

Animal Models and Their Role in Imaging-Assisted Co-Clinical Trials


 , ,
, ,  , ,
, ,  , , , ,
, , , ,  , , and add
Show full author list
, , and add
Show full author list
Abstract
1. Introduction
2. Co-Clinical Trials
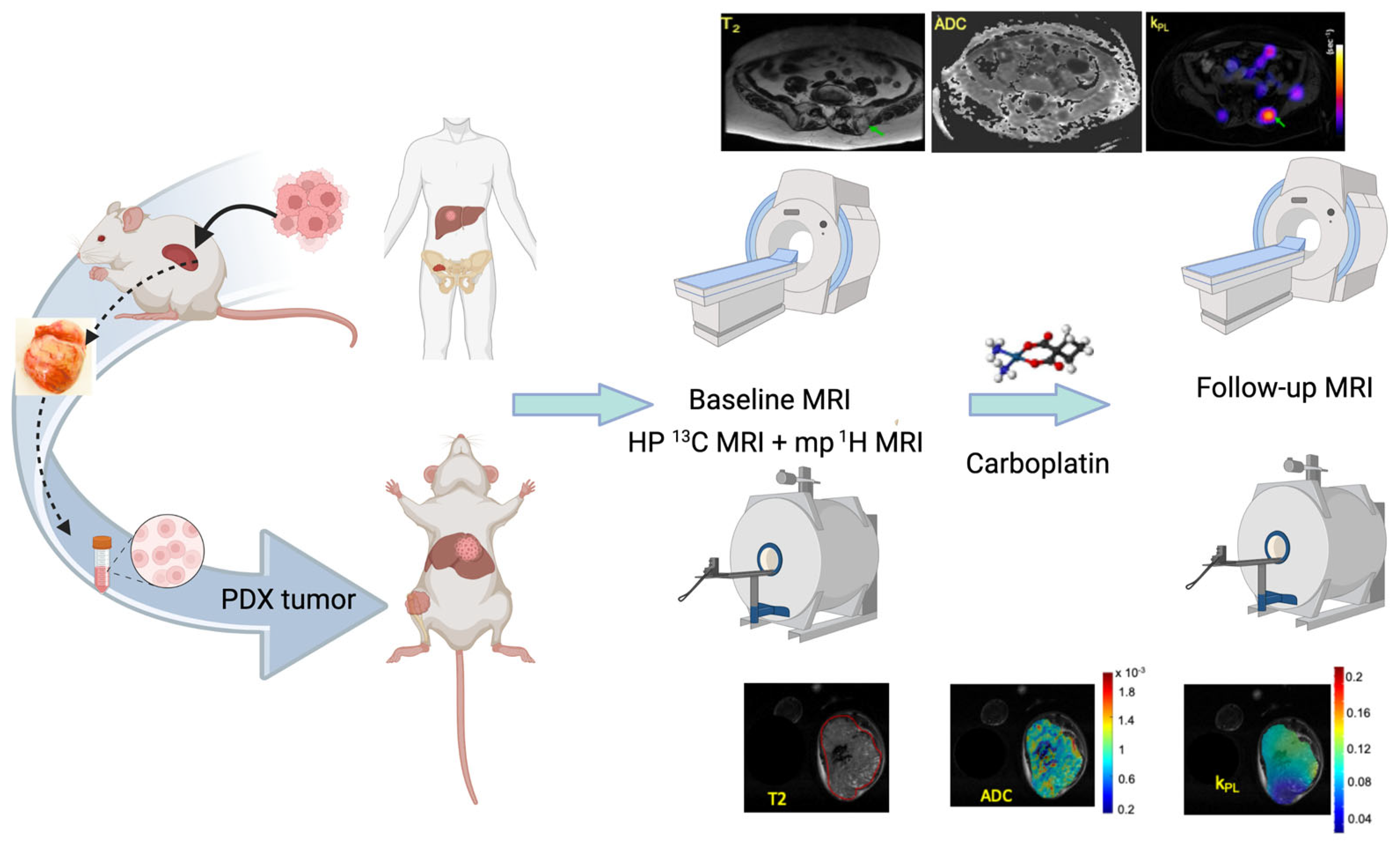
2.1. The UCSF Co-Clinical Quantitative Imaging of Small Cell Neuroendocrine Prostate Cancer Using Hyperpolarized 13C MRI
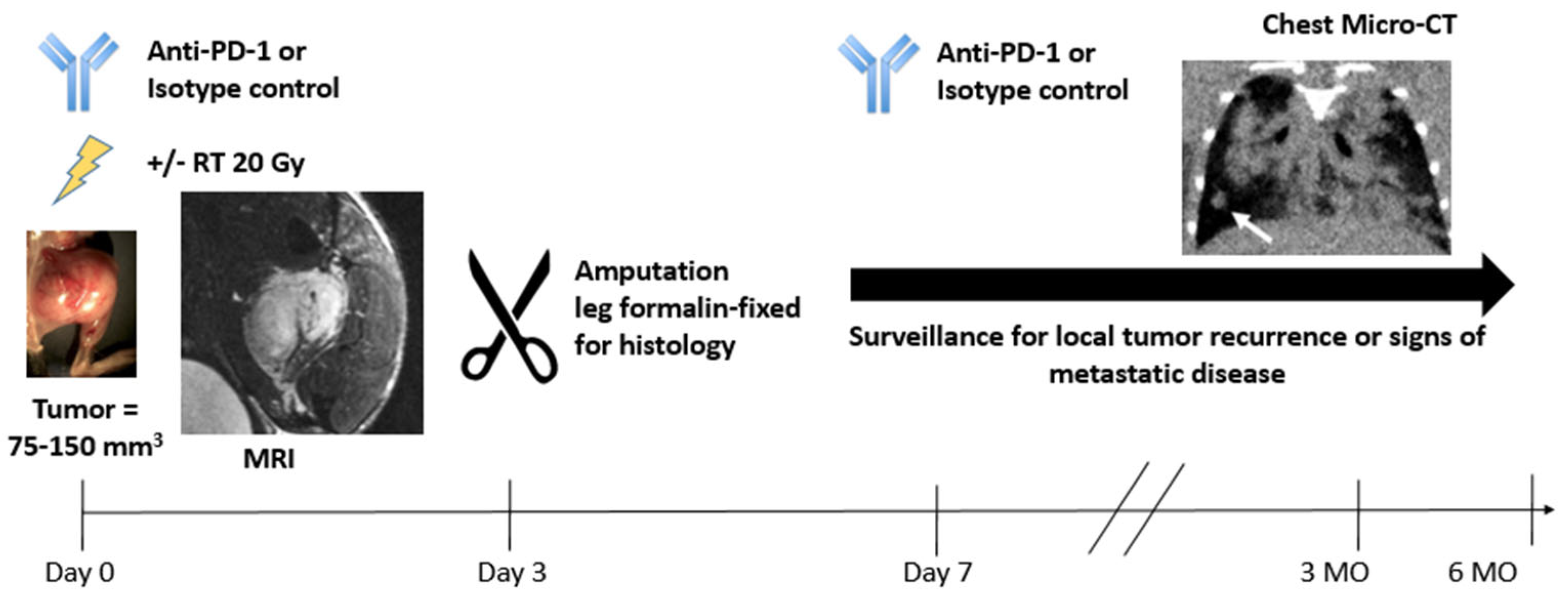
2.2. The Duke Preclinical Research Resources for Quantitative Imaging Biomarkers
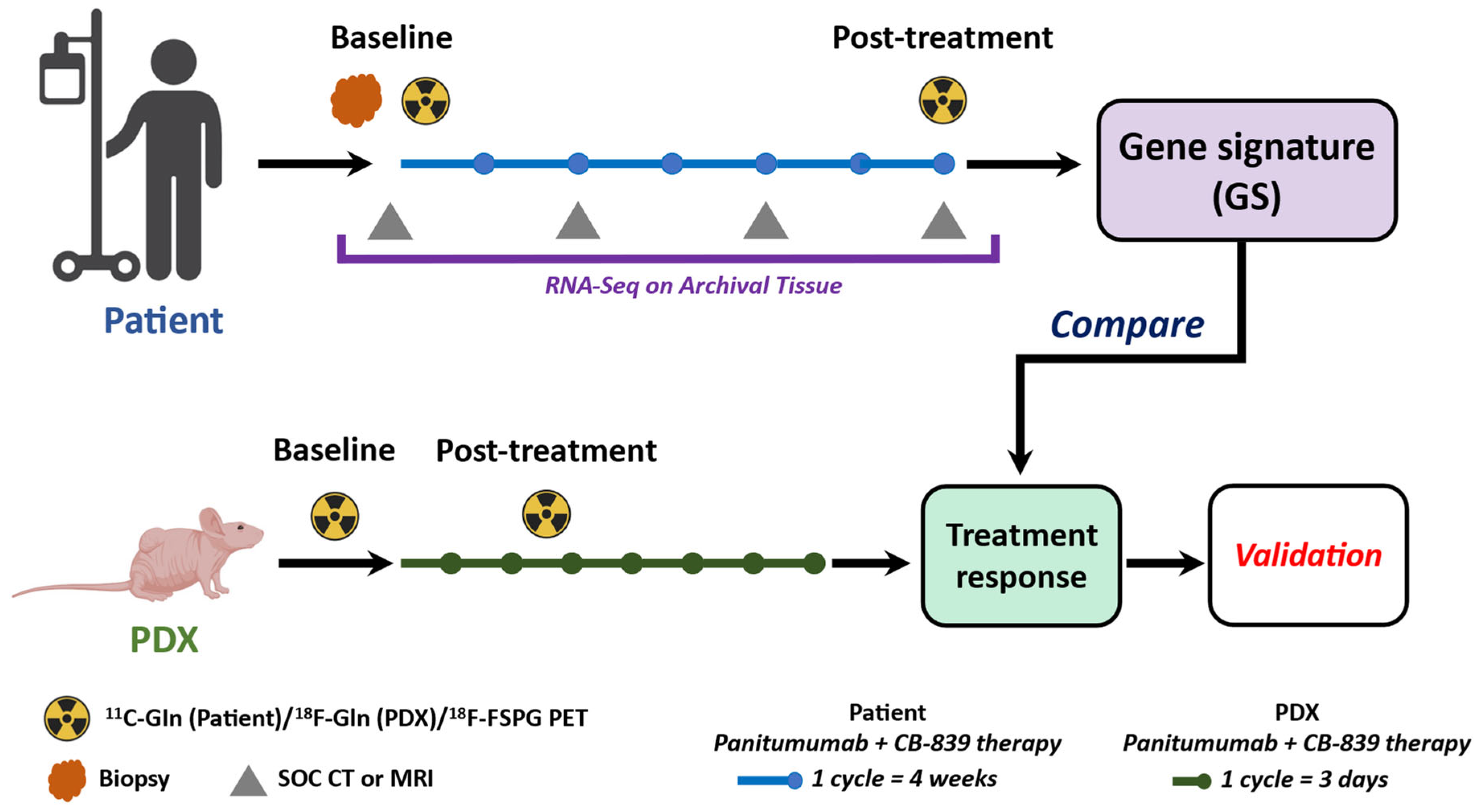
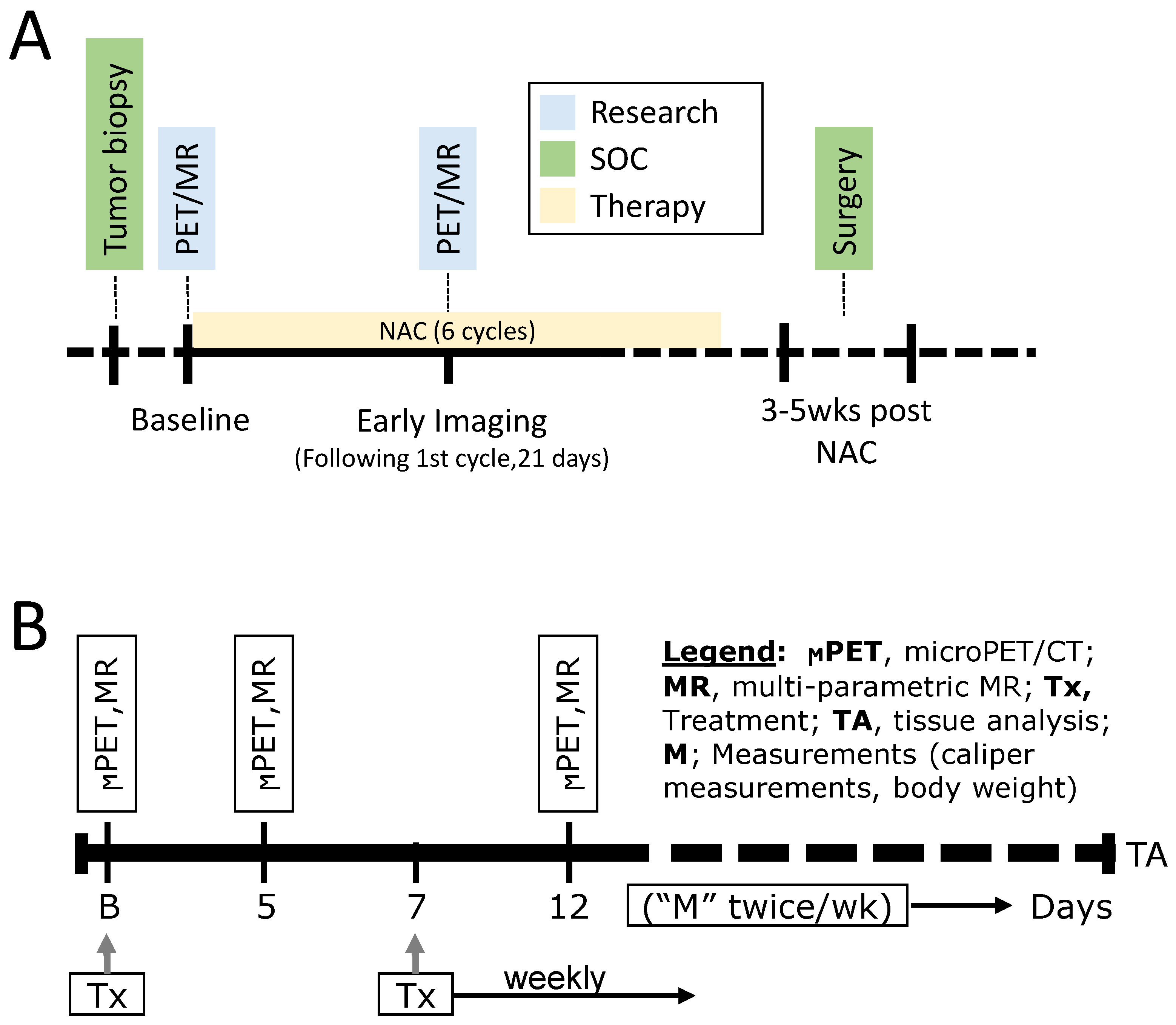
2.3. The MDACC PET Imaging Resource to Enhance Delivery of Individualized Cancer Therapeutics (PREDICT) for Wild-Type KRAS Colorectal Cancer
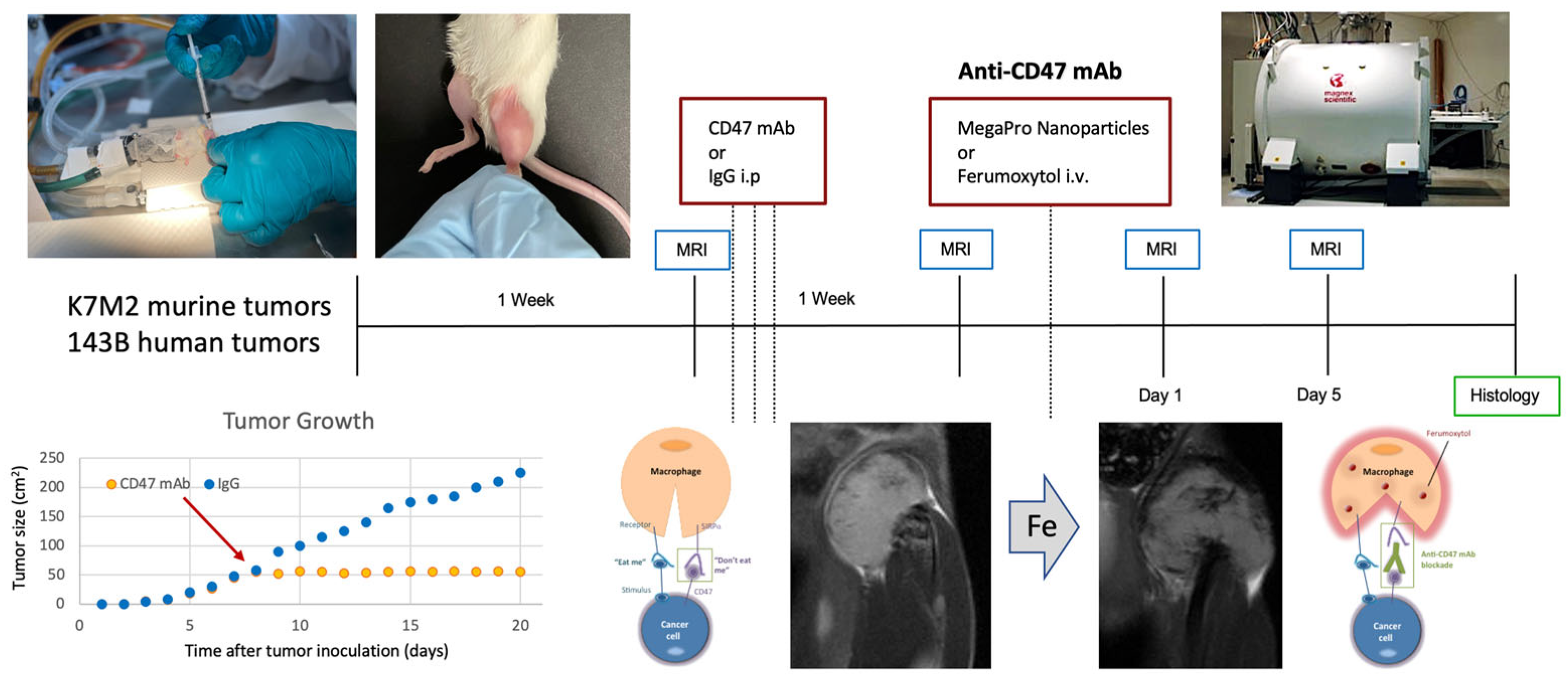
2.4. Stanford University Co-Clinical Research for Imaging Tumor-Associated Macrophages
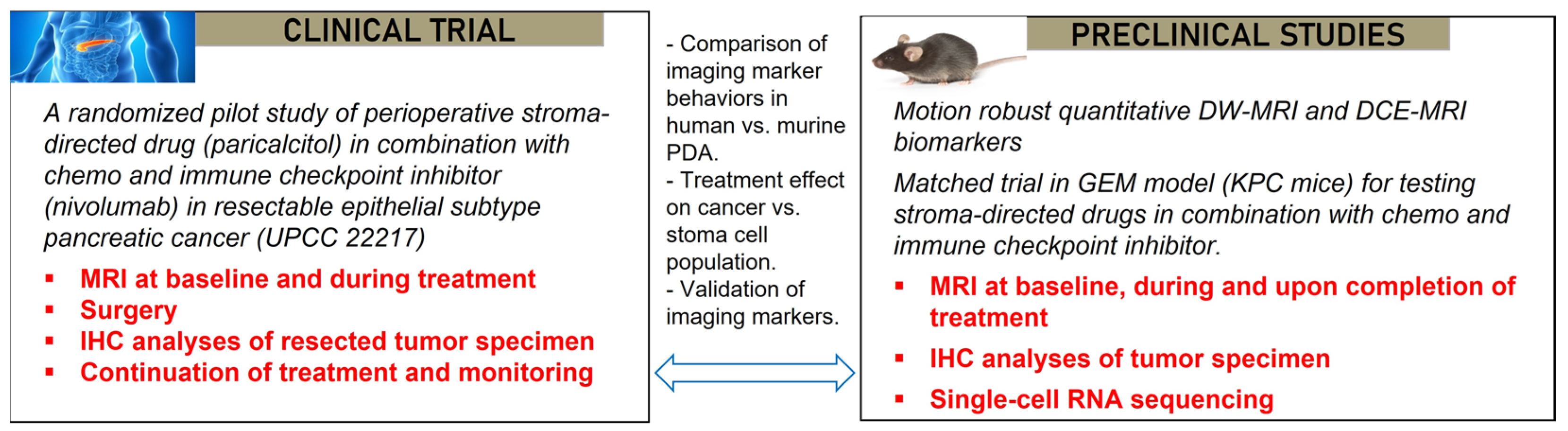
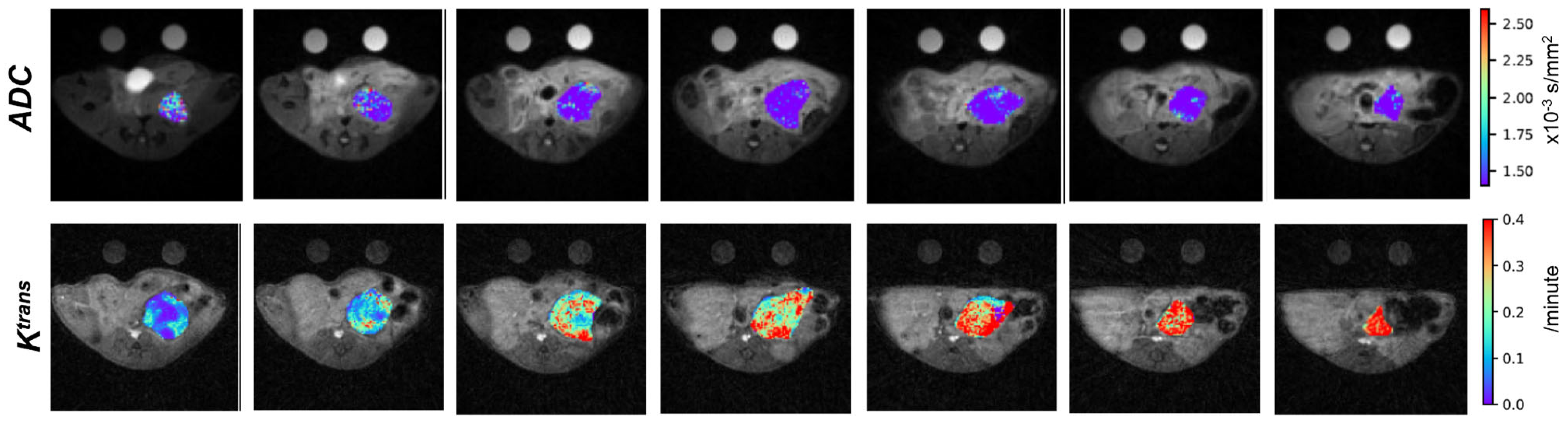
2.5. Penn Quantitative MRI Resource for Pancreatic Cancer
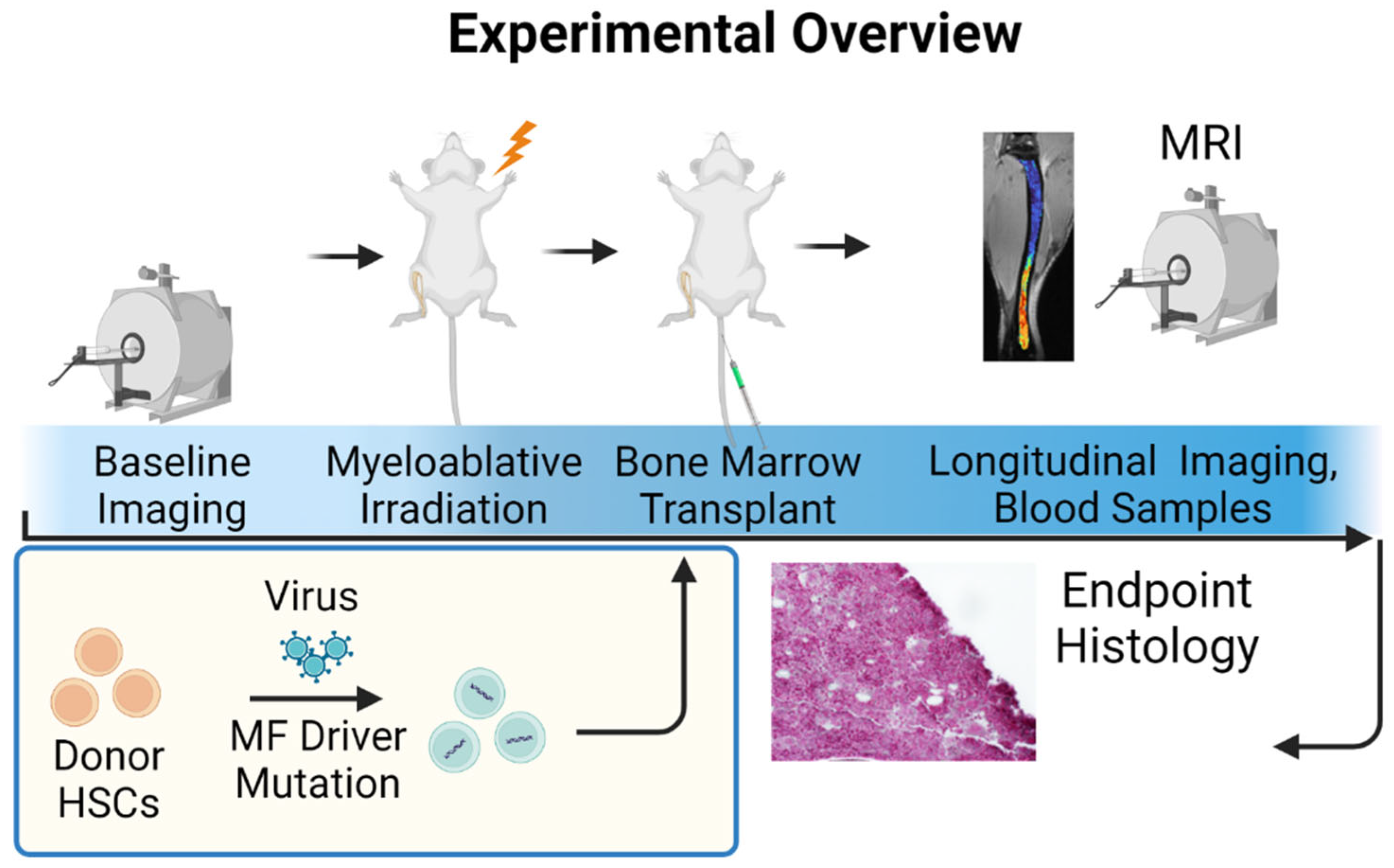
2.6. University of Michigan Quantitative Bone Marrow MRI in Myelofibrosis
2.7. Washington University St. Louis Co-Clinical Quantitative Imaging of Breast Cancer to Predict Response to Therapy
2.8. Baylor/UT Austin/Stanford University Integration of Omics and Quantitative Imaging Data in Co-Clinical Trials to Predict Treatment Response in Triple-Negative Breast Cancer
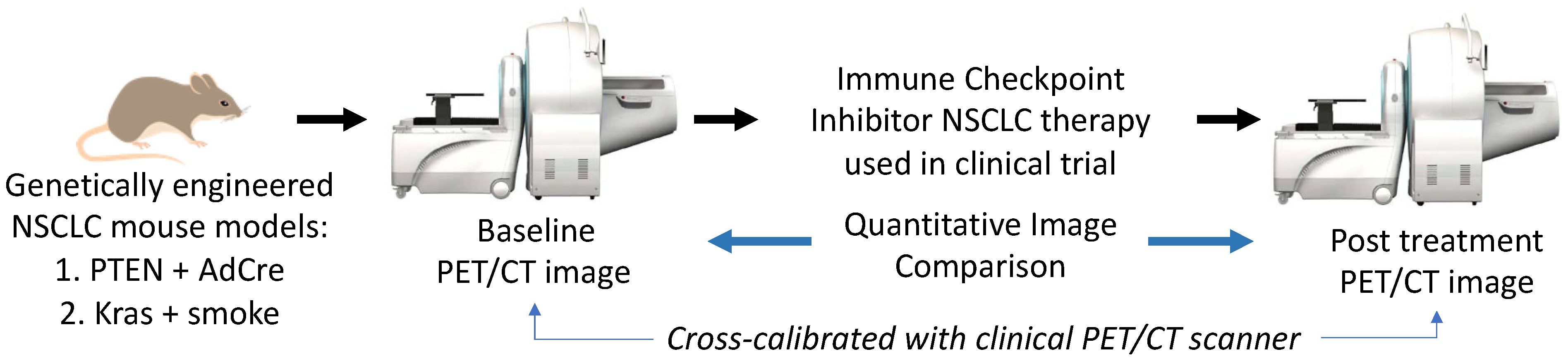
2.9. University of Washington/Fred Hutchinson Cancer Center Quantitative FDG PET Imaging of Non-Small Cell Lung Cancer in a Co-Clinical Immune Checkpoint Inhibitor Therapy Study
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Shoghi, K.I.; Badea, C.T.; Blocker, S.J.; Chenevert, T.L.; Laforest, R.; Lewis, M.T.; Luker, G.D.; Manning, H.C.; Marcus, D.S.; Mowery, Y.M.; et al. Co-Clinical Imaging Resource Program (CIRP): Bridging the Translational Divide to Advance Precision Medicine. Tomography 2020, 6, 273–287. [Google Scholar] [CrossRef]
- Zhou, J.; Ding, J.; Qi, J. Comparison of Typical Prostate Adenocarcinoma and Rare Histological Variant Prostate Cancer Showed Different Characteristics and Prognosis: A Surveillance, Epidemiology, and End Results Database Analysis. Eur. Urol. 2022, 82, 152–155. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Vessella, R.L.; Morrissey, C.; Brown, L.G.; Coleman, I.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Lam, H.-M.; et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease And Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017, 77, 654–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Wang, Y.; Lin, D.; Wang, Y. Patient-Derived Xenograft Models of Neuroendocrine Prostate Cancer. Cancer Lett. 2022, 525, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Klumpen, H.-J.; Gurney, H. Dose Calculation of Anticancer Drugs. Expert. Opin. Drug. Metab. Toxicol. 2008, 4, 1307–1319. [Google Scholar] [CrossRef]
- van Hennik, M.B.; van der Vijgh, W.J.; Klein, I.; Elferink, F.; Vermorken, J.B.; Winograd, B.; Pinedo, H.M. Comparative Pharmacokinetics of Cisplatin and Three Analogues in Mice and Humans. Cancer Res. 1987, 47, 6297–6301. [Google Scholar] [PubMed]
- Avanzini, S.; Antal, T. Cancer Recurrence Times from a Branching Process Model. PLoS Comput. Biol. 2019, 15, e1007423. [Google Scholar] [CrossRef]
- Singh, T.; Neal, A.; Dibernardo, G.; Raheseparian, N.; Moatamed, N.A.; Memarzadeh, S. Efficacy of Birinapant in Combination with Carboplatin in Targeting Platinum-resistant Epithelial Ovarian Cancers. Int. J. Oncol. 2022, 60, 35. [Google Scholar] [CrossRef]
- Godebu, E.; Muldong, M.; Strasner, A.; Wu, C.N.; Park, S.C.; Woo, J.R.; Ma, W.; Liss, M.A.; Hirata, T.; Raheem, O.; et al. PCSD1, a New Patient-Derived Model of Bone Metastatic Prostate Cancer, Is Castrate-Resistant in the Bone-Niche. J. Transl. Med. 2014, 12, 275. [Google Scholar] [CrossRef]
- Morrissey, C.; Dowell, A.; Koreckij, T.D.; Nguyen, H.; Lakely, B.; Fanslow, W.C.; True, L.D.; Corey, E.; Vessella, R.L. Inhibition of Angiopoietin-2 in LuCaP 23.1 Prostate Cancer Tumors Decreases Tumor Growth and Viability. Prostate 2010, 70, 1799–1808. [Google Scholar] [CrossRef]
- Halabi, S.; Kelly, W.K.; Ma, H.; Zhou, H.; Solomon, N.C.; Fizazi, K.; Tangen, C.M.; Rosenthal, M.; Petrylak, D.P.; Hussain, M.; et al. Meta-Analysis Evaluating the Impact of Site of Metastasis on Overall Survival in Men With Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2016, 34, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Baciarello, G.; Özgüroğlu, M.; Mundle, S.; Leitz, G.; Richarz, U.; Hu, P.; Feyerabend, S.; Matsubara, N.; Chi, K.N.; Fizazi, K. Impact of Abiraterone Acetate plus Prednisone in Patients with Castration-Sensitive Prostate Cancer and Visceral Metastases over Four Years of Follow-up: A Post-Hoc Exploratory Analysis of the LATITUDE Study. Eur. J. Cancer 2022, 162, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Saif, A.; Verbus, E.A.; Sarvestani, A.L.; Teke, M.E.; Lambdin, J.; Hernandez, J.M.; Kirsch, D.G. A Randomized Trial of Pembrolizumab & Radiotherapy Versus Radiotherapy in High-Risk Soft Tissue Sarcoma of the Extremity (SU2C-SARC032). Ann. Surg. Oncol. 2023, 30, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-L.; Mowery, Y.M.; Daniel, A.R.; Zhang, D.; Sibley, A.B.; Delaney, J.R.; Wisdom, A.J.; Qin, X.; Wang, X.; Caraballo, I.; et al. Mutational Landscape in Genetically Engineered, Carcinogen-Induced, and Radiation-Induced Mouse Sarcoma. JCI Insight 2019, 4, e128698. [Google Scholar] [CrossRef] [PubMed]
- Abeshouse, A.; Adebamowo, C.; Adebamowo, S.N.; Akbani, R.; Akeredolu, T.; Ally, A.; Anderson, M.L.; Anur, P.; Appelbaum, E.L.; Armenia, J.; et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017, 171, 950–965.e28. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, A.J.; Mowery, Y.M.; Hong, C.S.; Himes, J.E.; Nabet, B.Y.; Qin, X.; Zhang, D.; Chen, L.; Fradin, H.; Patel, R.; et al. Single Cell Analysis Reveals Distinct Immune Landscapes in Transplant and Primary Sarcomas That Determine Response or Resistance to Immunotherapy. Nat. Commun. 2020, 11, 6410. [Google Scholar] [CrossRef]
- Blocker, S.J.; Mowery, Y.M.; Holbrook, M.D.; Qi, Y.; Kirsch, D.G.; Johnson, G.A.; Badea, C.T. Bridging the Translational Gap: Implementation of Multimodal Small Animal Imaging Strategies for Tumor Burden Assessment in a Co-Clinical Trial. PLoS ONE 2019, 14, e0207555. [Google Scholar] [CrossRef]
- Patel, R.; Mowery, Y.M.; Qi, Y.; Bassil, A.M.; Holbrook, M.; Xu, E.S.; Hong, C.S.; Himes, J.E.; Williams, N.T.; Everitt, J.; et al. Neoadjuvant Radiation Therapy and Surgery Improves Metastasis-Free Survival over Surgery Alone in a Primary Mouse Model of Soft Tissue Sarcoma. Mol. Cancer Ther. 2023, 22, 112–122. [Google Scholar] [CrossRef]
- Janakiraman, M.; Vakiani, E.; Zeng, Z.; Pratilas, C.A.; Taylor, B.S.; Chitale, D.; Halilovic, E.; Wilson, M.; Huberman, K.; Ricarte Filho, J.C.; et al. Genomic and Biological Characterization of Exon 4 KRAS Mutations in Human Cancer. Cancer Res. 2010, 70, 5901–5911. [Google Scholar] [CrossRef]
- Wilson, P.M.; Labonte, M.J.; Lenz, H.-J. Molecular Markers in the Treatment of Metastatic Colorectal Cancer. Cancer J. 2010, 16, 262–272. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular Origins of Cancer: Molecular Basis of Colorectal Cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-Type BRAF Is Required for Response to Panitumumab or Cetuximab in Metastatic Colorectal Cancer. J. Clin. Oncol. 2008, 26, 5705–5712. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Köhne, C.-H.; Hitre, E.; Zaluski, J.; Chang Chien, C.-R.; Makhson, A.; D’Haens, G.; Pintér, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and Chemotherapy as Initial Treatment for Metastatic Colorectal Cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef]
- Rhoads, J.M.; Argenzio, R.A.; Chen, W.; Graves, L.M.; Licato, L.L.; Blikslager, A.T.; Smith, J.; Gatzy, J.; Brenner, D.A. Glutamine Metabolism Stimulates Intestinal Cell MAPKs by a CAMP-Inhibitable, Raf-Independent Mechanism. Gastroenterology 2000, 118, 90–100. [Google Scholar] [CrossRef]
- Cohen, A.S.; Geng, L.; Zhao, P.; Fu, A.; Schulte, M.L.; Graves-Deal, R.; Washington, M.K.; Berlin, J.; Coffey, R.J.; Manning, H.C. Combined Blockade of EGFR and Glutamine Metabolism in Preclinical Models of Colorectal Cancer. Transl. Oncol. 2020, 13, 100828. [Google Scholar] [CrossRef]
- Schulte, M.L.; Hight, M.R.; Ayers, G.D.; Liu, Q.; Shyr, Y.; Washington, M.K.; Manning, H.C. Non-Invasive Glutamine PET Reflects Pharmacological Inhibition of BRAFV600E In Vivo. Mol. Imaging Biol. 2017, 19, 421–428. [Google Scholar] [CrossRef]
- Cohen, A.S.; Grudzinski, J.; Smith, G.T.; Peterson, T.E.; Whisenant, J.G.; Hickman, T.L.; Ciombor, K.K.; Cardin, D.; Eng, C.; Goff, L.W.; et al. First-in-Human PET Imaging and Estimated Radiation Dosimetry of l-[5-11C]-Glutamine in Patients with Metastatic Colorectal Cancer. J. Nucl. Med. 2022, 63, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Kavanaugh, G.; Williams, J.; Morris, A.S.; Nickels, M.L.; Walker, R.; Koglin, N.; Stephens, A.W.; Washington, M.K.; Geevarghese, S.K.; Liu, Q.; et al. Utility of [18F]FSPG PET to Image Hepatocellular Carcinoma: First Clinical Evaluation in a US Population. Mol. Imaging Biol. 2016, 18, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.I.; Demo, S.D.; Dennison, J.B.; Chen, L.; Chernov-Rogan, T.; Goyal, B.; Janes, J.R.; Laidig, G.J.; Lewis, E.R.; Li, J.; et al. Antitumor Activity of the Glutaminase Inhibitor CB-839 in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2014, 13, 890–901. [Google Scholar] [CrossRef] [PubMed]
- McKinley, E.T.; Smith, R.A.; Tanksley, J.P.; Washington, M.K.; Walker, R.; Coffey, R.J.; Manning, H.C. [18F]FLT-PET to Predict Pharmacodynamic and Clinical Response to Cetuximab Therapy in Ménétrier’s Disease. Ann. Nucl. Med. 2012, 26, 757–763. [Google Scholar] [CrossRef]
- McKinley, E.T.; Zhao, P.; Coffey, R.J.; Washington, M.K.; Manning, H.C. 3′-Deoxy-3′-[18F]-Fluorothymidine PET Imaging Reflects PI3K-MTOR-Mediated pro-Survival Response to Targeted Therapy in Colorectal Cancer. PLoS ONE 2014, 9, e108193. [Google Scholar] [CrossRef]
- Bernstein, M.L.; Devidas, M.; Lafreniere, D.; Souid, A.-K.; Meyers, P.A.; Gebhardt, M.; Stine, K.; Nicholas, R.; Perlman, E.J.; Dubowy, R.; et al. Intensive Therapy with Growth Factor Support for Patients with Ewing Tumor Metastatic at Diagnosis: Pediatric Oncology Group/Children’s Cancer Group Phase II Study 9457--a Report from the Children’s Oncology Group. J. Clin. Oncol. 2006, 24, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Ladenstein, R.; Pötschger, U.; Le Deley, M.C.; Whelan, J.; Paulussen, M.; Oberlin, O.; van den Berg, H.; Dirksen, U.; Hjorth, L.; Michon, J.; et al. Primary Disseminated Multifocal Ewing Sarcoma: Results of the Euro-EWING 99 Trial. J. Clin. Oncol. 2010, 28, 3284–3291. [Google Scholar] [CrossRef]
- Miser, J.S.; Krailo, M.D.; Tarbell, N.J.; Link, M.P.; Fryer, C.J.H.; Pritchard, D.J.; Gebhardt, M.C.; Dickman, P.S.; Perlman, E.J.; Meyers, P.A.; et al. Treatment of Metastatic Ewing’s Sarcoma or Primitive Neuroectodermal Tumor of Bone: Evaluation of Combination Ifosfamide and Etoposide—A Children’s Cancer Group and Pediatric Oncology Group Study. J. Clin. Oncol. 2004, 22, 2873–2876. [Google Scholar] [CrossRef] [PubMed]
- Bakhshi, S.; Radhakrishnan, V. Prognostic Markers in Osteosarcoma. Expert Rev. Anticancer Ther. 2010, 10, 271–287. [Google Scholar] [CrossRef] [PubMed]
- Mialou, V.; Philip, T.; Kalifa, C.; Perol, D.; Gentet, J.-C.; Marec-Berard, P.; Pacquement, H.; Chastagner, P.; Defaschelles, A.-S.; Hartmann, O. Metastatic Osteosarcoma at Diagnosis: Prognostic Factors and Long-Term Outcome--the French Pediatric Experience. Cancer 2005, 104, 1100–1109. [Google Scholar] [CrossRef]
- Beird, H.C.; Bielack, S.S.; Flanagan, A.M.; Gill, J.; Heymann, D.; Janeway, K.A.; Livingston, J.A.; Roberts, R.D.; Strauss, S.J.; Gorlick, R. Osteosarcoma. Nat. Rev. Dis. Primers 2022, 8, 77. [Google Scholar] [CrossRef]
- Thompson, P.A.; Chintagumpala, M. Targeted Therapy in Bone and Soft Tissue Sarcoma in Children and Adolescents. Curr. Oncol. Rep. 2012, 14, 197–205. [Google Scholar] [CrossRef]
- Zhao, X.W.; van Beek, E.M.; Schornagel, K.; Van der Maaden, H.; Van Houdt, M.; Otten, M.A.; Finetti, P.; Van Egmond, M.; Matozaki, T.; Kraal, G.; et al. CD47-Signal Regulatory Protein-α (SIRPα) Interactions Form a Barrier for Antibody-Mediated Tumor Cell Destruction. Proc. Natl. Acad. Sci. USA 2011, 108, 18342–18347. [Google Scholar] [CrossRef]
- Chao, M.P.; Weissman, I.L.; Majeti, R. The CD47-SIRPα Pathway in Cancer Immune Evasion and Potential Therapeutic Implications. Curr. Opin. Immunol. 2012, 24, 225–232. [Google Scholar] [CrossRef]
- Majeti, R.; Chao, M.P.; Alizadeh, A.A.; Pang, W.W.; Jaiswal, S.; Gibbs, K.D.; van Rooijen, N.; Weissman, I.L. CD47 Is an Adverse Prognostic Factor and Therapeutic Antibody Target on Human Acute Myeloid Leukemia Stem Cells. Cell 2009, 138, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Myklebust, J.H.; Varghese, B.; Gill, S.; Jan, M.; Cha, A.C.; Chan, C.K.; Tan, B.T.; et al. Anti-CD47 Antibody Synergizes with Rituximab to Promote Phagocytosis and Eradicate Non-Hodgkin Lymphoma. Cell 2010, 142, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Edris, B.; Weiskopf, K.; Volkmer, A.K.; Volkmer, J.-P.; Willingham, S.B.; Contreras-Trujillo, H.; Liu, J.; Majeti, R.; West, R.B.; Fletcher, J.A.; et al. Antibody Therapy Targeting the CD47 Protein Is Effective in a Model of Aggressive Metastatic Leiomyosarcoma. Proc. Natl. Acad. Sci. USA 2012, 109, 6656–6661. [Google Scholar] [CrossRef]
- Chao, M.P.; Jaiswal, S.; Weissman-Tsukamoto, R.; Alizadeh, A.A.; Gentles, A.J.; Volkmer, J.; Weiskopf, K.; Willingham, S.B.; Raveh, T.; Park, C.Y.; et al. Calreticulin Is the Dominant Pro-Phagocytic Signal on Multiple Human Cancers and Is Counterbalanced by CD47. Sci. Transl. Med. 2010, 2, 63ra94. [Google Scholar] [CrossRef]
- Chao, M.P.; Alizadeh, A.A.; Tang, C.; Jan, M.; Weissman-Tsukamoto, R.; Zhao, F.; Park, C.Y.; Weissman, I.L.; Majeti, R. Therapeutic Antibody Targeting of CD47 Eliminates Human Acute Lymphoblastic Leukemia. Cancer Res. 2011, 71, 1374–1384. [Google Scholar] [CrossRef]
- Daldrup-Link, H.E.; Golovko, D.; Ruffell, B.; Denardo, D.G.; Castaneda, R.; Ansari, C.; Rao, J.; Tikhomirov, G.A.; Wendland, M.F.; Corot, C.; et al. MRI of Tumor-Associated Macrophages with Clinically Applicable Iron Oxide Nanoparticles. Clin. Cancer Res. 2011, 17, 5695–5704. [Google Scholar] [CrossRef]
- Daldrup-Link, H.; Coussens, L.M. MR Imaging of Tumor-Associated Macrophages. Oncoimmunology 2012, 1, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Aghighi, M.; Theruvath, A.J.; Pareek, A.; Pisani, L.L.; Alford, R.; Muehe, A.M.; Sethi, T.K.; Holdsworth, S.J.; Hazard, F.K.; Gratzinger, D.; et al. Magnetic Resonance Imaging of Tumor-Associated Macrophages: Clinical Translation. Clin. Cancer. Res. 2018, 24, 4110–4118. [Google Scholar] [CrossRef]
- Iv, M.; Samghabadi, P.; Holdsworth, S.; Gentles, A.; Rezaii, P.; Harsh, G.; Li, G.; Thomas, R.; Moseley, M.; Daldrup-Link, H.E.; et al. Quantification of Macrophages in High-Grade Gliomas by Using Ferumoxytol-Enhanced MRI: A Pilot Study. Radiology 2019, 290, 198–206. [Google Scholar] [CrossRef]
- Mohanty, S.; Yerneni, K.; Theruvath, J.L.; Graef, C.M.; Nejadnik, H.; Lenkov, O.; Pisani, L.; Rosenberg, J.; Mitra, S.; Cordero, A.S.; et al. Nanoparticle Enhanced MRI Can Monitor Macrophage Response to CD47 MAb Immunotherapy in Osteosarcoma. Cell Death. Dis. 2019, 10, 36. [Google Scholar] [CrossRef]
- Mohanty, S.; Aghighi, M.; Yerneni, K.; Theruvath, J.L.; Daldrup-Link, H.E. Improving the Efficacy of Osteosarcoma Therapy: Combining Drugs That Turn Cancer Cell “don’t Eat Me” Signals off and “Eat Me” Signals On. Mol. Oncol. 2019, 13, 2049–2061. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D Cooperate to Promote Chromosomal Instability and Widely Metastatic Pancreatic Ductal Adenocarcinoma in Mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and Invasive Ductal Pancreatic Cancer and Its Early Detection in the Mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Song, H.K.; Yang, H.; Castillo, V.; Chen, J.; Clendenin, C.; Rosen, M.; Zhou, R.; Pickup, S. Respiratory Motion Mitigation and Repeatability of Two Diffusion-Weighted MRI Methods Applied to a Murine Model of Spontaneous Pancreatic Cancer. Tomography 2021, 7, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Pickup, S.; Romanello, M.; Gupta, M.; Song, H.K.; Zhou, R. Dynamic Contrast-Enhanced MRI in the Abdomen of Mice with High Temporal and Spatial Resolution Using Stack-of-Stars Sampling and KWIC Reconstruction. Tomography 2022, 8, 2113–2128. [Google Scholar] [CrossRef]
- Romanello Joaquim, M.; Furth, E.E.; Fan, Y.; Song, H.K.; Pickup, S.; Cao, J.; Choi, H.; Gupta, M.; Cao, Q.; Shinohara, R.; et al. DWI Metrics Differentiating Benign Intraductal Papillary Mucinous Neoplasms from Invasive Pancreatic Cancer: A Study in GEM Models. Cancers 2022, 14, 4017. [Google Scholar] [CrossRef]
- Ross, B.D.; Jang, Y.; Welton, A.; Bonham, C.A.; Palagama, D.S.W.; Heist, K.; Boppisetti, J.; Imaduwage, K.P.; Robison, T.; King, L.R.; et al. A Lymphatic-Absorbed Multi-Targeted Kinase Inhibitor for Myelofibrosis Therapy. Nat. Commun. 2022, 13, 4730. [Google Scholar] [CrossRef]
- Robison, T.H.; Solipuram, M.; Heist, K.; Amouzandeh, G.; Lee, W.Y.; Humphries, B.A.; Buschhaus, J.M.; Bevoor, A.; Zhang, A.; Luker, K.E.; et al. Multiparametric MRI to Quantify Disease and Treatment Response in Mice with Myeloproliferative Neoplasms. JCI Insight 2022, 7, e161457. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Pietenpol, J.A. Identification and Use of Biomarkers in Treatment Strategies for Triple-Negative Breast Cancer Subtypes. J. Pathol. 2014, 232, 142–150. [Google Scholar] [CrossRef]
- Savaikar, M.A.; Whitehead, T.; Roy, S.; Strong, L.; Fettig, N.; Prmeau, T.; Luo, J.; Li, S.; Wahl, R.L.; Shoghi, K.I. Preclinical PERCIST and 25% of SUVmax Threshold: Precision Imaging of Response to Therapy in Co-Clinical 18F-FDG PET Imaging of Triple-Negative Breast Cancer Patient-Derived Tumor Xenografts. J. Nucl. Med. 2020, 61, 842–849. [Google Scholar] [CrossRef]
- Ge, X.; Quirk, J.D.; Engelbach, J.A.; Bretthorst, G.L.; Li, S.; Shoghi, K.I.; Garbow, J.R.; Ackerman, J.J.H. Test-Retest Performance of a 1-Hour Multiparametric MR Image Acquisition Pipeline With Orthotopic Triple-Negative Breast Cancer Patient-Derived Tumor Xenografts. Tomography 2019, 5, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Whitehead, T.D.; Quirk, J.D.; Salter, A.; Ademuyiwa, F.O.; Li, S.; An, H.; Shoghi, K.I. Optimal Co-Clinical Radiomics: Sensitivity of Radiomic Features to Tumour Volume, Image Noise and Resolution in Co-Clinical T1-Weighted and T2-Weighted Magnetic Resonance Imaging. EBioMedicine 2020, 59, 102963. [Google Scholar] [CrossRef] [PubMed]
- Dutta, K.; Roy, S.; Whitehead, T.D.; Luo, J.; Jha, A.K.; Li, S.; Quirk, J.D.; Shoghi, K.I. Deep Learning Segmentation of Triple-Negative Breast Cancer (TNBC) Patient Derived Tumor Xenograft (PDX) and Sensitivity of Radiomic Pipeline to Tumor Probability Boundary. Cancers 2021, 13, 3795. [Google Scholar] [CrossRef]
- Roy, S.; Whitehead, T.D.; Li, S.; Ademuyiwa, F.O.; Wahl, R.L.; Dehdashti, F.; Shoghi, K.I. Co-Clinical FDG-PET Radiomic Signature in Predicting Response to Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Altekruse, S.F.; Li, C.I.; Chen, V.W.; Clarke, C.A.; Ries, L.A.G.; Cronin, K.A. US Incidence of Breast Cancer Subtypes Defined by Joint Hormone Receptor and HER2 Status. J. Natl. Cancer Inst. 2014, 106, dju055. [Google Scholar] [CrossRef]
- Rubin, D.L.; Ugur Akdogan, M.; Altindag, C.; Alkim, E. EPAD: An Image Annotation and Analysis Platform for Quantitative Imaging. Tomography 2019, 5, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Vasaikar, S.V.; Straub, P.; Wang, J.; Zhang, B. LinkedOmics: Analyzing Multi-Omics Data within and across 32 Cancer Types. Nucleic Acids Res. 2018, 46, D956–D963. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Claerhout, S.; Prat, A.; Dobrolecki, L.E.; Petrovic, I.; Lai, Q.; Landis, M.D.; Wiechmann, L.; Schiff, R.; Giuliano, M.; et al. A Renewable Tissue Resource of Phenotypically Stable, Biologically and Ethnically Diverse, Patient-Derived Human Breast Cancer Xenograft Models. Cancer Res. 2013, 73, 4885–4897. [Google Scholar] [CrossRef]
- Petrosyan, V.; Dobrolecki, L.E.; Thistlethwaite, L.; Lewis, A.N.; Sallas, C.; Srinivasan, R.R.; Lei, J.T.; Kovacevic, V.; Obradovic, P.; Ellis, M.J.; et al. Identifying Biomarkers of Differential Chemotherapy Response in TNBC Patient-Derived Xenografts with a CTD/WGCNA Approach. iScience 2023, 26, 105799. [Google Scholar] [CrossRef]
- Lv, X.; Dobrolecki, L.E.; Ding, Y.; Rosen, J.M.; Lewis, M.T.; Chen, X. Orthotopic Transplantation of Breast Tumors as Preclinical Models for Breast Cancer. J. Vis. Exp. 2020, 159, e61173. [Google Scholar] [CrossRef]
- Yankeelov, T.E.; Luci, J.J.; Lepage, M.; Li, R.; Debusk, L.; Lin, P.C.; Price, R.R.; Gore, J.C. Quantitative Pharmacokinetic Analysis of DCE-MRI Data without an Arterial Input Function: A Reference Region Model. Magn. Reson. Imaging 2005, 23, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Mark, N.M.; Kargl, J.; Busch, S.E.; Yang, G.H.Y.; Metz, H.E.; Zhang, H.; Hubbard, J.J.; Pipavath, S.N.J.; Madtes, D.K.; Houghton, A.M. Chronic Obstructive Pulmonary Disease Alters Immune Cell Composition and Immune Checkpoint Inhibitor Efficacy in Non-Small Cell Lung Cancer. Am. J. Respir. Crit. Care Med. 2018, 197, 325–336. [Google Scholar] [CrossRef] [PubMed]
- McFadden, D.G.; Politi, K.; Bhutkar, A.; Chen, F.K.; Song, X.; Pirun, M.; Santiago, P.M.; Kim-Kiselak, C.; Platt, J.T.; Lee, E.; et al. Mutational Landscape of EGFR-, MYC-, and Kras-Driven Genetically Engineered Mouse Models of Lung Adenocarcinoma. Proc. Natl. Acad. Sci. USA 2016, 113, E6409–E6417. [Google Scholar] [CrossRef] [PubMed]
- Kargl, J.; Busch, S.E.; Yang, G.H.Y.; Kim, K.-H.; Hanke, M.L.; Metz, H.E.; Hubbard, J.J.; Lee, S.M.; Madtes, D.K.; McIntosh, M.W.; et al. Neutrophils Dominate the Immune Cell Composition in Non-Small Cell Lung Cancer. Nat. Commun. 2017, 8, 14381. [Google Scholar] [CrossRef]
- Kargl, J.; Zhu, X.; Zhang, H.; Yang, G.H.Y.; Friesen, T.J.; Shipley, M.; Maeda, D.Y.; Zebala, J.A.; McKay-Fleisch, J.; Meredith, G.; et al. Neutrophil Content Predicts Lymphocyte Depletion and Anti-PD1 Treatment Failure in NSCLC. JCI Insight 2019, 4, e130850. [Google Scholar] [CrossRef]
- Byrd, D.W.; Doot, R.K.; Allberg, K.C.; MacDonald, L.R.; McDougald, W.A.; Elston, B.F.; Linden, H.M.; Kinahan, P.E. Evaluation of Cross-Calibrated 68Ge/68Ga Phantoms for Assessing PET/CT Measurement Bias in Oncology Imaging for Single- and Multicenter Trials. Tomography 2016, 2, 353–360. [Google Scholar] [CrossRef]
- Jackson, S.J.; Thomas, G.J. Human Tissue Models in Cancer Research: Looking beyond the Mouse. Dis. Model. Mech. 2017, 10, 939–942. [Google Scholar] [CrossRef]
- Willyard, C. The Mice with Human Tumours: Growing Pains for a Popular Cancer Model. Nature 2018, 560, 156–157. [Google Scholar] [CrossRef]
- Ortmann, J.; Rampášek, L.; Tai, E.; Mer, A.S.; Shi, R.; Stewart, E.L.; Mascaux, C.; Fares, A.; Pham, N.-A.; Beri, G.; et al. Assessing Therapy Response in Patient-Derived Xenografts. Sci. Transl. Med. 2021, 13, eabf4969. [Google Scholar] [CrossRef]
- Evrard, Y.A.; Srivastava, A.; Randjelovic, J.; Doroshow, J.H.; Dean, D.A.; Morris, J.S.; Chuang, J.H. NCI PDXNet Consortium Systematic Establishment of Robustness and Standards in Patient-Derived Xenograft Experiments and Analysis. Cancer Res. 2020, 80, 2286–2297. [Google Scholar] [CrossRef] [PubMed]
- Serkova, N.J.; Glunde, K.; Haney, C.R.; Farhoud, M.; De Lille, A.; Redente, E.F.; Simberg, D.; Westerly, D.C.; Griffin, L.; Mason, R.P. Preclinical Applications of Multi-Platform Imaging in Animal Models of Cancer. Cancer Res. 2021, 81, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- de Jong, M.; Essers, J.; van Weerden, W.M. Imaging Preclinical Tumour Models: Improving Translational Power. Nat. Rev. Cancer 2014, 14, 481–493. [Google Scholar] [CrossRef]
- O’Connor, J.P.B.; Aboagye, E.O.; Adams, J.E.; Aerts, H.J.W.L.; Barrington, S.F.; Beer, A.J.; Boellaard, R.; Bohndiek, S.E.; Brady, M.; Brown, G.; et al. Imaging Biomarker Roadmap for Cancer Studies. Nat. Rev. Clin. Oncol. 2017, 14, 169–186. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Institution | Cancer/Disease | Model | Site | Disease Development | Therapy | Imaging |
|---|---|---|---|---|---|---|
| Baylor/UT Austin/Stanford | Triple-negative breast cancer | PDX | Orthotopic | 2–6 weeks | Chemotherapy | mpMRI |
| Duke | Soft tissue sarcoma | GEMM | Orthotopic | Median 54 days | Immunotherapy Radiation Surgery | mpMRI CT |
| MDACC | Colorectal cancer | PDX | Subcutaneous | 3 weeks | Targeted therapy | PET |
| Stanford | Osteosarcoma | xenografts | Orthotopic | 2–3 weeks | Immunotherapy | T2-weighted MRI |
| UCSF | Small cell neuroendocrine prostate cancer | PDX | Bone, liver | 1–4.5 months to reach 0.3 cc | Chemotherapy | HP MRI, mpMRI |
| U Mich | Myelofibrosis | GEMM | Orthotopic | 21 days | Targeted therapy | mpMRI |
| U Penn | Pancreatic ductal adenocarcinoma | GEMM | Orthotopic | 17–19 weeks | Chemoimmuno& stromal- targeted therapy | mpMRI |
| U Wash | Non-small cell lung cancer | GEMM | Orthotopic | 20–30 weeks | Immunotherapy | PET/CT |
| WUSTL | Triple-negative breast cancer | PDX | Orthotopic | 4 weeks–6 months | Chemotherapy | PET/MRI |
| WUSTL | ER+ breast cancer | PDX | Orthotopic | ~3–4 months | Endocrine therapy Targeted therapy | PET |
| Drug | Vendor | Catalog Number | Dose (mg/kg) | Concentration (mg/mL) | Vehicle | Route | Schedule |
|---|---|---|---|---|---|---|---|
| Carboplatin (Carbo) | McKesson (Teva) | 740278 | 50 | 10 | 10 mg Mannitol per 1 mL Water | IP | Weekly |
| Paclitaxel (Pac) | Millipore Sigma | T7402 | 33 | 1 | 90% Saline/5% Kolliphor/5% Ethanol | IP | Weekly |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peehl, D.M.; Badea, C.T.; Chenevert, T.L.; Daldrup-Link, H.E.; Ding, L.; Dobrolecki, L.E.; Houghton, A.M.; Kinahan, P.E.; Kurhanewicz, J.; Lewis, M.T.; et al. Animal Models and Their Role in Imaging-Assisted Co-Clinical Trials. Tomography 2023, 9, 657-680. https://doi.org/10.3390/tomography9020053
Peehl DM, Badea CT, Chenevert TL, Daldrup-Link HE, Ding L, Dobrolecki LE, Houghton AM, Kinahan PE, Kurhanewicz J, Lewis MT, et al. Animal Models and Their Role in Imaging-Assisted Co-Clinical Trials. Tomography. 2023; 9(2):657-680. https://doi.org/10.3390/tomography9020053
Chicago/Turabian StylePeehl, Donna M., Cristian T. Badea, Thomas L. Chenevert, Heike E. Daldrup-Link, Li Ding, Lacey E. Dobrolecki, A. McGarry Houghton, Paul E. Kinahan, John Kurhanewicz, Michael T. Lewis, and et al. 2023. "Animal Models and Their Role in Imaging-Assisted Co-Clinical Trials" Tomography 9, no. 2: 657-680. https://doi.org/10.3390/tomography9020053
APA StylePeehl, D. M., Badea, C. T., Chenevert, T. L., Daldrup-Link, H. E., Ding, L., Dobrolecki, L. E., Houghton, A. M., Kinahan, P. E., Kurhanewicz, J., Lewis, M. T., Li, S., Luker, G. D., Ma, C. X., Manning, H. C., Mowery, Y. M., O'Dwyer, P. J., Pautler, R. G., Rosen, M. A., Roudi, R., ... Zhou, R. (2023). Animal Models and Their Role in Imaging-Assisted Co-Clinical Trials. Tomography, 9(2), 657-680. https://doi.org/10.3390/tomography9020053