Performance of a Three-Tier (IRT-DNA-IRT) Cystic Fibrosis Screening Algorithm in British Columbia

Abstract
1. Introduction
2. Materials and Methods
2.1. Data Sources
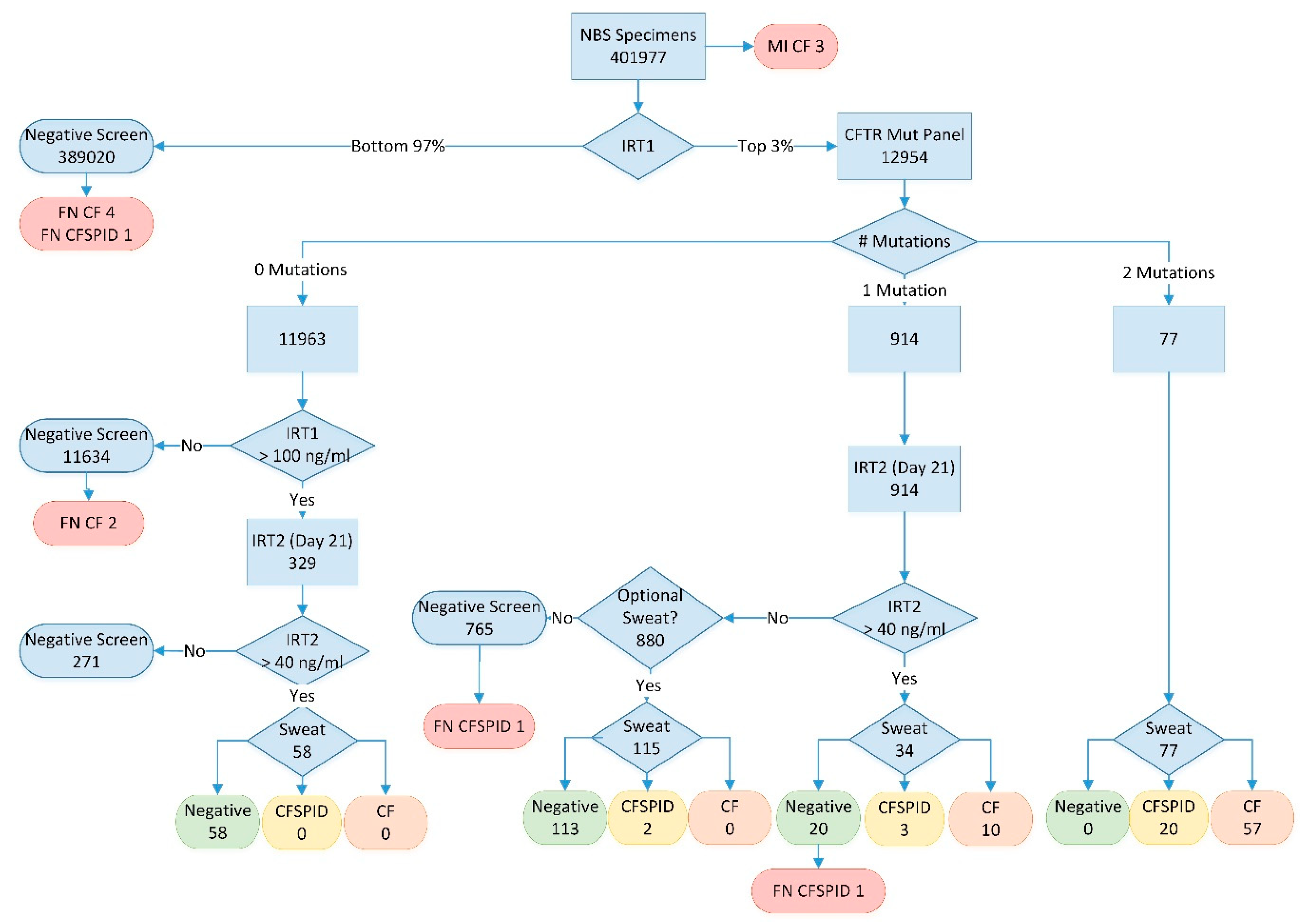
2.2. Screening Algorithm
3. Results
4. Discussion
4.1. Birth Incidence
4.2. Program Performance
4.3. False Negatives
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Therrell, B.L.; Hannon, W.H.; Hoffman, G.; Ojodu, J.; Farrell, P.M. Immunoreactive Trypsinogen (IRT) as a Biomarker for Cystic Fibrosis: Challenges in Newborn Dried Blood Spot Screening. Mol. Genet. Metab. 2012, 106, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Barben, J.; Castellani, C.; Dankert-Roelse, J.; Gartner, S.; Kashirskaya, N.; Linnane, B.; Mayell, S.; Munck, A.; Sands, D.; Sommerburg, O.; et al. The Expansion and Performance of National Newborn Screening Programmes for Cystic Fibrosis in Europe. J. Cyst. Fibros. 2017, 16, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Currier, R.J.; Sciortino, S.; Liu, R.; Bishop, T.; Alikhani Koupaei, R.; Feuchtbaum, L.; Koupaei, R.A.; Feuchtbaum, L. Genomic Sequencing in Cystic Fibrosis Newborn Screening: What Works Best, Two-Tier Predefined CFTR Mutation Panels or Second-Tier CFTR Panel Followed by Third-Tier Sequencing? Genet. Med. 2017, 19, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.; Rosenberg, M.; Hoffman, G.; Anstead, M.; Farrell, P.M. A Decision-Tree Approach to Cost Comparison of Newborn Screening Strategies for Cystic Fibrosis. Pediatrics 2012, 129, 339. [Google Scholar] [CrossRef] [PubMed]
- Sontag, M.K.; Lee, R.; Wright, D.; Freedenberg, D.; Sagel, S.D. Improving the Sensitivity and Positive Predictive Value in a Cystic Fibrosis Newborn Screening Program Using a Repeat Immunoreactive Trypsinogen and Genetic Analysis. J. Pediatr. 2016, 175, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Werbrouck, A.; Verhaeghe, N.; De Wachter, E.; Simoens, S.; Annemans, L.; Putman, K. Strategies for Newborn Screening for Cystic Fibrosis: A Systematic Review of Health Economic Evaluations. J. Cyst. Fibros. 2018, 17, 306–315. [Google Scholar] [CrossRef]
- Farrell, P.M.; White, T.B.; Ren, C.L.; Hempstead, S.E.; Accurso, F.; Derichs, N.; Howenstine, M.; McColley, S.A.; Rock, M.; Rosenfeld, M.; et al. Diagnosis of Cystic Fibrosis: Consensus Guidelines from the Cystic Fibrosis Foundation. J. Pediatr. 2017, 181, S4–S15. [Google Scholar] [CrossRef]
- Castellani, C.; Duff, A.J.A.; Bell, S.C.; Heijerman, H.G.M.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS Best Practice Guidelines: The 2018 Revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef]
- Barben, J.; Southern, K.W. Cystic Fibrosis Screen Positive, Inconclusive Diagnosis. Curr. Opin. Pulm. Med. 2016, 22, 617–622. [Google Scholar] [CrossRef][Green Version]
- Steinraths, M.; Vallance, H.D.; Davidson, A.G.F. Delays in Diagnosing Cystic Fibrosis: Can We Find Ways to Diagnose It Earlier? Can. Fam. Physician 2008, 54, 877–883. [Google Scholar]
- Hale, J.E.; Parad, R.B.; Comeau, A.M. Newborn Screening Showing Decreasing Incidence of Cystic Fibrosis. N. Engl. J. Med. 2008, 358, 973–974. [Google Scholar] [CrossRef] [PubMed]
- Scotet, V.; Assael, B.; Dugueperoux, I.; Tamanini, A.; Audrezet, M.; Ferec, C.; Catellani, C. Time Trends in Birth Incidence of Cystic Fibrosis in Two European Areas: Data from Newborn Screening Programs. J. Pediatr. 2008, 152, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Census Profile, 2016 Census. Available online: https://www12.statcan.gc.ca/census-recensement/2016/dp-pd/prof/index.cfm?Lang=E (accessed on 13 February 2020).
- Data Tables, 1991 Census. Available online: https://www12.statcan.gc.ca/English/census91/data/tables (accessed on 13 February 2020).
- Hayeems, R.Z.; Miller, F.A.; Vermeulen, M.; Potter, B.K.; Chakraborty, P.; Davies, C.; Carroll, J.C.; Ratjen, F.; Guttmann, A. False-Positive Newborn Screening for Cystic Fibrosis and Health Care Use. Pediatrics 2017, 140, 20170604. [Google Scholar] [CrossRef] [PubMed]
- Burgess, C.A.; McMahon, V.R.; Schellenberg, A.; Sinclair, G.; Nelson, T.; Vallance, H.D.; Chilvers, M.A. The Optional Sweat Test: Why Do Parents Want It? Pediatr. Pulmonol. 2015, 50, 503. [Google Scholar]
- Mak, D.Y.F.; Sykes, J.; Stephenson, A.L.; Lands, L.C. The Benefits of Newborn Screening for Cystic Fibrosis: The Canadian Experience. J. Cyst. Fibros. 2016, 15, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.T.C.; Wallis, C.; Price, J.F.; Carr, S.B.; Chavasse, R.J.; Shankar, A.; Seddon, P.; Balfour-Lynn, I.M. Diagnosis of Cystic Fibrosis in London and South East England before and after the Introduction of Newborn Screening. Arch. Dis. Child. 2014, 99, 197–202. [Google Scholar] [CrossRef]
- Sanders, D.B.; Lai, H.J.; Rock, M.J.; Farrell, P.M. Comparing Age of Cystic Fibrosis Diagnosis and Treatment Initiation after Newborn Screening with Two Common Strategies. J. Cyst. Fibros. 2012, 11, 150–153. [Google Scholar] [CrossRef][Green Version]
- Kay, D.M.; Langfelder-Schwind, E.; Decelie-Germana, J.; Sharp, J.K.; Maloney, B.; Tavakoli, N.P.; Saavedra-Matiz, C.A.; Krein, L.M.; Caggana, M.; Kier, C. Utility of a Very High IRT/No Mutation Referral Category in Cystic Fibrosis Newborn Screening. Pediatr. Pulmonol. 2015, 50, 771–780. [Google Scholar] [CrossRef]
- Munck, A.; Delmas, D.; Audrézet, M.P.; Lemonnier, L.; Cheillan, D.; Roussey, M. Optimization of the French Cystic Fibrosis Newborn Screening Programme by a Centralized Tracking Process. J. Med. Screen. 2018, 25, 6–12. [Google Scholar] [CrossRef]
- Kharrazi, M.; Yang, J.; Bishop, T.; Lessing, S.; Young, S.; Graham, S.; Pearl, M.; Chow, H.; Ho, T.; Currier, R.; et al. Newborn Screening for Cystic Fibrosis in California. Pediatrics 2015, 136, 1062–1072. [Google Scholar] [CrossRef]
- Weidler, S.; Stopsack, K.H.; Hammermann, J.; Sommerburg, O.; Mall, M.A.; Hoffmann, G.F.; Kohlmüller, D.; Okun, J.G.; Macek, M.F.; Votava, F.; et al. A Product of Immunoreactive Trypsinogen and Pancreatitis-Associated Protein as Second-Tier Strategy in Cystic Fibrosis Newborn Screening. J. Cyst. Fibros. 2016, 15, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Dankert-Roelse, J.E.; Bouva, M.J.; Jakobs, B.S.; Janssens, H.M.; De Winter-De Groot, K.M.; Schönbeck, Y.; Gille, J.J.P.; Gulmans, V.A.M.; Verschoof-Puite, R.K.; Schielen, P.C.J.I.; et al. Newborn Blood Spot Screening for Cystic Fibrosis with a Four-Step Screening Strategy in the Netherlands. J. Cyst. Fibros. 2018, 18, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Krulišová, V.; Balaščaková, M.; Skalická, V.; Piskáčková, T.; Holubová, A.; Paděrová, J.; Křenková, P.; Dvořáková, L.; Zemková, D.; Kračmar, P.; et al. Prospective and Parallel Assessments of Cystic Fibrosis Newborn Screening Protocols in the Czech Republic: IRT/DNA/IRT versus IRT/PAP and IRT/PAP/DNA DNA Deoxyribonucleic Acid IRT Immunoreactive Trypsinogen NBS Newborn Screening PAP Pancreatitis-Associate. Eur. J. Pediatr. 2012, 171, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Doull, I. Devil in the Detail of Newborn Screening for Cystic Fibrosis. Arch. Dis. Child. 2019, 104, 938–940. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Results | CF | CF + CFSPID | ||
|---|---|---|---|---|
| Cases Detected | PPV | Cases Detected | PPV | |
| Total Cases (Incidence) | 76 (1/5289) | 104 (1/3865) | ||
| True Positive | 70 | 95 | ||
| Meconium Ileus | 3 | 3 | ||
| 2 Mutations | 57 | 74% | 77 | 100% |
| 1 Mutation and High IRT2 | 10 | 29% | 13 | 38% |
| 1 Mutation and Normal IRT2 (optional sweat test) | 0 | 0% | 2 1 | 1.7% |
| No Mutations and Top 0.1% IRT1 and High IRT2 | 0 | 0% | 0 | 0% |
| False Negative | 6 | 9 | ||
| Normal IRT1 | 4 | 5 | ||
| No mutations and IRT1 NOT top 0.1% | 2 | 2 | ||
| 1 Mutation and Normal IRT2 (no sweat) | 0 | 1 | ||
| 1 Mutation and High IRT2 (Normal Sweat) | 0 | 1 | ||
| Time to First CF Clinic Contact (Days) | ||
|---|---|---|
| Case Definition (Total) | Median | Range |
| 2 mutations (77) | 22 | 10–50 |
| 1 Mutation and High IRT2 (13) | 31 | 12–39 |
| Overall (90) | 23 | 10–50 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinclair, G.; McMahon, V.; Schellenberg, A.; Nelson, T.N.; Chilvers, M.; Vallance, H. Performance of a Three-Tier (IRT-DNA-IRT) Cystic Fibrosis Screening Algorithm in British Columbia. Int. J. Neonatal Screen. 2020, 6, 46. https://doi.org/10.3390/ijns6020046
Sinclair G, McMahon V, Schellenberg A, Nelson TN, Chilvers M, Vallance H. Performance of a Three-Tier (IRT-DNA-IRT) Cystic Fibrosis Screening Algorithm in British Columbia. International Journal of Neonatal Screening. 2020; 6(2):46. https://doi.org/10.3390/ijns6020046
Chicago/Turabian StyleSinclair, Graham, Vanessa McMahon, Amy Schellenberg, Tanya N. Nelson, Mark Chilvers, and Hilary Vallance. 2020. "Performance of a Three-Tier (IRT-DNA-IRT) Cystic Fibrosis Screening Algorithm in British Columbia" International Journal of Neonatal Screening 6, no. 2: 46. https://doi.org/10.3390/ijns6020046
APA StyleSinclair, G., McMahon, V., Schellenberg, A., Nelson, T. N., Chilvers, M., & Vallance, H. (2020). Performance of a Three-Tier (IRT-DNA-IRT) Cystic Fibrosis Screening Algorithm in British Columbia. International Journal of Neonatal Screening, 6(2), 46. https://doi.org/10.3390/ijns6020046