Newborn Screening for SCD in the USA and Canada


{kind=link}
Abstract
:1. Introduction
2. Epidemiology
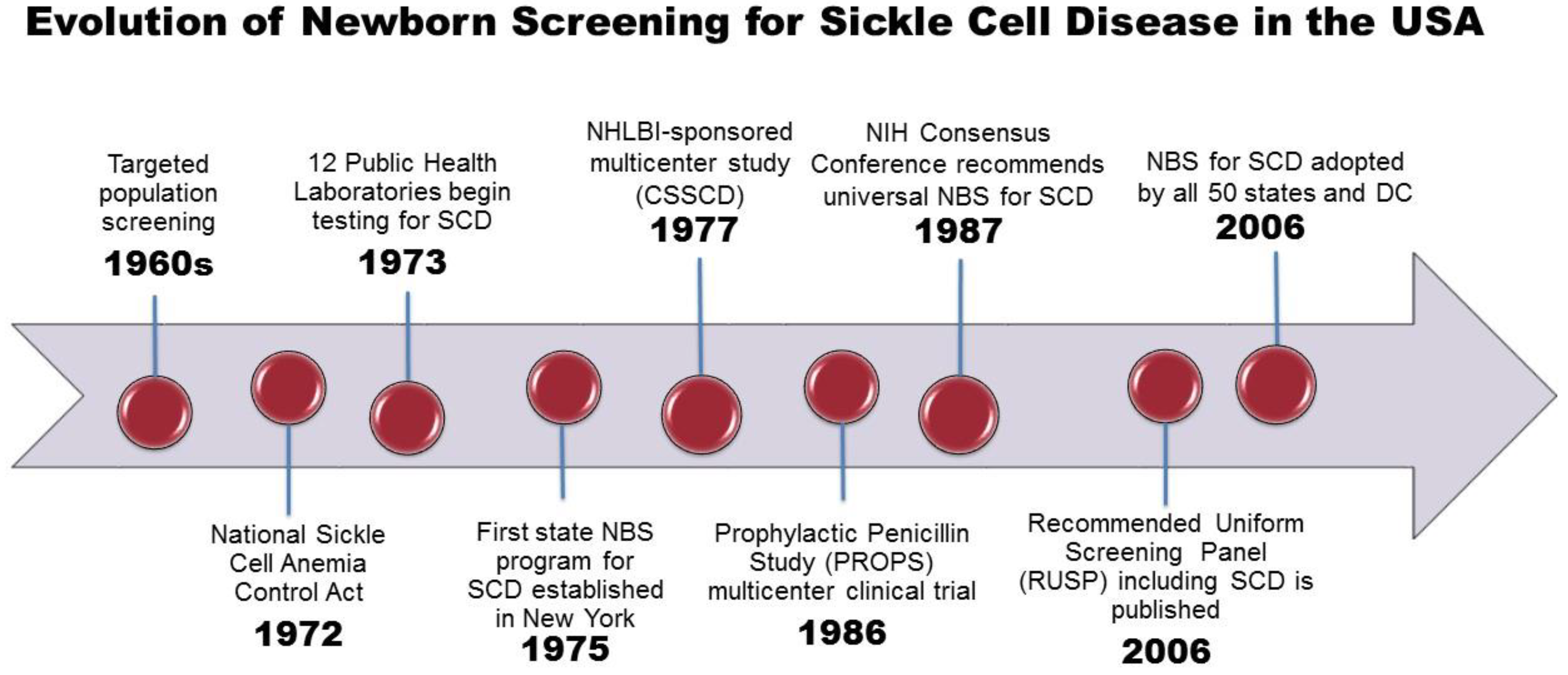
3. History of NBS for SCD
4. Components of Newborn Screening for SCD
5. Screening
5.1. Specimen Collection
5.2. Specimen Submission
5.3. Testing Methods
6. Short Term Follow up
6.1. Primary Screening Results Reporting
6.2. Confirmatory Testing
7. Long-term Follow-up
8. Challenges of NBS for SCD
9. Conclusion
Author Contributions
Funding
Conflicts of Interest
References
- Brittenham, G.M.; Schechter, A.N.; Noguchi, C.T. Hemoglobin S polymerization: Primary determinant of the hemolytic and clinical severity of the sickling syndromes. Blood 1985, 65, 183–189. [Google Scholar] [PubMed]
- Frenette, P.S. Sickle cell vaso-occlusion: Multistep and multicellular paradigm. Curr. Opin. Hematol. 2002, 9, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Gill, F.M.; Sleeper, L.A.; Weiner, S.J.; Brown, A.K.; Bellevue, R.; Grover, R.; Pegelow, C.H.; Vichinsky, E. Clinical events in the first decade in a cohort of infants with sickle cell disease. Cooperative Study of Sickle Cell Disease. Blood 1995, 86, 776–783. [Google Scholar] [PubMed]
- Gaston, M.H.; Verter, J.I.; Woods, G.; Pegelow, C.; Kelleher, J.; Presbury, G.; Zarkowsky, H.; Vichinsky, E.; Iyer, R.; Lobel, J.S.; et al. Prophylaxis with oral penicillin in children with sickle cell anemia. A randomized trial. N. Engl. J. Med. 1986, 314, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Vichinsky, E.; Hurst, D.; Earles, A.; Kleman, K.; Lubin, B. Newborn screening for sickle cell disease: Effect on mortality. Pediatrics 1988, 81, 749–755. [Google Scholar] [PubMed]
- Frempong, T.; Pearson, H.A. Newborn screening coupled with comprehensive follow-up reduced early mortality of sickle cell disease in Connecticut. Connect. Med. 2007, 71, 9–12. [Google Scholar]
- Emond, A.M.; Collis, R.; Darvill, D.; Higgs, D.R.; Maude, G.H.; Serjeant, G.R. Acute splenic sequestration in homozygous sickle cell disease: Natural history and management. J. Pediatr. 1985, 107, 201–206. [Google Scholar] [CrossRef]
- Vichinsky, E.P. Comprehensive care in sickle cell disease: Its impact on morbidity and mortality. Semin. Hematol. 1991, 28, 220–226. [Google Scholar] [PubMed]
- Piel, F.B. The Present and future global burden of the inherited disorders of hemoglobin. Hematol. Oncol. Clin. North. Am. 2016, 30, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Piel, F.B.; Tatem, A.J.; Huang, Z.; Gupta, S.; Williams, T.N.; Weatherall, D.J. Global migration and the changing distribution of sickle haemoglobin: A quantitative study of temporal trends between 1960 and 2000. Lancet Glob. Health 2014, 2, e80–e89. [Google Scholar] [CrossRef]
- Hassell, K.L. Population estimates of sickle cell disease in the U.S. Am. J. Prev. Med. 2010, 38 (Suppl. 4), S512–S521. [Google Scholar] [CrossRef] [PubMed]
- Therrell, B.L., Jr.; Lloyd-Puryear, M.A.; Eckman, J.R.; Mann, M.Y. Newborn screening for sickle cell diseases in the United States: A review of data spanning 2 decades. Semin. Perinatol. 2015, 39, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Robitaille, N.; Delvin, E.E.; Hume, H.A. Newborn screening for sickle cell disease: A 1988-2003 Quebec experience. Paediatr. Child Health 2006, 11, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Potter, B.K.; Khangura, M.; Hawken, S.; Hawken, J. Epidemiology and health system impact of hemoglobinopathy: Results from Newborn Screening Ontario. 2015 Canadian Newborn and Child Screening Symposium, Ottawa, ON, Canada, 30 April–1 May 2015. [Google Scholar]
- Benson, J.M.; Therrell, B.L., Jr. History and current status of newborn screening for hemoglobinopathies. Semin. Perinatol. 2010, 34, 134–144. [Google Scholar] [CrossRef] [PubMed]
- National Sickle Cell Anemia Control Act; US Congress: Washington, DC, USA, 1972; Volume 86, p. 136.
- Garrick, M.D.; Dembure, P.; Guthrie, R. Sickle-cell anemia and other hemoglobinopathies. Procedures and strategy for screening employing spots of blood on filter paper as specimens. N. Engl. J. Med. 1973, 288, 1265–1268. [Google Scholar] [CrossRef] [PubMed]
- Grover, R.; Shahidi, S.; Fisher, B.; Goldberg, D.; Wethers, D. Current sickle cell screening program for newborns in New York City, 1979–1980. Am. J. Public Health 1983, 73, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.M. Hemoglobinopathy screening: Approaches to diagnosis, education and counseling. Am. J. Public Health 1974, 64, 799–804. [Google Scholar] [CrossRef] [PubMed]
- Laboratories, A.O.P.H. Hemoglobinopathies: Current Practices for Screening, Confirmation and Follow up; Centers for Disease Control: Atlanta, GA, USA, 2015.
- Selekman, J. Update: New guidelines for the treatment of infants with sickle cell disease. Agency for Health Care Policy and Research. Pediatr. Nurs. 1993, 19, 600–605. [Google Scholar] [PubMed]
- Consensus conference. Newborn screening for sickle cell disease and other hemoglobinopathies. JAMA 1987, 258, 1205–1209. [Google Scholar] [CrossRef]
- Smith, J.A.; Kinney, T.R. Sickle cell disease: Screening and management in newborns and infants. Agency for Health Care Policy and Research. Am. Fam. Phys. 1993, 48, 95–102. [Google Scholar]
- Newborn screening: Toward a uniform screening panel and system-executive summary. Pediatrics 2006, 117, S296–S307. [CrossRef] [PubMed]
- Calonge, N.; Green, N.S.; Rinaldo, P.; Lloyd-Puryear, M.; Dougherty, D.; Boyle, C.; Watson, M.; Trotter, T.; Terry, S.F.; Howell, R.R.; et al. Committee report: Method for evaluating conditions nominated for population-based screening of newborns and children. Genet. Med. 2010, 12, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Knapp, A.A.; Metterville, D.R.; Kemper, A.R.; Perrin, J.M. Newborn Screening for Hemoglobin H Disease: A Summary of the Evidence and Advisory Committee Decision; Health Resources and Services Administration: Rockville, MD, USA, 2010. [Google Scholar]
- Hiller, E.H.; Landenburger, G.; Natowicz, M.R. Public participation in medical policy-making and the status of consumer autonomy: The example of newborn-screening programs in the United States. Am. J. Public Health 1997, 87, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Newborn Blood Spot Screening for Galactosemia, Tyrosinemia Type 1, Homocystinuria, Sickle Cell Anemia, Sickle Cell/Beta-Thallassemia, Sickle Cell/Hemoglobin C Disease and Severe Combined Immunodeficiency: Costs and Cost Analysis; 2016-03; Institute of Health Economics: Edmonton, AB, Canada, 2016.
- Therrell, B.L.; Adams, J. Newborn screening in North America. J. Inherit. Metab. Dis. 2007, 30, 447–465. [Google Scholar] [CrossRef] [PubMed]
- Therrell, B.L.; Hannon, W.H. National evaluation of US newborn screening system components. Ment. Retard. Dev. Disabil. Res. Rev. 2006, 12, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Campbell, M.; Henthorn, J.S.; Davies, S.C. Evaluation of cation-exchange HPLC compared with isoelectric focusing for neonatal hemoglobinopathy screening. Clin. Chem. 1999, 45, 969–975. [Google Scholar] [PubMed]
- CLSI Document NBS01-A6. Blood Collection on Filter Paper for Newborn Screening Programs, 6th ed.; Approved Standard; CLSI: Wayne, PA, USA, 2013. [Google Scholar]
- Adam, B.W.; Haynes, C.A.; Chafin, D.L.; De Jesus, V.R. Stabilities of intact hemoglobin molecules and hemoglobin peptides in dried blood samples. Clin. Chim. Acta 2014, 429, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huisman, T.H. Separation of hemoglobins and hemoglobin chains by high-performance liquid chromatography. J. Chromatogr. 1987, 418, 277–304. [Google Scholar] [CrossRef]
- Boemer, F.; Ketelslegers, O.; Minon, J.M.; Bours, V.; Schoos, R. Newborn screening for sickle cell disease using tandem mass spectrometry. Clin. Chem. 2008, 54, 2036–2041. [Google Scholar] [CrossRef] [PubMed]
- Newborn Screening Ontario. Regional Treatment Centres. Available online: https://www.newbornscreening.on.ca/en/health-care-providers/regional-treatment-centres (accessed on 28 September 2018).
- Feuchtbaum, L.; Dowray, S.; Lorey, F. The context and approach for the California newborn screening short- and long-term follow-up data system: Preliminary findings. Genet. Med. 2010, 12 (Suppl. 12), S242–S250. [Google Scholar] [CrossRef]
- Kavanagh, P.L.; Wang, C.J.; Therrell, B.L.; Sprinz, P.G.; Bauchner, H. Communication of positive newborn screening results for sickle cell disease and sickle cell trait: Variation across states. Am. J. Med. Genet. Part C Semin. Med. Genet. 2008, 148C, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Hinton, C.F.; Feuchtbaum, L.; Kus, C.A.; Kemper, A.R.; Berry, S.A.; Levy-Fisch, J.; Luedtke, J.; Kaye, C.; Boyle, C.A. What questions should newborn screening long-term follow-up be able to answer? A statement of the US Secretary for Health and Human Services’ Advisory Committee on Heritable Disorders in Newborns and Children. Genet. Med. 2011, 13, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Kemper, A.R.; Boyle, C.A.; Aceves, J.; Dougherty, D.; Figge, J.; Fisch, J.L.; Hinman, A.R.; Greene, C.L.; Kus, C.A.; Miller, J.; et al. Long-term follow-up after diagnosis resulting from newborn screening: Statement of the US Secretary of Health and Human Services’ Advisory Committee on Heritable Disorders and Genetic Diseases in Newborns and Children. Genet. Med. Off. J. Am. Coll. Med. Genet. 2008, 10, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease. Prevention, Good laboratory practices for biochemical genetic testing and newborn screening for inherited metabolic disorders. MMWR. Recomm. Rep. Morb. Mortal. Wkly. Rep. Recomm. Rep. 2012, 61, 1–44. [Google Scholar]
- Sontag, M.K.; Sarkar, D.; Comeau, A.M.; Hassell, K.; Botto, L.D.; Parad, R.; Rose, S.R.; Wintergerst, K.A.; Smith-Whitley, K.; Singh, S.; et al. Case definitions for conditions identified by newborn screening public health surveillance. Int. J. Neonatal Screen. 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Paulukonis, S.T.; Harris, W.T.; Coates, T.D.; Neumayr, L.; Treadwell, M.; Vichinsky, E.; Feuchtbaum, L.B. Population based surveillance in sickle cell disease: Methods, findings and implications from the California registry and surveillance system in hemoglobinopathies project (RuSH). Pediatr. Blood Cancer 2014, 61, 2271–2276. [Google Scholar] [CrossRef] [PubMed]
- Sox, H.C. Resolving the tension between population health and individual health care. JAMA 2013, 310, 1933–1934. [Google Scholar] [CrossRef] [PubMed]
- Wharton, M.; Chorba, T.L.; Vogt, R.L.; Morse, D.L.; Buehler, J.W. Case Definitions for Public Health Surveillance; U.S. Department of Health and Human Services: Atlanta, GA, USA, 1990; pp. 1–43.
- Wang, Y.; Caggana, M.; Sango-Jordan, M.; Sun, M.; Druschel, C.M. Long-term follow-up of children with confirmed newborn screening disorders using record linkage. Genet. Med. 2011, 13, 881–886. [Google Scholar] [CrossRef] [PubMed]
- DiMartino, L.D.; Baumann, A.A.; Hsu, L.L.; Kanter, J.; Gordeuk, V.R.; Glassberg, J.; Treadwell, M.J.; Melvin, C.L.; Telfair, J.; Klesges, L.M.; et al. The sickle cell disease implementation consortium: Translating evidence-based guidelines into practice for sickle cell disease. Am. J. Hematol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hassell, T.; Hennis, A. Chronic Disease Challenges in the Caribbean. Glob. Heart 2016, 11, 437–438. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
El-Haj, N.; Hoppe, C.C. Newborn Screening for SCD in the USA and Canada. Int. J. Neonatal Screen. 2018, 4, 36. https://doi.org/10.3390/ijns4040036
El-Haj N, Hoppe CC. Newborn Screening for SCD in the USA and Canada. International Journal of Neonatal Screening. 2018; 4(4):36. https://doi.org/10.3390/ijns4040036
Chicago/Turabian StyleEl-Haj, Nura, and Carolyn C. Hoppe. 2018. "Newborn Screening for SCD in the USA and Canada" International Journal of Neonatal Screening 4, no. 4: 36. https://doi.org/10.3390/ijns4040036
APA StyleEl-Haj, N., & Hoppe, C. C. (2018). Newborn Screening for SCD in the USA and Canada. International Journal of Neonatal Screening, 4(4), 36. https://doi.org/10.3390/ijns4040036