Incorporation of Second-Tier Biomarker Testing Improves the Specificity of Newborn Screening for Mucopolysaccharidosis Type I

 , , ,
, , , {kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Analytical Methods and Reference Population
2.2. Study Population
2.3. Confirmatory Testing
2.4. Post-Analytical Interpretation
3. Results
3.1. Patient Cohorts
3.1.1. Group 1
3.1.2. Group 2
3.1.3. Group 3
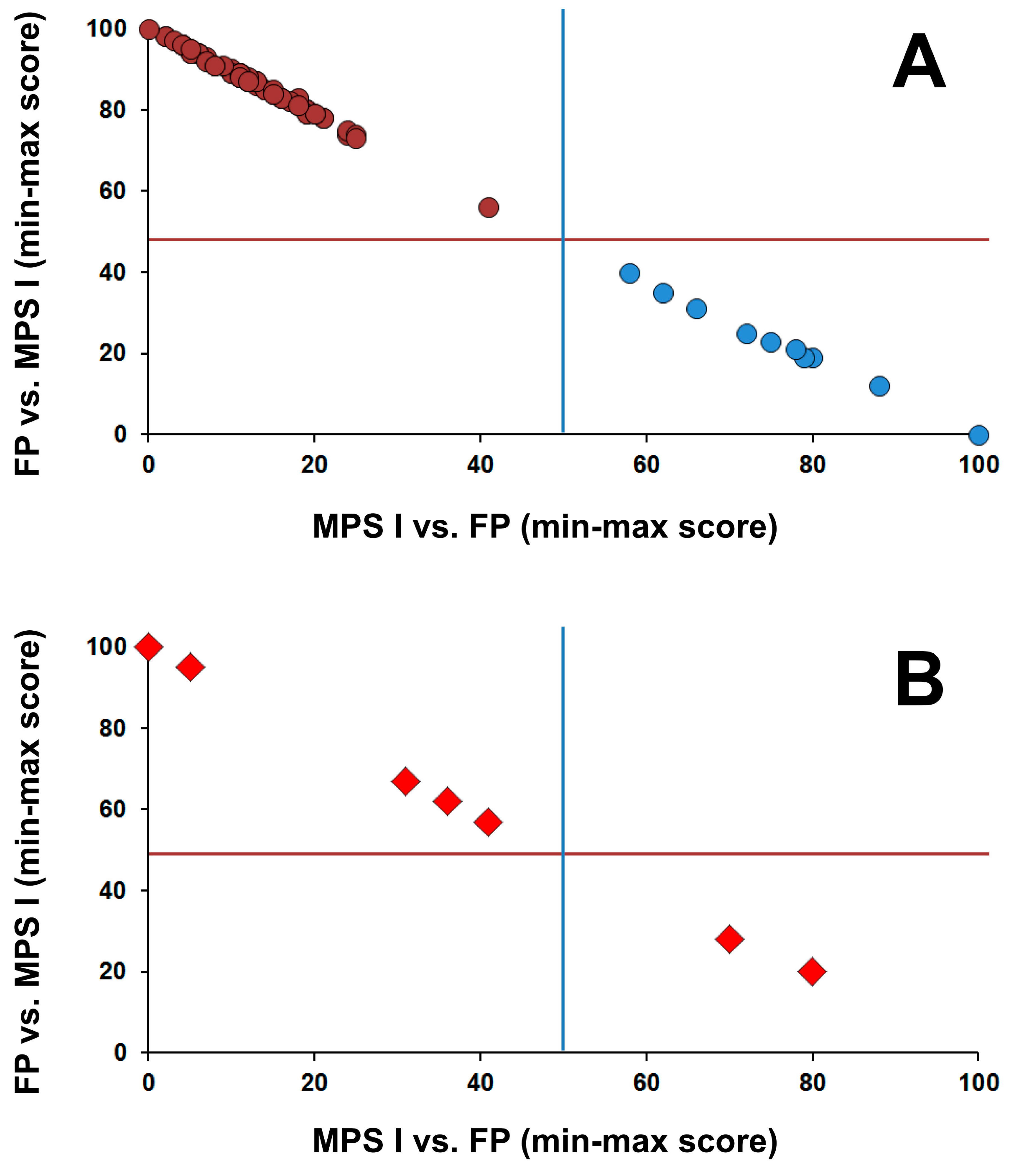
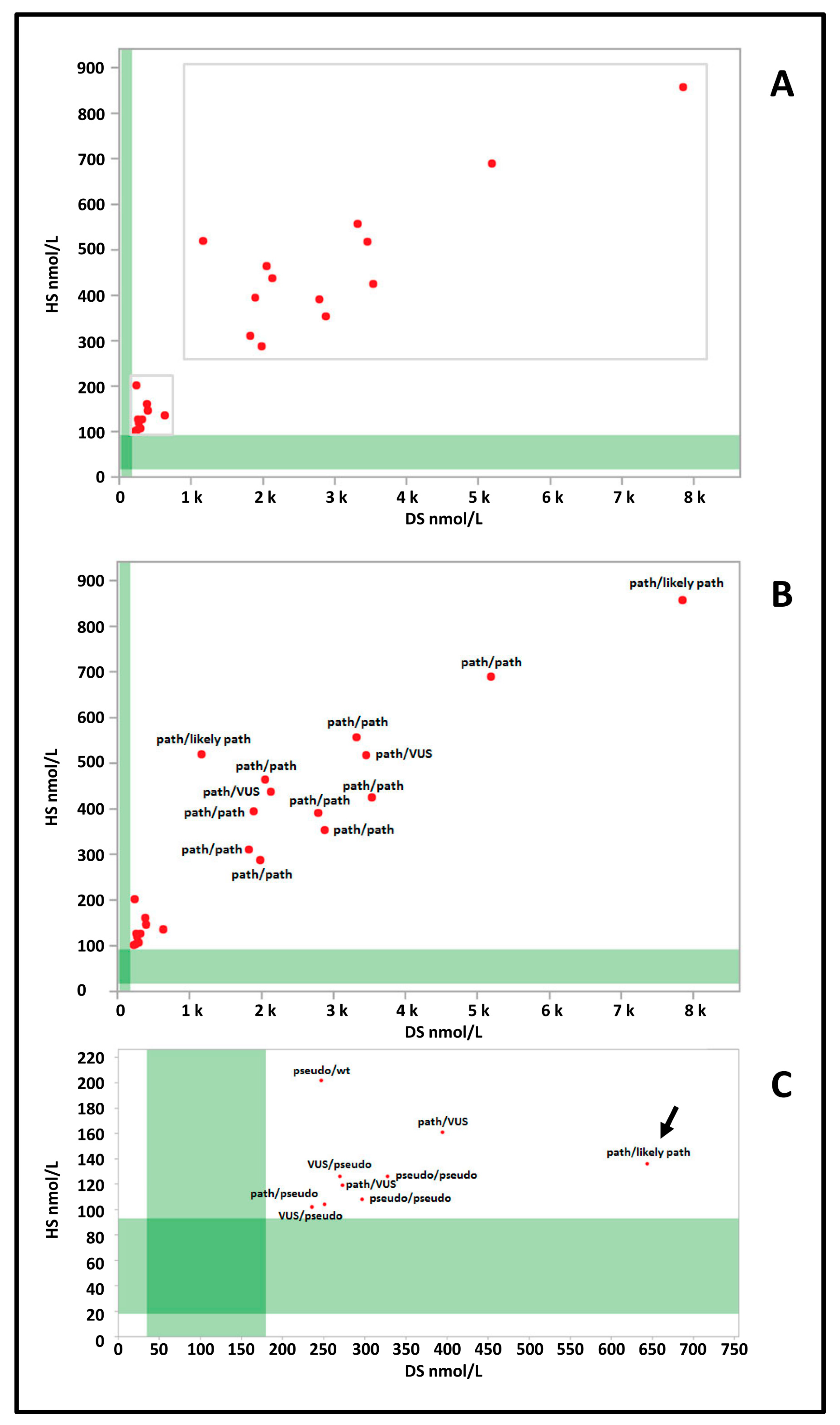
3.2. Postanalytical Interpretation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chamoles, N.A.; Blanco, M.; Gaggioli, D. Diagnosis of alpha-L-iduronidase deficiency in dried blood spots on filter paper: The possibility of newborn diagnosis. Clin. Chem. 2001, 47, 780–781. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Clarke, L.A.; Beck, M.; Kolodny, E.H.; Pastores, G.M.; Muenzer, J.; Braakman, T. Enzyme replacement therapy for mucopolysaccharidosis I: A randomized, double-blinded, placebo-controlled, multinational study of recombinant human alpha-L-iduronidase (laronidase). J. Pediatr. 2004, 144, 581–588. [Google Scholar] [CrossRef] [PubMed]
- Aldenhoven, M.; Jones, S.A.; Bonney, D.; Borrill, R.E.; Coussons, M.; Mercer, J.; van der Ploeg, A.T. Hematopoietic cell transplantation for mucopolysaccharidosis patients is safe and effective: Results after implementation of international guidelines. Biol. Blood Marrow Transpl. 2015, 21, 1106–1109. [Google Scholar] [CrossRef] [PubMed]
- Poe, M.D.; Chagnon, S.L.; Escolar, M.L. Early treatment is associated with improved cognition in Hurler syndrome. Ann. Neurol. 2014, 76, 747–753. [Google Scholar] [CrossRef]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Waggoner, D.; Tinkle, B.; Braddock, S.R.; Schneider, M.; Grange, D.K.; Nash, C.; Shryock, H.; et al. Newborn screening for lysosomal storage disorders in illinois: The Initial 15-month experience. J. Pediatr. 2017, 190, 130–135. [Google Scholar] [CrossRef]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in Missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef]
- Taylor, J.L.; Clinard, K.; Powell, C.M.; Rehder, C.; Young, S.P.; Bali, D.; Beckloff, S.E.; Gehtland, L.M.; Kemper, A.R.; Lee, S.; et al. The North Carolina experience with mucopolysaccharidosis type 1 newborn screening. J. Pediatr. 2019, 211, 193–200. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Jahic, A.; Günther, S.; Muschol, N.; Fossøy Stadheim, B.; Braaten, Ø.; Kjensli Hyldebrandt, H.; Kuiper, G.-A.; Tylee, K.; Wijburg, F.A.; Beetz, C. “Missing mutations” in MPS I: Identification of two novel copy number variations by an IDUA-specific in house MLPA assay. Mol. Genet. Genom. Med. 2019, 7, e00615. [Google Scholar] [CrossRef]
- Minter Baerg, M.M.; Stoway, S.D.; Hart, J.; Mott, L.; Peck, D.S.; Nett, S.L.; Eckerman, J.S.; Lacey, J.M.; Turgeon, C.T.; Gavrilov, D.; et al. Precision newborn screening for lysosomal disorders. Genet. Med. 2018, 20, 847–854. [Google Scholar] [CrossRef]
- Gatti, R.; Borrone, C.; Filocamo, M.; Pannone, N.; Di Natale, P. Prenatal diagnosis of mucopolysaccharidosis I: A special difficulty arising from an unusually low enzyme activity in mother’s cells. Prenat. Diagn. 1985, 5, 149–154. [Google Scholar] [CrossRef] [PubMed]
- De Ruijter, J.; de Ru, M.H.; Wagemans, T.; Ijlst, L.; Lund, A.M.; Orchard, P.J.; Schaefer, G.B.; Wijburg, F.A.; van Vlies, N. Heparan sulfate and dermatan sulfate derived disaccharides are sensitive markers for newborn screening for mucopolysaccharidoses types I, II and III. Mol. Genet. Metab. 2012, 107, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Kubaski, F.; Mason, R.W.; Nakatomi, A.; Shintaku, H.; Xie, L.; van Vlies, N.N.; Church, H.; Giugliani, R.; Kobayashi, H.; Yamaguchi, S.; et al. Newborn screening for mucopolysaccharidoses: A pilot study of measurement of glycosaminoglycans by tandem mass spectrometry. J. Inherit. Metab. Dis. 2017, 40, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Fujii, T.; Fukushi, M.; Oguma, T.; Shimada, T.; Maeda, M.; Kida, K.; Shibata, Y.; Futatsumori, H.; Montano, A.M.; et al. Newborn screening and diagnosis of mucopolysaccharidoses. Mol. Genet. Metab. 2013, 110, 42–53. [Google Scholar] [CrossRef]
- Martin, J.J.; Ceuterick, C. Prenatal pathology in mucopolysaccharidoses: A comparison with postnatal cases. Clin. Neuropathol. 1983, 2, 122–127. [Google Scholar]
- Matern, D.; Tortorelli, S.; Oglesbee, D.; Gavrilov, D.; Rinaldo, P. Reduction of the false-positive rate in newborn screening by implementation of MS/MS-based second-tier tests: The Mayo Clinic experience (2004–2007). J. Inherit. Metab. Dis. 2007, 30, 585–592. [Google Scholar] [CrossRef]
- Osago, H.; Shibata, T.; Hara, N.; Kuwata, S.; Kono, M.; Uchio, Y.; Tsuchiya, M. Quantitative analysis of glycosaminoglycans, chondroitin/dermatan sulfate, hyaluronic acid, heparan sulfate, and keratan sulfate by liquid chromatography-electrospray ionization-tandem mass spectrometry. Anal. Biochem. 2014, 467, 62–74. [Google Scholar] [CrossRef]
- Tortorelli, S.; Lacey, J.; Raymond, K.; Gavrilov, D.; Oglesbee, D.; Rinaldo, P.; Matern, D. A two-tier biochemical genetic approach to newborn screening for mucopolysaccharidosis type I. J. Inherit. Metab. Dis. 2016, 39, S77. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Tortorelli, S.; Turgeon, C.T.; Gavrilov, D.K.; Oglesbee, D.; Raymond, K.M.; Rinaldo, P.; Matern, D. Simultaneous Testing for 6 Lysosomal Storage Disorders and X-Adrenoleukodystrophy in Dried Blood Spots by Tandem Mass Spectrometry. Clin. Chem. 2016, 62, 1248–1254. [Google Scholar] [CrossRef]
- Hall, P.L.; Marquardt, G.; McHugh, D.M.; Currier, R.J.; Tang, H.; Stoway, S.D.; Rinaldo, P. Postanalytical tools improve performance of newborn screening by tandem mass spectrometry. Genet. Med. 2014, 16, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, G.; Currier, R.; McHugh, D.M.; Gavrilov, D.; Magera, M.J.; Matern, D.; Oglesbee, D.; Raymond, K.; Rinaldo, P.; Smith, E.H.; et al. Enhanced interpretation of newborn screening results without analyte cutoff values. Genet. Med. 2012, 14, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Poletto, E.; Pasqualim, G.; Giugliani, R.; Matte, U.; Baldo, G. Worldwide distribution of common IDUA pathogenic variants. Clin. Genet. 2018, 94, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Bravo, H.; Neto, E.C.; Schulte, J.; Pereira, J.; Filho, C.S.; Bittencourt, F.; Sebastiao, F.; Bender, F.; de Magalhaes, A.P.S.; Guidobono, R.; et al. Investigation of newborns with abnormal results in a newborn screening program for four lysosomal storage diseases in Brazil. Mol. Genet. Metab. Rep. 2017, 12, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.J.; Liao, H.C.; Gelb, M.H.; Chuang, C.K.; Liu, M.Y.; Chen, H.J.; Kao, S.M.; Lin, H.Y.; Huang, Y.H.; Kumar, A.B.; et al. Taiwan national newborn screening program by tandem mass spectrometry for mucopolysaccharidoses types I, II, and VI. J. Pediatr. 2019, 205, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Chuang, C.K.; Lin, H.Y.; Wang, T.J.; Huang, Y.H.; Chan, M.J.; Liao, H.C.; Lo, Y.T.; Wang, L.Y.; Tu, R.Y.; Fang, Y.Y.; et al. Status of newborn screening and follow up investigations for Mucopolysaccharidoses I and II in Taiwan. Orphanet J. Rare Dis. 2018, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Donati, M.A.; Pasquini, E.; Spada, M.; Polo, G.; Burlina, A. Newborn screening in mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 126. [Google Scholar] [CrossRef]
- Lin, S.P.; Lin, H.Y.; Wang, T.J.; Chang, C.Y.; Lin, C.H.; Huang, S.F.; Tsai, C.C.; Liu, H.L.; Keutzer, J.; Chuang, C.K. A pilot newborn screening program for Mucopolysaccharidosis type I in Taiwan. Orphanet J. Rare Dis. 2013, 8, 147. [Google Scholar] [CrossRef]
- Burlina, A.B.P.; Burlina, A.B.P.G.; Rubert, L.; Gueraldi, D.; Cazzorla, C.; Duro Galviati, L.; Burlina, A.P. Implementation of second-tier tests in newborn screening for lysosomal disorders in North Eastern Italy. Int. J. Neonatal. Screen 2019, 5, 24–35. [Google Scholar] [CrossRef]
- Lawrence, R.; Brown, J.R.; Al-Mafraji, K.; Lamanna, W.C.; Beitel, J.R.; Boons, G.J.; Esko, J.D.; Crawford, B.E. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat. Chem. Biol. 2012, 8, 197–204. [Google Scholar] [CrossRef]
- Clarke, L.A.; Giugliani, R.; Guffon, N.; Jones, S.A.; Keenan, H.A.; Munoz-Rojas, M.V.; Okuyama, T.; Viskochil, D.; Whitley, C.B.; Wijburg, F.A.; et al. Genotype-phenotype relationships in mucopolysaccharidosis type I (MPS I): Insights from the International MPS I Registry. Clin. Genet. 2019, 96, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Hoganson, G.E.; Fleischer, J.; Grange, D.K.; Braddock, S.R.; Hickey, R.; Hitchins, L.; Groepper, D.; Christensen, K.M.; Kirby, A.; et al. Population-based newborn screening for mucopolysaccharidosis type II in Illinois: The first year experience. J. Pediatr. 2019, 214, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Karaceper, M.D.; Chakraborty, P.; Coyle, D.; Wilson, K.; Kronick, J.B.; Hawken, S.; Davies, C.; Brownell, M.; Dodds, L.; Feigenbaum, A.; et al. The health system impact of false positive newborn screening results for medium-chain acyl-CoA dehydrogenase deficiency: A cohort study. Orphanet J. Rare Dis. 2016, 11, 12. [Google Scholar] [CrossRef] [PubMed]
- Tu, W.J.; He, J.; Chen, H.; Shi, X.D.; Li, Y. Psychological effects of false-positive results in expanded newborn screening in China. PLoS ONE 2012, 7, e36235. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peck, D.S.; Lacey, J.M.; White, A.L.; Pino, G.; Studinski, A.L.; Fisher, R.; Ahmad, A.; Spencer, L.; Viall, S.; Shallow, N.; et al. Incorporation of Second-Tier Biomarker Testing Improves the Specificity of Newborn Screening for Mucopolysaccharidosis Type I. Int. J. Neonatal Screen. 2020, 6, 10. https://doi.org/10.3390/ijns6010010
Peck DS, Lacey JM, White AL, Pino G, Studinski AL, Fisher R, Ahmad A, Spencer L, Viall S, Shallow N, et al. Incorporation of Second-Tier Biomarker Testing Improves the Specificity of Newborn Screening for Mucopolysaccharidosis Type I. International Journal of Neonatal Screening. 2020; 6(1):10. https://doi.org/10.3390/ijns6010010
Chicago/Turabian StylePeck, Dawn S., Jean M. Lacey, Amy L. White, Gisele Pino, April L. Studinski, Rachel Fisher, Ayesha Ahmad, Linda Spencer, Sarah Viall, Natalie Shallow, and et al. 2020. "Incorporation of Second-Tier Biomarker Testing Improves the Specificity of Newborn Screening for Mucopolysaccharidosis Type I" International Journal of Neonatal Screening 6, no. 1: 10. https://doi.org/10.3390/ijns6010010
APA StylePeck, D. S., Lacey, J. M., White, A. L., Pino, G., Studinski, A. L., Fisher, R., Ahmad, A., Spencer, L., Viall, S., Shallow, N., Siemon, A., Hamm, J. A., Murray, B. K., Jones, K. L., Gavrilov, D., Oglesbee, D., Raymond, K., Matern, D., Rinaldo, P., & Tortorelli, S. (2020). Incorporation of Second-Tier Biomarker Testing Improves the Specificity of Newborn Screening for Mucopolysaccharidosis Type I. International Journal of Neonatal Screening, 6(1), 10. https://doi.org/10.3390/ijns6010010