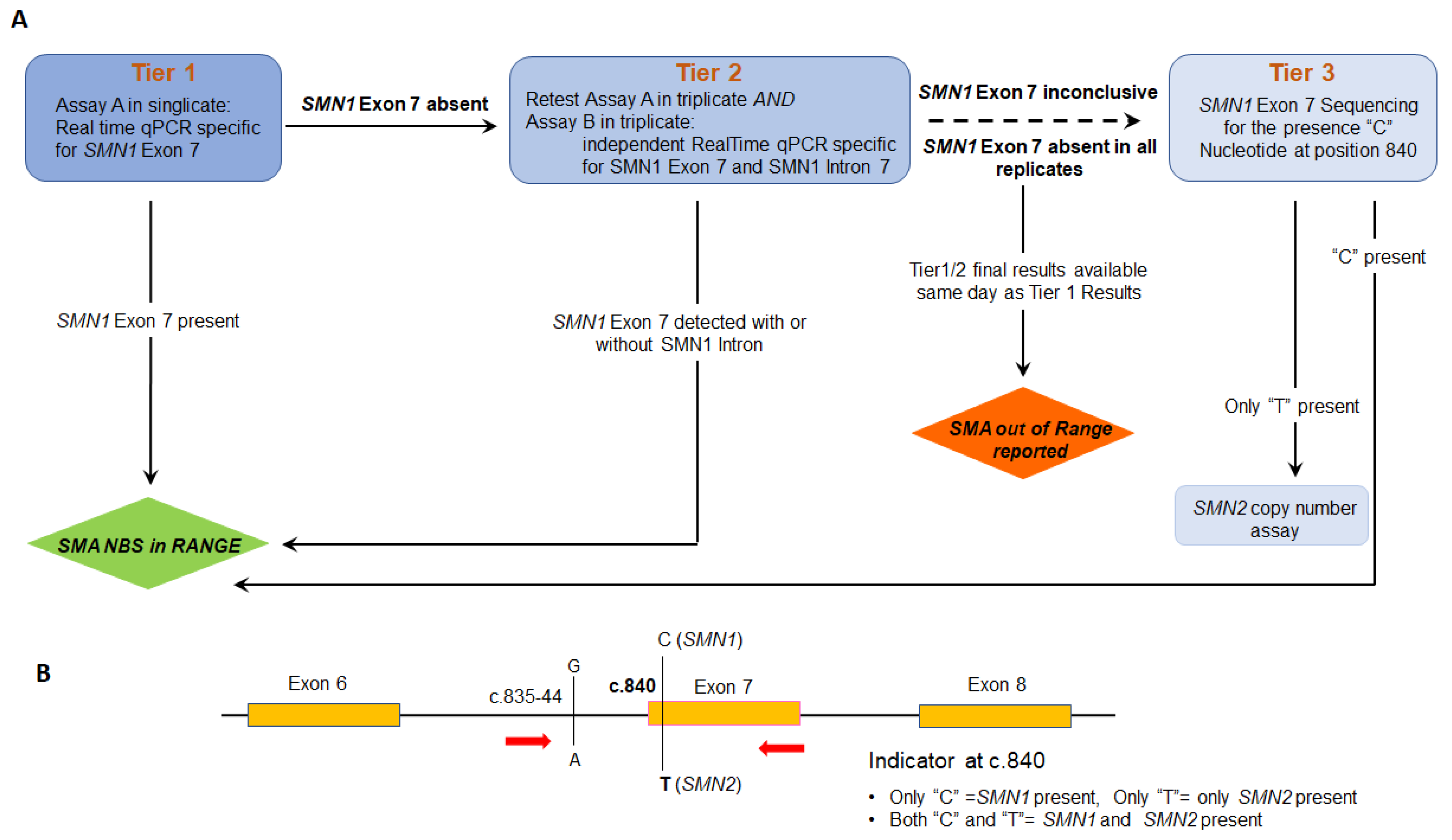
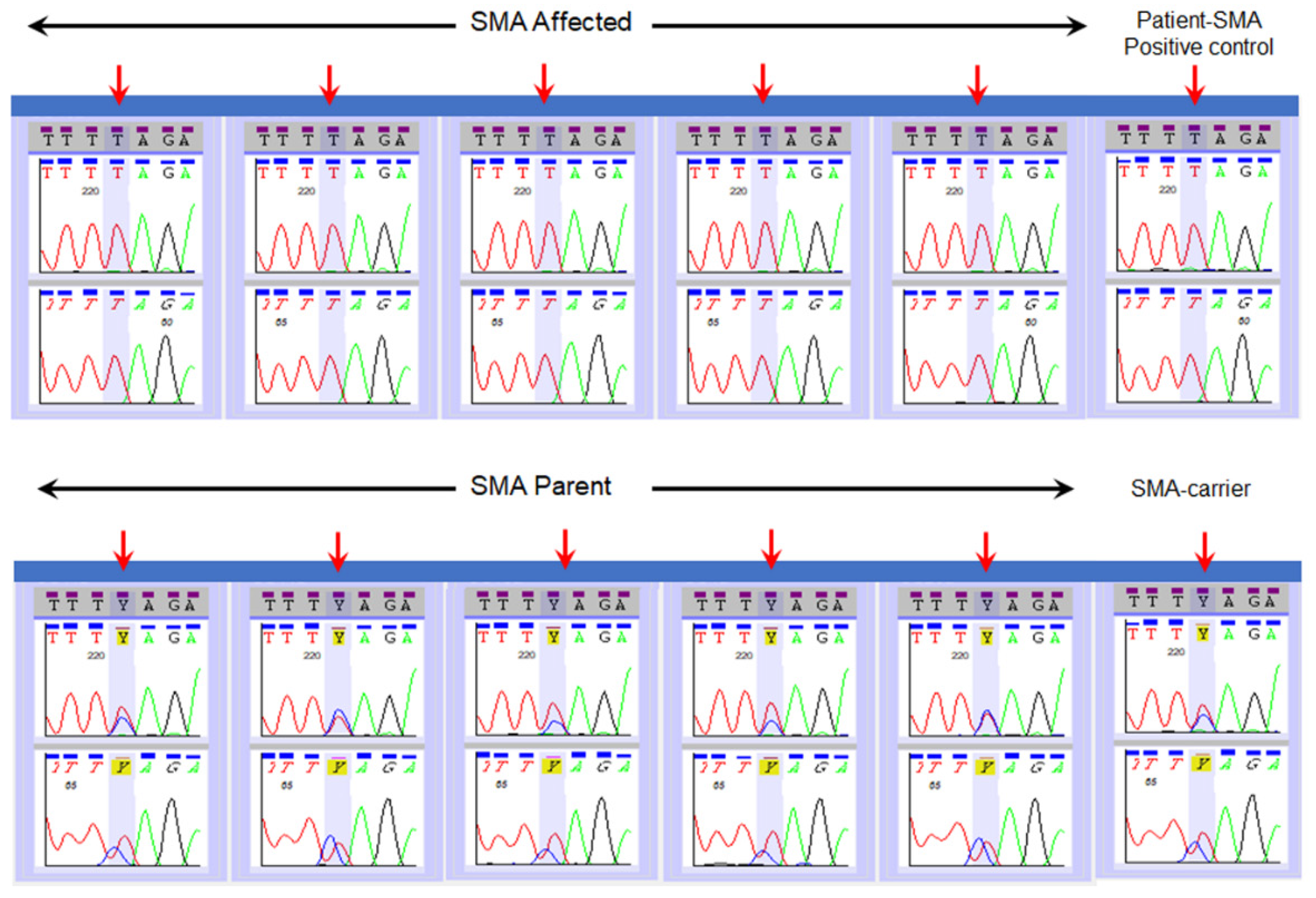
2.4. Validation of SMN1 Sequencing Assay
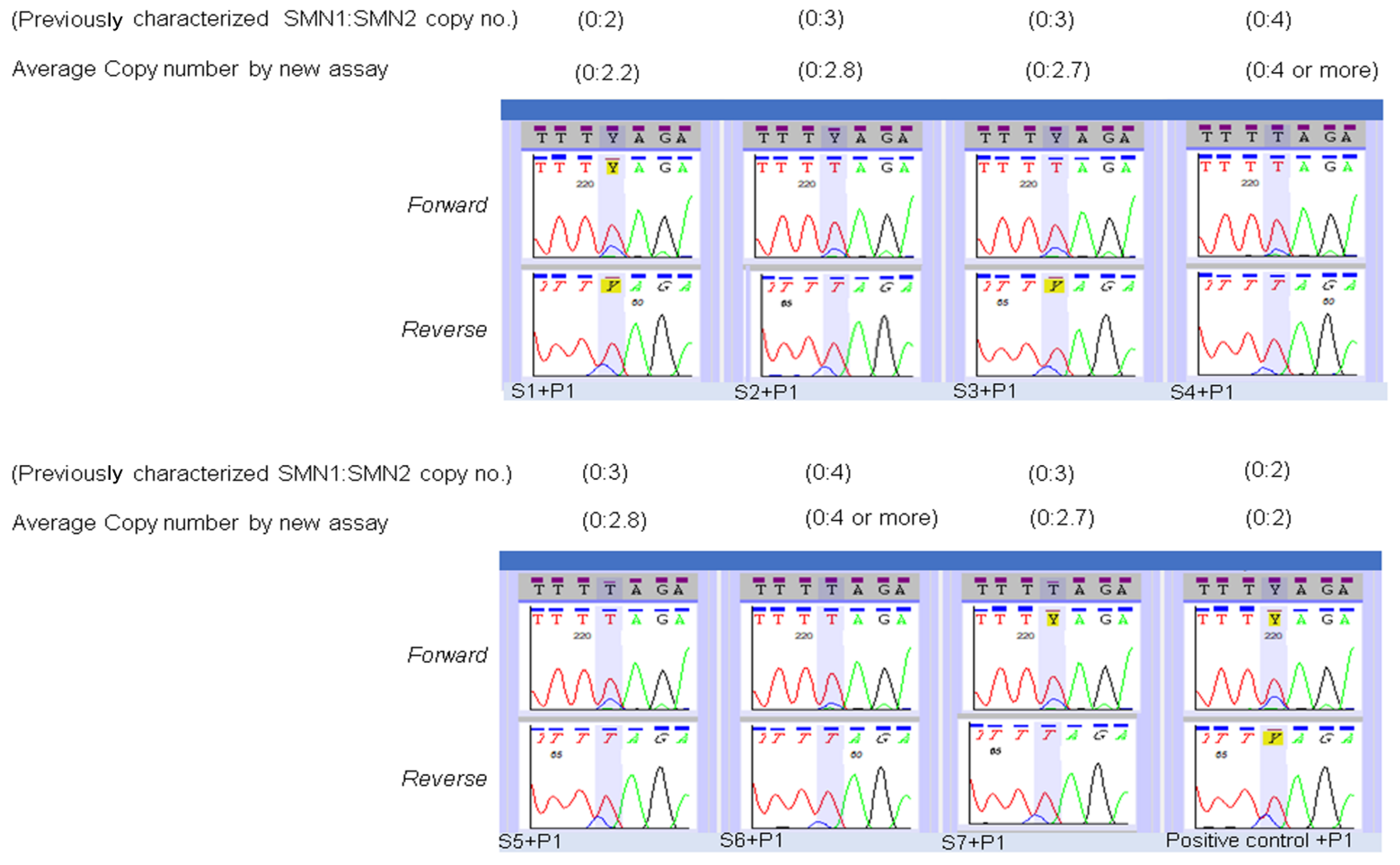
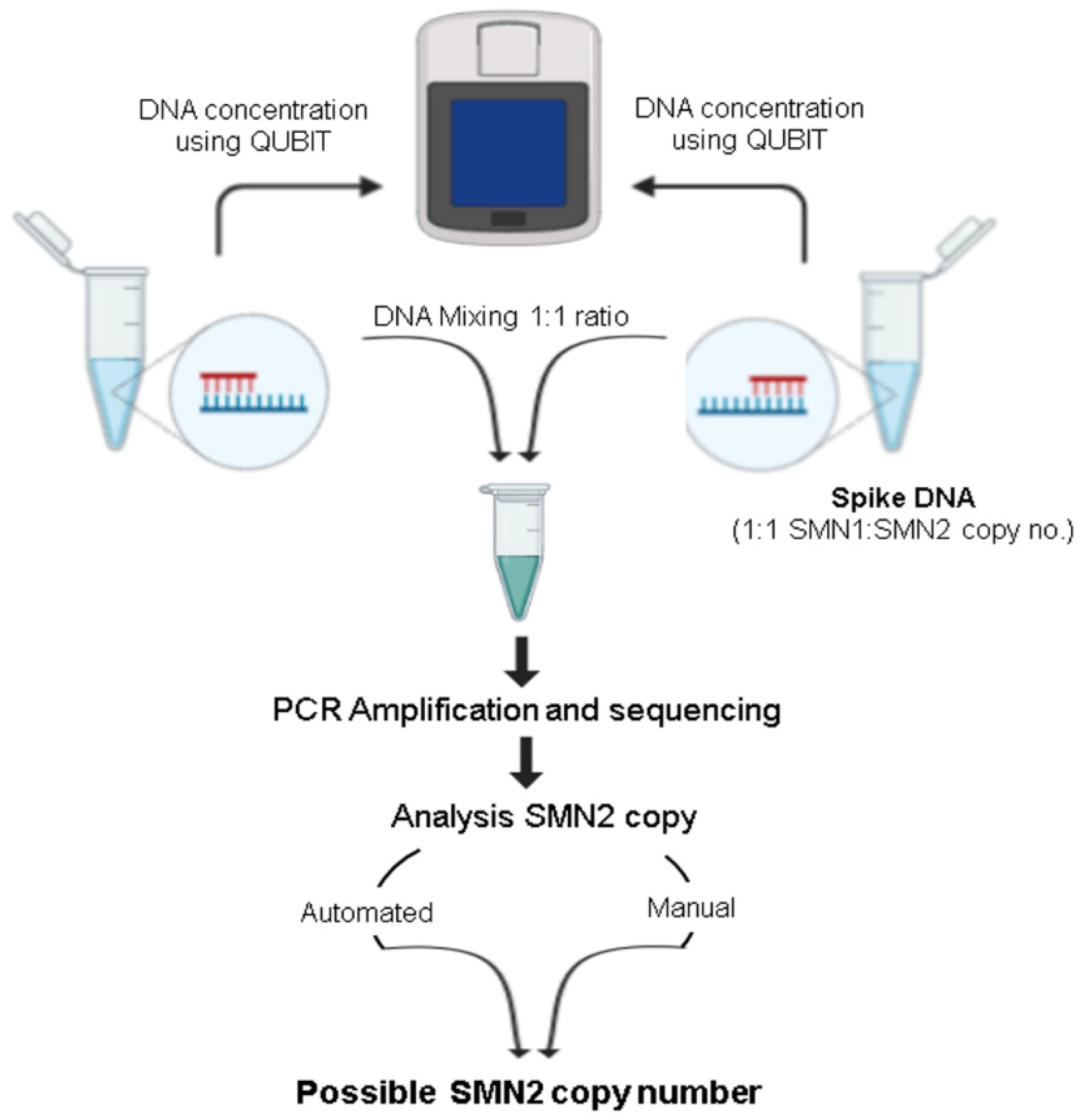
2.5. Modification of Sequencing Assay for Determination of SMN2 Copy Number
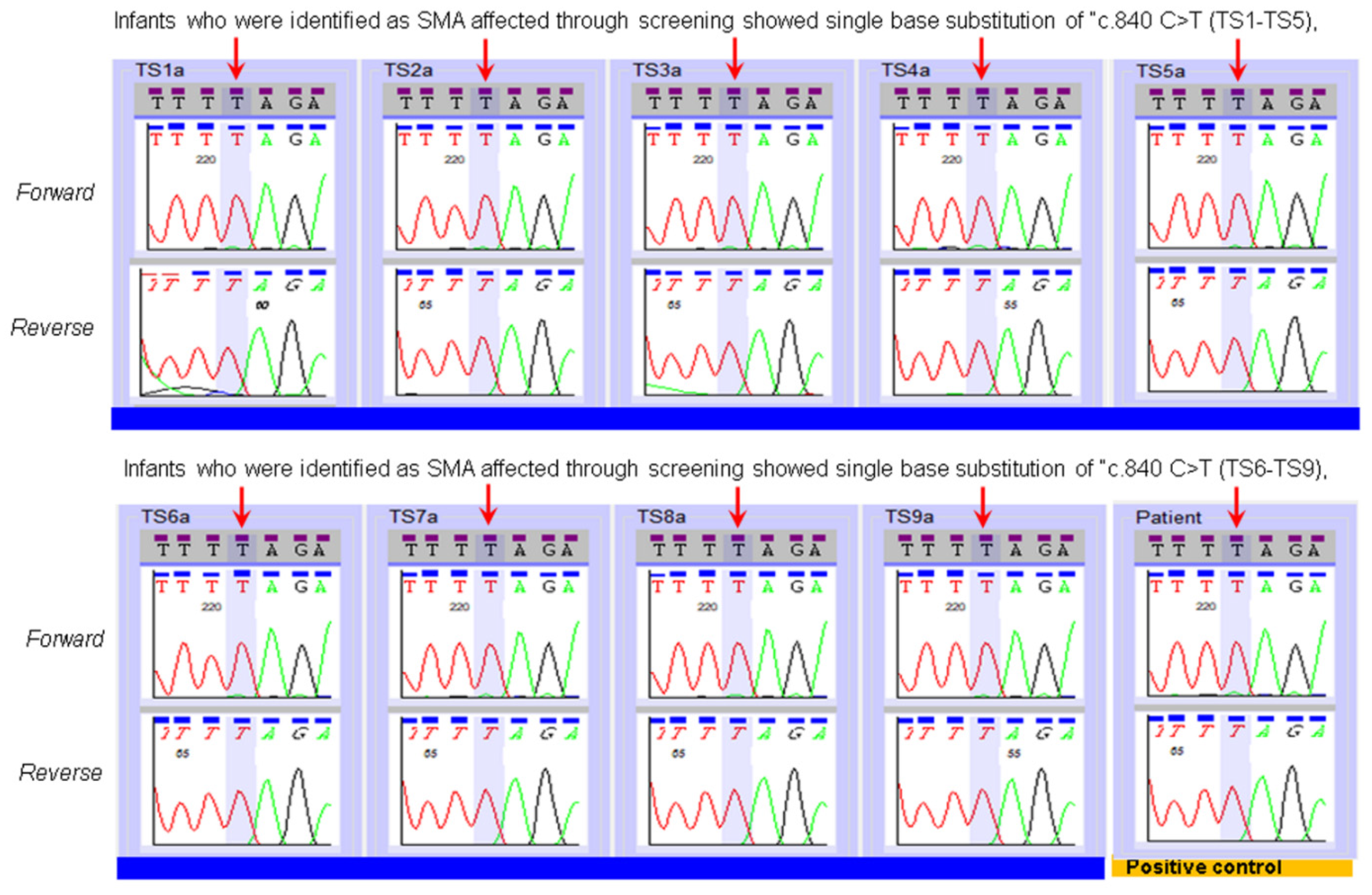
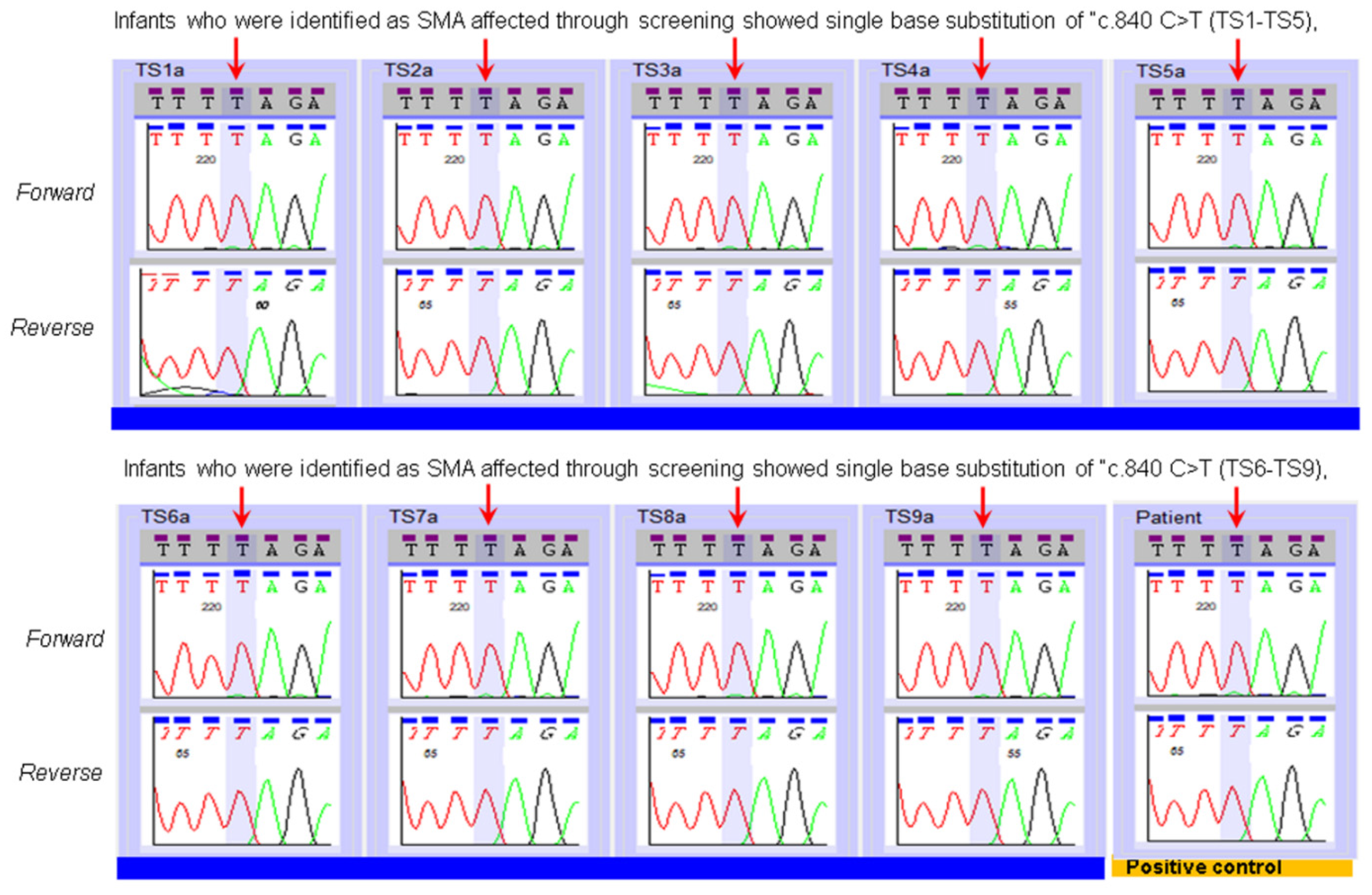
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kolb, S.J.; Kissel, J.T. Spinal Muscular Atrophy. Neurol Clin. 2015, 33, 831–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogino, S.; Wilson, R.B. Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum. Genet. 2002, 111, 477–500. [Google Scholar] [CrossRef] [PubMed]
- Wirth, B. An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum. Mutat. 2000, 15, 228–237. [Google Scholar] [CrossRef]
- Nurputra, D.K.; Lai, P.S.; Harahap, N.I.; Morikawa, S.; Yamamoto, T.; Nishimura, N.; Kubo, Y.; Takeuchi, A.; Saito, T. Spinal muscular atrophy: From gene discovery to clinical trials. Ann. Hum. Genet. 2013, 77, 435–463. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Wilson, R.B.; Gold, B. New insight on the evolution of the SMN1 and SMN2 region: Simulation and meta-analysis for allele and haplotype frequency calculations. Eur J. Hum. Genet. 2004, 12, 1015–1023. [Google Scholar] [CrossRef]
- Mailman, M.D.; Heinz, J.W.; Papp, A.C.; Snyder, P.J.; Sedra, M.S.; Wirth, B.; Burghes, A.H.M.; Prior, T.W. Molecular analysis of spinal muscular atrophy and modification of the phenotype by SMN2. Genet. Med. 2002, 4, 20–26. [Google Scholar] [CrossRef] [Green Version]
- Prior, T.W.; Nagan, N.; Sugarman, E.A.; Batish, S.D.; Braastad, C. Technical standards and guidelines for spinal muscular atrophy testing. Genet. Med. 2011, 13, 686–694. [Google Scholar] [CrossRef] [Green Version]
- Prior, T.W.; Krainer, A.R.; Hua, Y.; Swoboda, K.J.; Snyder, P.C.; Bridgeman, S.J.; Kissel, J.T. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am. J. Hum. Genet. 2009, 85, 408–413. [Google Scholar] [CrossRef] [Green Version]
- Calucho, M.; Bernal, S.; Alías, L.; March, F.; Venceslá, A.; Rodríguez-Álvarez, F.J.; Aller, E.; Fernández, R.M.; Borrego, S.; Millán, J.M.; et al. Correlation between SMA type and SMN2 copy number revisited: An analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscular Disord. 2018, 28, 208–215. [Google Scholar] [CrossRef]
- Feldkötter, M.; Schwarzer, V.; Wirth, R.; Wienker, T.F.; Wirth, B. Quantitative analyses of SMN1 and SMN2 based on real-time lightcycler PCR: Fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002, 70, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.L.; Lee, F.K.; Yazdanpanah, G.K.; Staropoli, J.F.; Liu, M.; Carulli, J.P.; Sun, C.; Dobrowolski, S.F.; Hannon, W.H.; Vogt, R.F. Newborn blood spot screening test using multiplexed real-time PCR to simultaneously screen for spinal muscular atrophy and severe combined immunodeficiency. Clin. Chem 2015, 61, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Lee, F.K. Newborn Screening for Spinal Muscular Atrophy (SMA) in the US. Presented at Spinal Muscular Atrophy: Overview 324 of Available Screening Methods, Webinar. 28 June 2018. Available online: https://www.newsteps.org/sites/default/files/re-325sources/download/aphl_sma_webinar_slides_webinarslides_june2018_kh.pdf (accessed on 27 April 2021).
- Mercer, K. Newborn screening for spinal muscular atrophy. In Proceedings of the Newborn Screening and Genetic Testing Sym-321 posium, New Orleans, LA, USA, 10–13 September 2017; Available online: https://www.aphl.org/conferences/proceedings/Doc-322uments/Mercer.pdf (accessed on 27 April 2021).
- Hale, J.E.; Darras, B.T.; Swoboda, K.J.; Estrella, E.; Chen, J.Y.H.; Abbott, M.A.; Hay, B.N.; Kumar, B.; Counihan, A.M.; Gerstel-Thompson, J.; et al. Massachusetts’ findings from statewide newborn screening for spinal muscular atrophy. Int J. Neonatal Screen. 2021, 23, 26. [Google Scholar] [CrossRef]
- Chen, T.H.; Tzeng, C.C.; Wang, C.C.; Wu, S.M.; Chang, J.G.; Yang, S.N.; Hung, C.H.; Jong, Y.J. Identification of bidirectional gene conversion between SMN1 and SMN2 by simultaneous analysis of SMN dosage and hybrid genes in a Chinese population. J. Neurol Sci. 2011, 308, 83–87. [Google Scholar] [CrossRef]
- Niba, E.T.E.; Nishio, H.; Wijaya, Y.O.S.; Lai, P.S.; Tozawa, T.; Chiyonobu, T.; Yamadera, M.; Okamoto, K.; Awano, H.; Takeshima, Y.; et al. Clinical phenotypes of spinal muscular atrophy patients with hybrid SMN gene. Brain Dev. 2021, 43, 294–302. [Google Scholar] [CrossRef]
- Velasco, E.; Valero, C.; Valero, A.; Moreno, F.; Hernández-Chico, C. Molecular analysis of the SMN and NAIP genes in Spanish spinal muscular atrophy (SMA) families and correlation between number of copies of c BCD541and SMA phenotype. Hum. Mol. Genet. 1996, 5, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, Y.H.; Chiang, S.C.; Weng, W.C.; Lee, N.C.; Lin, C.J.; Hsieh, W.S.; Lee, W.T.; Jong, Y.J.; Ko, T.M.; Hwu, W.L. Presymptomatic Diagnosis of Spinal Muscular Atrophy Through Newborn Screening. J. Pediatr. 2017, 190, 124–129. [Google Scholar] [CrossRef]
- Huang, C.H.; Chang, Y.Y.; Chen, C.H.; Kuo, Y.S.; Hwu, W.L.; Gerdes, T.; Ko, T.M. Copy number analysis of survival motor neuron genes by multiplex ligation-dependent probe amplification. Genet. Med. 2007, 9, 241–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstel-Thompson, J.L.; Wilkey, J.F.; Baptiste, J.C.; Navas, J.S.; Pai, S.; Pass, K.A.; Eaton, R.B.; Comeau, A.M. High-throughput multiplexed T-cell-receptor excision circle quantitative PCR assay with internal controls for detection of severe combined immunodeficiency in population-based newborn screening. Clin. Chem. 2010, 56, 1466–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glascock, J.; Sampson, J.; Connolly, A.M.; Darras, B.T.; Day, J.W.; Finkel, R.; Jarecki, J. Revised recommendations for the treatment of infants diagnosed with spinal muscular atrophy via newborn screening who have 4 copies of SMN2. J. Neuromuscul. Dis. 2020, 7, 97–100. [Google Scholar] [CrossRef] [Green Version]
- Glascock, J.; Sampson, J.; Haidet-Phillips, A.; Connolly, A.; Darras, B.; Day, J.; Jarecki, J. Treatment algorithm for infants diagnosed with spinal muscular atrophy through newborn screening. J. Neuromuscul. Dis. 2018, 5, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Schorling, D.C.; Becker, J.; Pechmann, A.; Langer, T.; Wirth, B.; Kirschner, J. Discrepancy in redetermination of SMN2 copy numbers in children with SMA. Neurology 2019, 93, 267–269. [Google Scholar] [CrossRef]
- Dangouloff, T.; Boemer, F.; Dideberg, V.; Caberg, J.H.; Servais, L. Reader response: Discrepancy in redetermination of SMN2 copy numbers in children with SMA. Neurology 2020, 95, 144–145. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, J.; Becker, J.; Schorling, D.; Pechmann, A.; Wirth, B. Author response: Discrepancy in redetermination of SMN2 copy numbers in children with SMA. Neurology 2020, 95, 145. [Google Scholar] [CrossRef] [PubMed]
- Aravind Ganesh, A.; Steven Galetta, S. Editors’ note: Discrepancy in redetermination of SMN2 copy numbers in children with SMA. Neurology 2020, 95, 144. [Google Scholar]
- Blasco-Pérez, L.; Paramonov, I.; Leno, J.; Bernal, S.; Alias, L.; Fuentes-Prior, P.; Cuscó, I.; Tizzano, E.F. Beyond copy number: A new, rapid, and versatile method for sequencing the entire SMN2 gene in SMA patients. Hum. Mutat. 2021, 42, 787–795. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Reference Specimens | Number of Specimens | NENSP Detection of Nucleotide at c.840 of SMN1 |
|---|---|---|
| Affected | n = 14 | “T” |
| Parents | n = 16 | “C” and “T” |
| Carrier | “C” and “T” | |
| Positive control (known SMA) | “T” |
| SMN2 Copy Number Assay Applied to the Specimens of Infants Identified by NBS as SMA Affected. | |||
|---|---|---|---|
| MA Patient Specimens | SMA Status | SMN2 Copy Number Prediction by NENSP from DBS | SMN2 Copy Number Prediction by Diagnostic Lab Test of an Independent Specimen |
| TS1 | Affected | 3 | 2 |
| TS2 | Affected | More than 3 copies | 4 |
| TS3 | Affected | 2 | 2 |
| TS4 | Affected | 3 | 2 |
| TS5 | Affected | 2 | 2 |
| TS6 | Affected | 2 | 2 |
| TS7 | Affected | 4 | 4 |
| TS8 | Affected | 3 | 2 |
| TS9 | Affected | 2 | 2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, B.; Barton, S.; Kordowska, J.; Eaton, R.B.; Counihan, A.M.; Hale, J.E.; Comeau, A.M. Novel Modification of a Confirmatory SMA Sequencing Assay that Can Be Used to Determine SMN2 Copy Number. Int. J. Neonatal Screen. 2021, 7, 47. https://doi.org/10.3390/ijns7030047
Kumar B, Barton S, Kordowska J, Eaton RB, Counihan AM, Hale JE, Comeau AM. Novel Modification of a Confirmatory SMA Sequencing Assay that Can Be Used to Determine SMN2 Copy Number. International Journal of Neonatal Screening. 2021; 7(3):47. https://doi.org/10.3390/ijns7030047
Chicago/Turabian StyleKumar, Binod, Samantha Barton, Jolanta Kordowska, Roger B. Eaton, Anne M. Counihan, Jaime E. Hale, and Anne Marie Comeau. 2021. "Novel Modification of a Confirmatory SMA Sequencing Assay that Can Be Used to Determine SMN2 Copy Number" International Journal of Neonatal Screening 7, no. 3: 47. https://doi.org/10.3390/ijns7030047
APA StyleKumar, B., Barton, S., Kordowska, J., Eaton, R. B., Counihan, A. M., Hale, J. E., & Comeau, A. M. (2021). Novel Modification of a Confirmatory SMA Sequencing Assay that Can Be Used to Determine SMN2 Copy Number. International Journal of Neonatal Screening, 7(3), 47. https://doi.org/10.3390/ijns7030047