Performance of the Four-Plex Tandem Mass Spectrometry Lysosomal Storage Disease Newborn Screening Test: The Necessity of Adding a 2nd Tier Test for Pompe Disease

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Screening Assay
2.3. Confirmatory Testing
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hirschhorn, R.; Reuser, A. Glycogen storage disease type II: Acid α-glucosidase (acid maltase) deficiency. In The Metabolic and Molecular Bases of Inherited Disease; Scriver, C., Beaudet, A., Sly, W., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; pp. 3389–3420. [Google Scholar]
- Hwu, W.L.; Chien, Y.H.; Lee, N.C.; Chiang, S.C.; Dobrovolny, R.; Huang, A.C.; Yeh, H.Y.; Chao, M.C.; Lin, S.J.; Kitagawa, T.; et al. Newborn screening for fabry disease in taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A). Hum. Mutat. 2009, 30, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Lee, N.C.; Chen, C.A.; Tsai, F.J.; Tsai, W.H.; Shieh, J.Y.; Huang, H.J.; Hsu, W.C.; Tsai, T.H.; Hwu, W.L. Long-term prognosis of patients with infantile-onset pompe disease diagnosed by newborn screening and treated since birth. J. Pediatr. 2015, 166, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Chiang, S.C.; Zhang, X.K.; Keutzer, J.; Lee, N.C.; Huang, A.C.; Chen, C.A.; Wu, M.H.; Huang, P.H.; Tsai, F.J.; et al. Early detection of pompe disease by newborn screening is feasible: Results from the taiwan screening program. Pediatrics 2008, 122, e39–e45. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.C.; Hwu, W.L.; Lee, N.C.; Hsu, L.W.; Chien, Y.H. Algorithm for pompe disease newborn screening: Results from the taiwan screening program. Mol. Genet. Metab. 2012, 106, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Lee, N.C.; Chiang, S.C.; Desnick, R.J.; Hwu, W.L. Fabry disease: Incidence of the common later-onset α-galactosidase a IVS4+919G>A mutation in taiwanese newborns—Superiority of DNA-based to enzyme-based newborn screening for common mutations. Mol. Med. 2012, 18, 780–784. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.H.; Lee, N.C.; Thurberg, B.L.; Chiang, S.C.; Zhang, X.K.; Keutzer, J.; Huang, A.C.; Wu, M.H.; Huang, P.H.; Tsai, F.J.; et al. Pompe disease in infants: Improving the prognosis by newborn screening and early treatment. Pediatrics 2009, 124, e1116–e1125. [Google Scholar] [CrossRef] [PubMed]
- Labrousse, P.; Chien, Y.H.; Pomponio, R.J.; Keutzer, J.; Lee, N.C.; Akmaev, V.R.; Scholl, T.; Hwu, W.L. Genetic heterozygosity and pseudodeficiency in the Pompe disease newborn screening pilot program. Mol. Genet. Metab. 2010, 99, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Wasserstein, M.P.; Caggana, M.; Bailey, S.M.; Desnick, R.J.; Edelmann, L.; Estrella, L.; Holzman, I.; Kelly, N.R.; Kornreich, R.; Kupchik, S.G.; et al. The New York pilot newborn screening program for lysosomal storage diseases: Report of the first 65,000 infants. Genet. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, P.V.; Campbell, C.; Klug, T.; Rogers, S.; Raburn-Miller, J.; Kiesling, J. Lysosomal storage disorder screening implementation: Findings from the first six months of full population pilot testing in missouri. J. Pediatr. 2015, 166, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Burton, B.K.; Charrow, J.; Hoganson, G.E.; Waggoner, D.; Tinkle, B.; Braddock, S.R.; Schneider, M.; Grange, D.K.; Nash, C.; Shryock, H.; et al. Newborn screening for lysosomal storage disorders in illinois: The initial 15-month experience. J. Pediatr. 2017, 190, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.; Buroker, N.; Cournoyer, J.J.; Potier, A.M.; Trometer, J.D.; Elbin, C.; Schermer, M.J.; Kantola, J.; Boyce, A.; Turecek, F.; et al. Dataset and standard operating procedure for newborn screening of six lysosomal storage diseases: By tandem mass spectrometry. Data Brief 2016, 8, 915–924. [Google Scholar] [CrossRef] [PubMed]
- Burlina, A.B.; Polo, G.; Salviati, L.; Duro, G.; Zizzo, C.; Dardis, A.; Bembi, B.; Cazzorla, C.; Rubert, L.; Zordan, R.; et al. Newborn screening for lysosomal storage disorders by tandem mass spectrometry in north east italy. J. Inherit. Metab. Dis. 2018, 41, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.C.; Chan, M.J.; Yang, C.F.; Chiang, C.C.; Niu, D.M.; Huang, C.K.; Gelb, M.H. Mass spectrometry but not fluorimetry distinguishes affected and pseudodeficiency patients in newborn screening for pompe disease. Clin. Chem. 2017, 63, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Tortorelli, S.; Eckerman, J.S.; Orsini, J.J.; Stevens, C.; Hart, J.; Hall, P.L.; Alexander, J.J.; Gavrilov, D.; Oglesbee, D.; Raymond, K.; et al. Moonlighting newborn screening markers: The incidental discovery of a second-tier test for pompe disease. Genet. Med. 2017, 20, 840–846. [Google Scholar] [CrossRef] [PubMed]
- Minter Baerg, M.M.; Stoway, S.D.; Hart, J.; Mott, L.; Peck, D.S.; Nett, S.L.; Eckerman, J.S.; Lacey, J.M.; Turgeon, C.T.; Gavrilov, D.; et al. Precision newborn screening for lysosomal disorders. Genet. Med. 2017, 20, 847–854. [Google Scholar] [CrossRef] [PubMed]
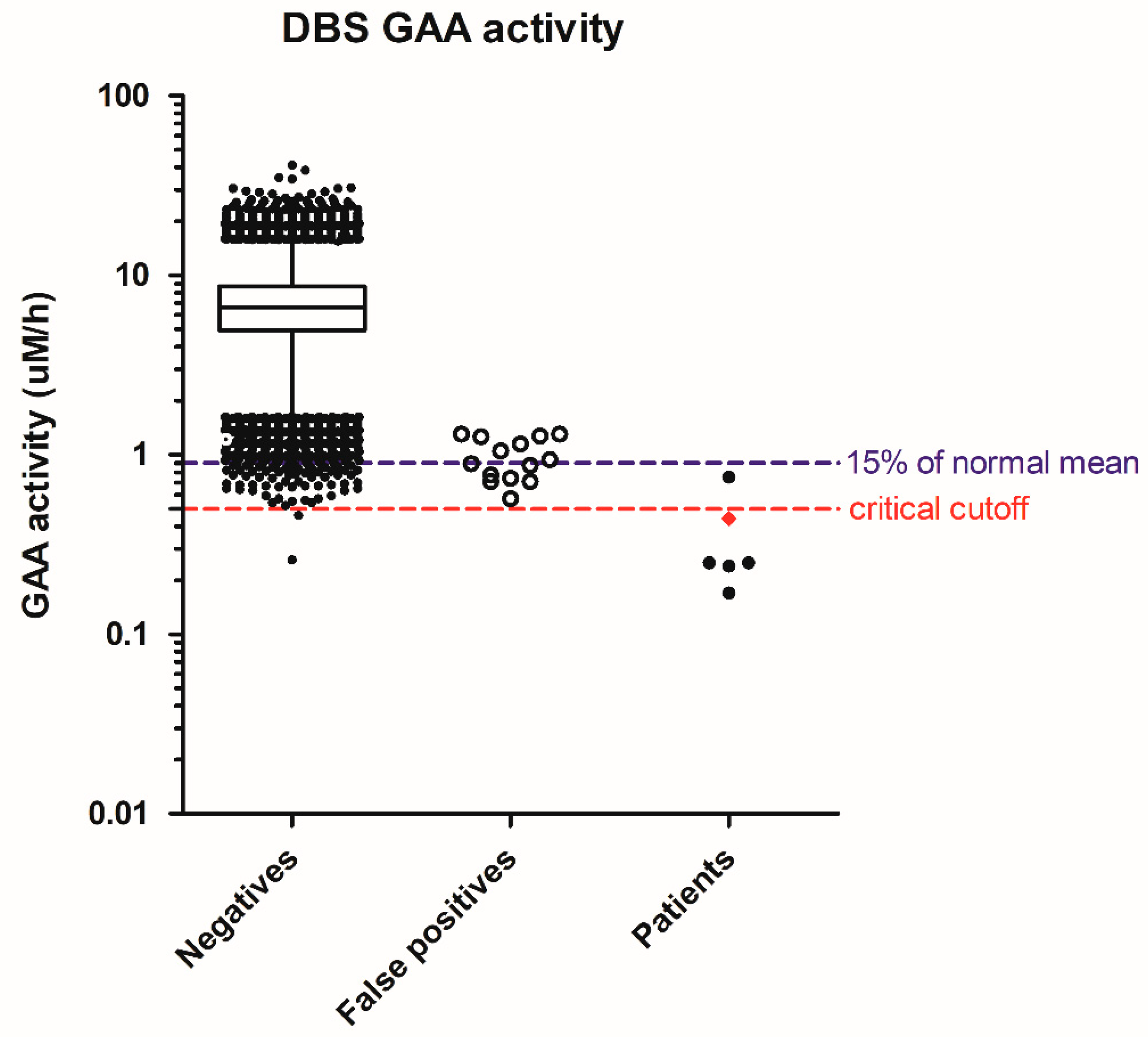

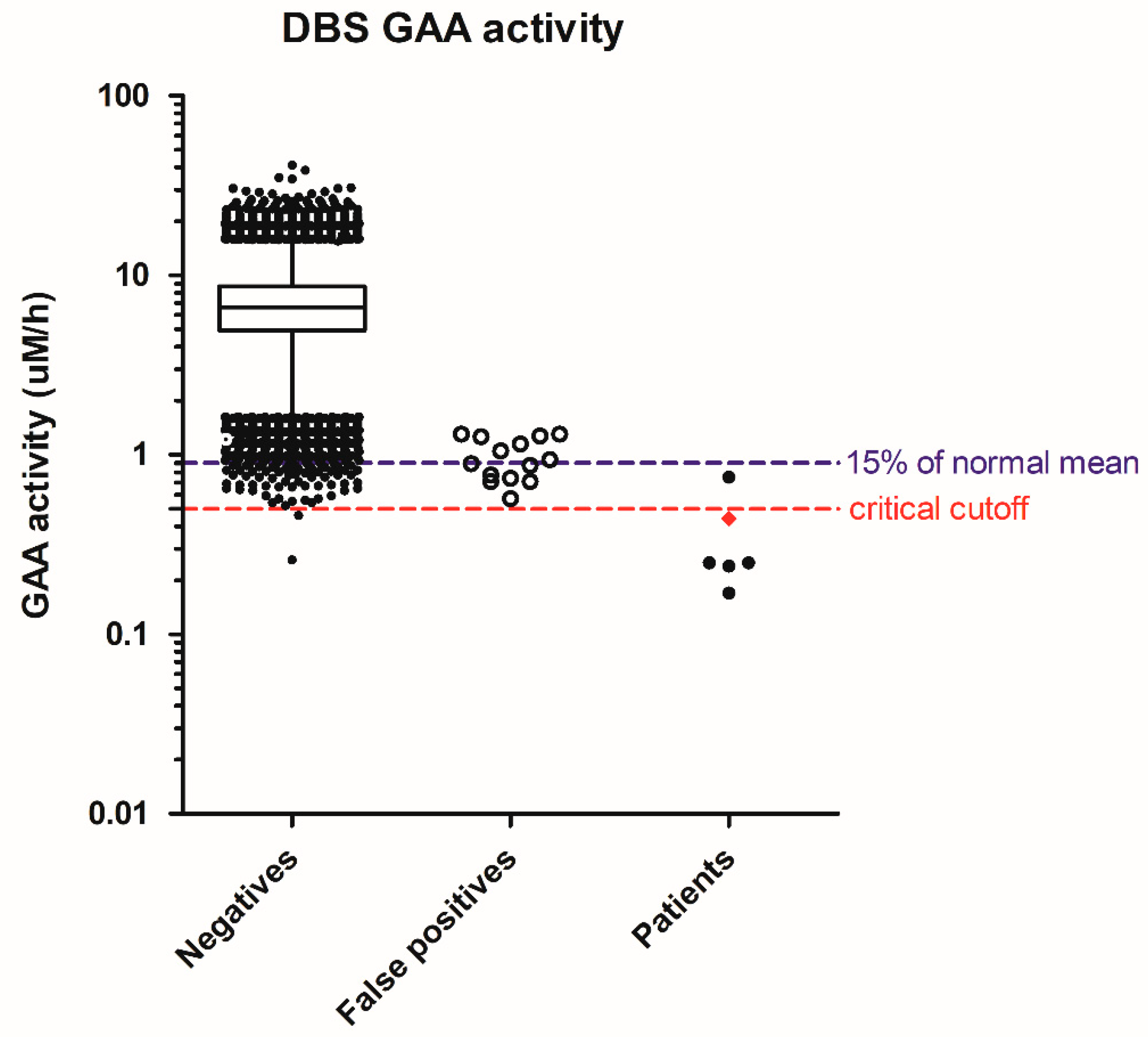{kind=link}
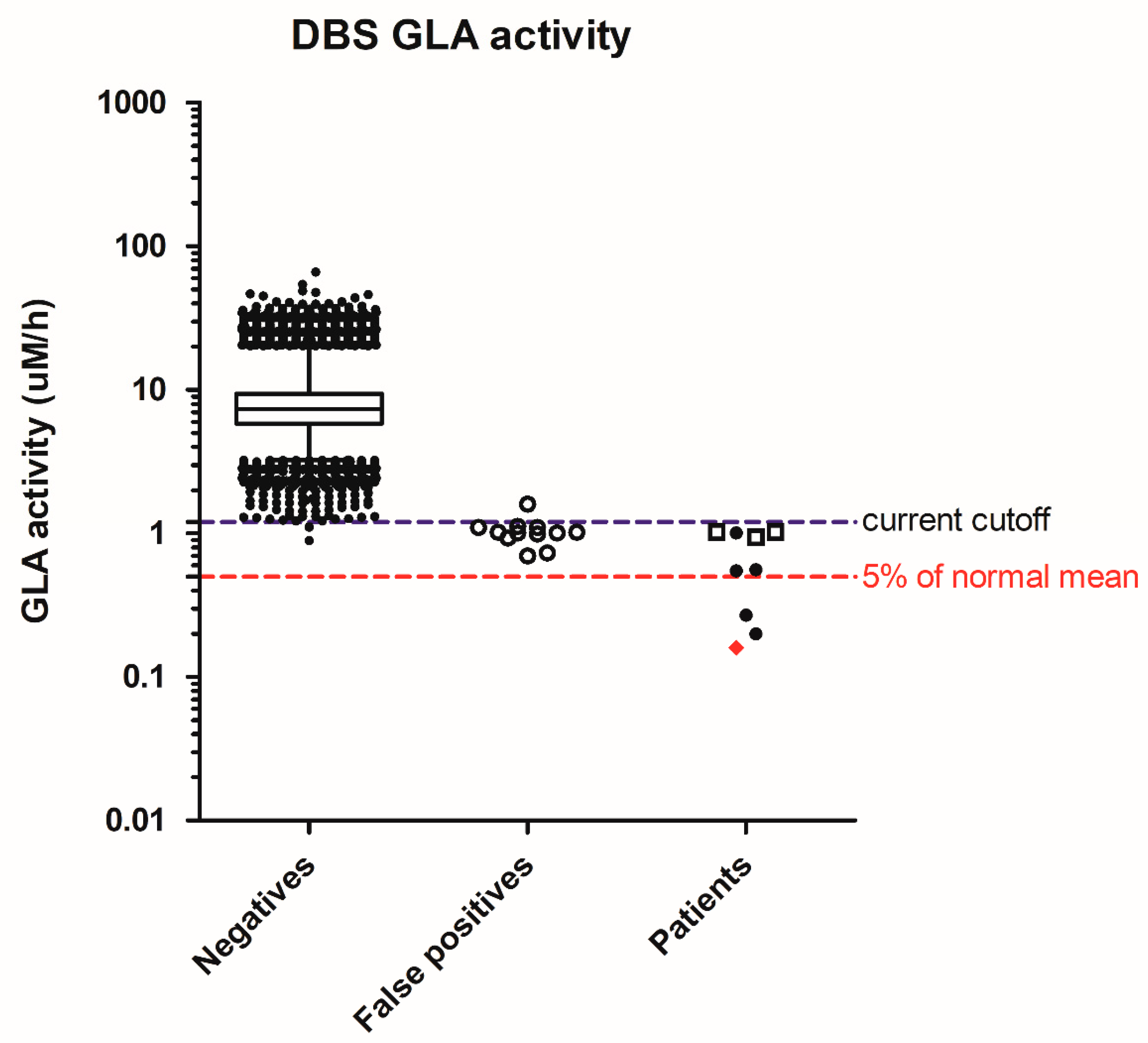
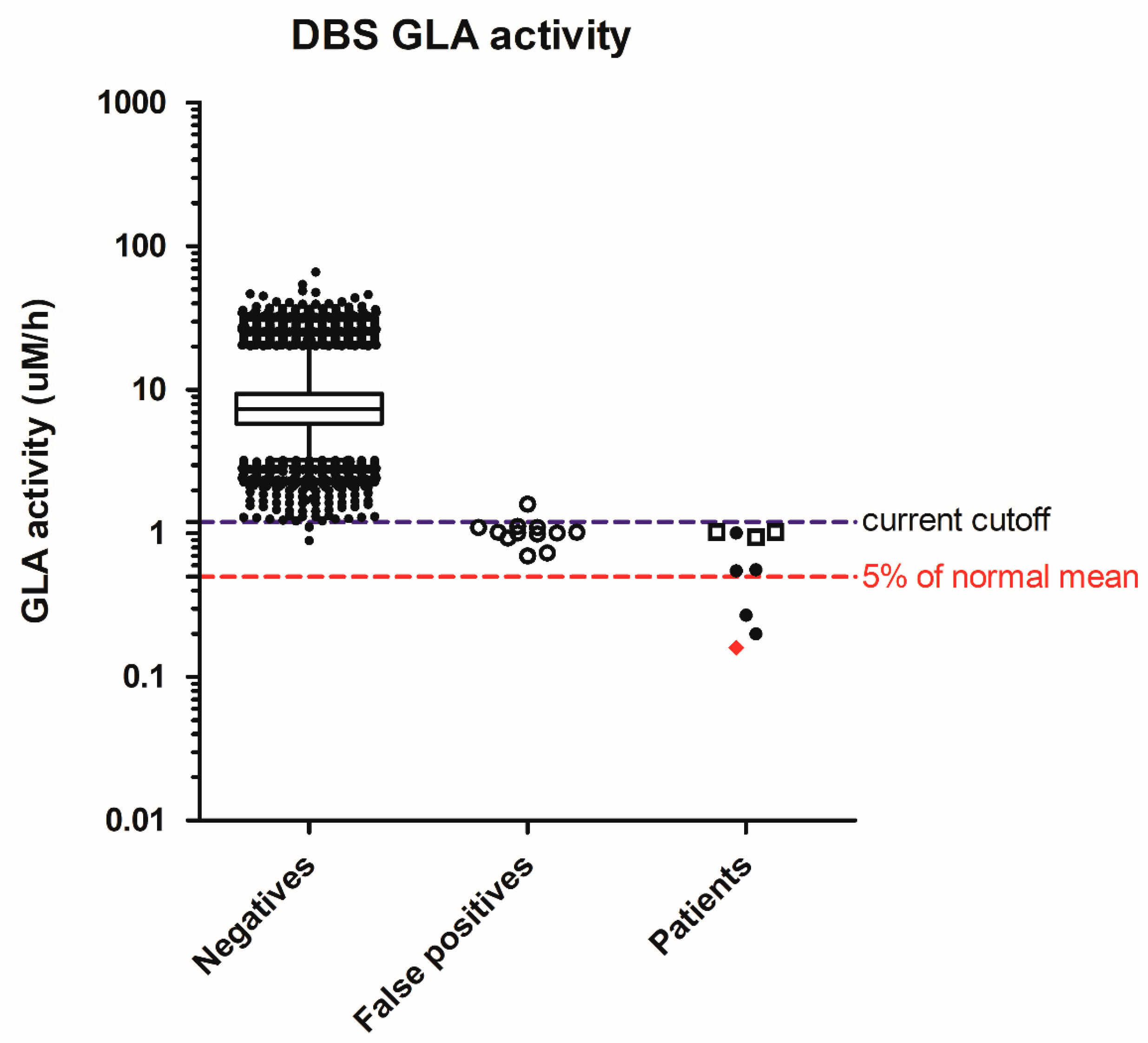{kind=link}
| LSD | Repeat NBS (on 2nd DBS) | False Positives | Patients | Early Onset | Later-Onset |
|---|---|---|---|---|---|
| Pompe | - | 14 | 6 | 1 | 5 |
| Fabry | 21 | 0 | 9 | 1 | 8 |
| MPS I | 34 | 0 | 0 | 0 | 0 |
| Gaucher | 10 | 0 | 0 | 0 | 0 |
| Disorders Suspected | GAA | GLA | ABG | ABG/GAA | ABG/GLA | Inhibition (%) # | % of mean DBS Activity | WBC Activity * | Diagnosis |
|---|---|---|---|---|---|---|---|---|---|
| Pompe | 0.44 | 11.0 | 22.1 | 50.4 | 2.00 | - | 6% | 0.11 | IOPD |
| Pompe | 0.17 | 8.62 | 15.3 | 88.4 | 1.77 | - | 2% | 0.93 | LOPD |
| Pompe | 0.24 | 8.16 | 8.16 | 33.7 | 1.00 | - | 3% | 0.74 | LOPD |
| Pompe | 0.25 | 11.1 | 19.8 | 77.9 | 1.78 | - | 4% | 0.50 | LOPD |
| Pompe | 0.25 | 8.29 | 10.2 | 40.0 | 1.23 | - | 4% | 0.30 | LOPD |
| Pompe | 0.75 | 11.5 | 13.6 | 18.1 | 1.18 | 88.0 | 13% | 0.76 | LOPD |
| Pompe | 0.57 | 3.63 | 4.72 | 8.28 | 1.30 | 87.0 | 8% | 5.56 | Not affected |
| Pompe | 0.71 | 9.87 | 16.8 | 23.6 | 1.70 | - | 10% | 4.43 | Not affected |
| Pompe | 0.71 | 11.7 | 14.5 | 20.3 | 1.23 | - | 10% | 1.16 | Not affected |
| Pompe | 0.74 | 5.93 | 9.97 | 13.5 | 1.68 | 89.5 | 11% | 4.07 | Not affected |
| Pompe | 0.77 | 6.60 | 11.7 | 15.2 | 1.78 | 87.4 | 11% | 3.58 | Not affected |
| Pompe | 0.87 | 3.32 | 11.1 | 12.8 | 3.34 | 87.7 | 12% | 2.11 | Not affected |
| Pompe | 0.89 | 5.83 | 9.10 | 10.2 | 1.56 | 89.0 | 13% | 3.54 | Not affected |
| Pompe | 0.94 | 3.67 | 11.7 | 12.4 | 3.20 | 87.1 | 13% | 3.86 | Not affected |
| Pompe | 1.05 | 4.69 | 8.49 | 8.06 | 1.81 | 87.8 | 15% | - | Not affected |
| Pompe | 1.15 | 5.11 | 8.59 | 7.45 | 1.68 | 87.4 | 16% | 4.08 | Not affected |
| Pompe | 1.26 | 3.47 | 10.5 | 8.34 | 3.02 | 87.5 | 18% | 3.98 | Not affected |
| Pompe | 1.27 | 8.74 | 14.6 | 11.5 | 1.67 | 88.3 | 18% | - | Not affected |
| Pompe | 1.30 | 7.78 | 12.0 | 9.26 | 1.55 | 87.2 | 19% | 4.56 | Not affected |
| Pompe | 1.30 | 6.74 | 17.9 | 13.8 | 2.66 | 87.3 | 19% | 3.91 | Not affected |
| Fabry | 3.57 | 0.16 | 7.26 | 2.03 | 44.1 | - | 2% | 0.42 | T1FD |
| Fabry | 8.20 | 0.27 | 9.05 | 1.10 | 33.9 | - | 3% | 1.00 | (S) T1FD |
| Fabry | 5.10 | 0.20 | 16.8 | 3.29 | 85.9 | - | 2% | 0.88 | T2FD |
| Fabry | 2.42 | 0.55 | 6.89 | 2.85 | 12.6 | - | 7% | 4.62 | (S) T2FD |
| Fabry | 6.32 | 0.56 | 16.4 | 2.60 | 29.4 | - | 7% | - | (S) T2FD |
| Fabry | 6.85 | 1.02 | 9.09 | 1.33 | 8.89 | - | 13% | 4.53 | IVS4 + 919A |
| Fabry | 5.64 | 1.02 | 14.5 | 2.57 | 15.0 | - | 13% | 3.01 | IVS4 + 919A |
| Fabry | 10.5 | 1.01 | 9.22 | 0.88 | 9.14 | - | 13% | 6.50 | IVS4 + 919A |
| Fabry | 8.86 | 1.01 | 13.3 | 1.50 | 15.0 | - | 13% | 6.24 | Benign |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiang, S.-C.; Chen, P.-W.; Hwu, W.-L.; Lee, A.-J.; Chen, L.-C.; Lee, N.-C.; Chiou, L.-Y.; Chien, Y.-H. Performance of the Four-Plex Tandem Mass Spectrometry Lysosomal Storage Disease Newborn Screening Test: The Necessity of Adding a 2nd Tier Test for Pompe Disease. Int. J. Neonatal Screen. 2018, 4, 41. https://doi.org/10.3390/ijns4040041
Chiang S-C, Chen P-W, Hwu W-L, Lee A-J, Chen L-C, Lee N-C, Chiou L-Y, Chien Y-H. Performance of the Four-Plex Tandem Mass Spectrometry Lysosomal Storage Disease Newborn Screening Test: The Necessity of Adding a 2nd Tier Test for Pompe Disease. International Journal of Neonatal Screening. 2018; 4(4):41. https://doi.org/10.3390/ijns4040041
Chicago/Turabian StyleChiang, Shu-Chuan, Pin-Wen Chen, Wuh-Liang Hwu, An-Ju Lee, Li-Chu Chen, Ni-Chung Lee, Li-Yan Chiou, and Yin-Hsiu Chien. 2018. "Performance of the Four-Plex Tandem Mass Spectrometry Lysosomal Storage Disease Newborn Screening Test: The Necessity of Adding a 2nd Tier Test for Pompe Disease" International Journal of Neonatal Screening 4, no. 4: 41. https://doi.org/10.3390/ijns4040041