




Feasibility of [18F]FSPG PET for Early Response Assessment to Combined Blockade of EGFR and Glutamine Metabolism in Wild-Type KRAS Colorectal Cancer

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sequencing Data Acquisition
2.2. In Vivo Tumor Studies
2.3. Preclinical [18F]FSPG PET Imaging
2.4. Histological Evaluation
2.5. Immunohistochemistry (IHC)
2.6. Statistical Analysis
3. Results
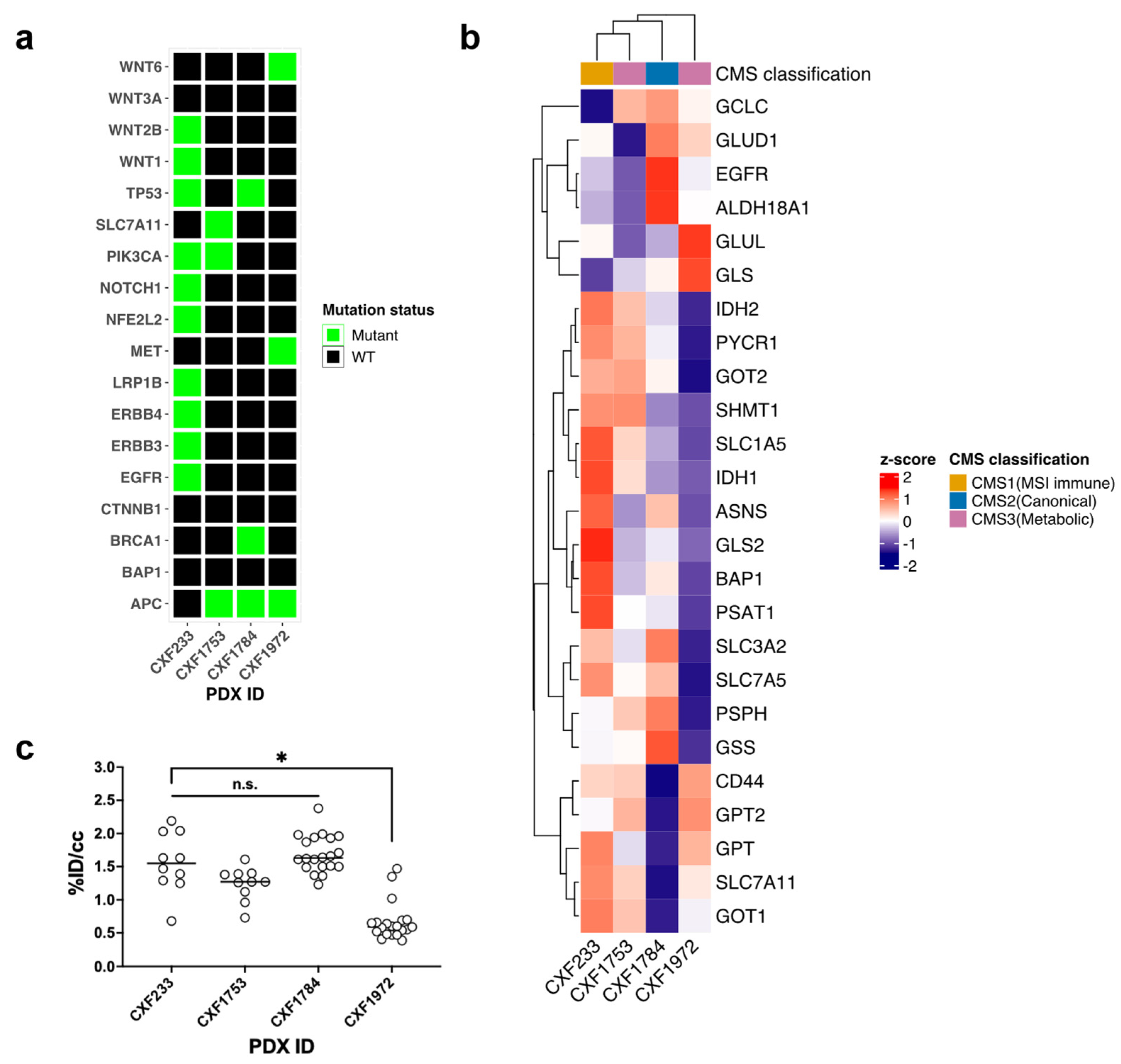
3.1. Characterization of Wild-Type KRAS PDX Tumors
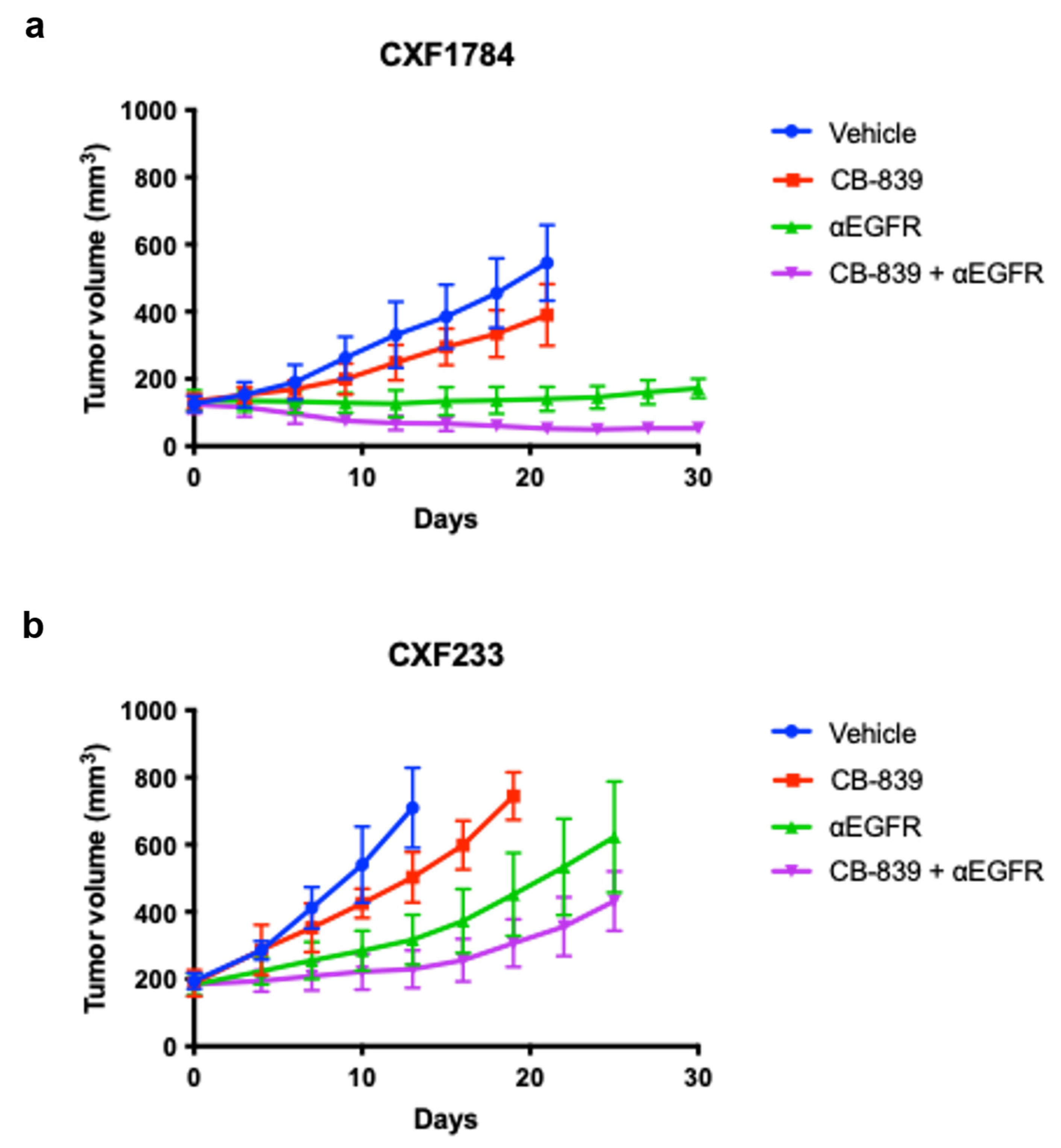
3.2. In Vivo Evaluation of Treatment
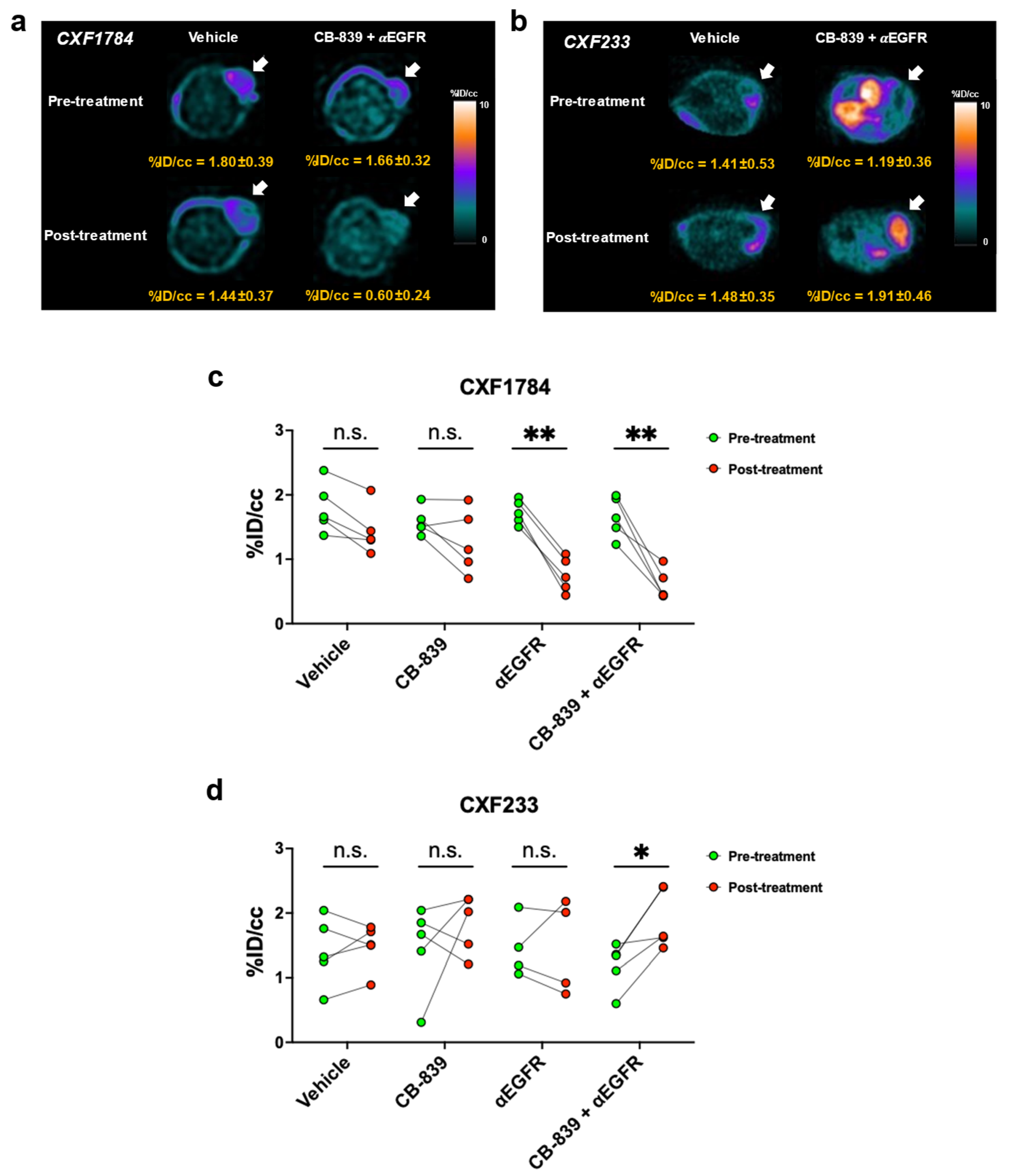
3.3. [18F]FSPG PET of PDX Tumors
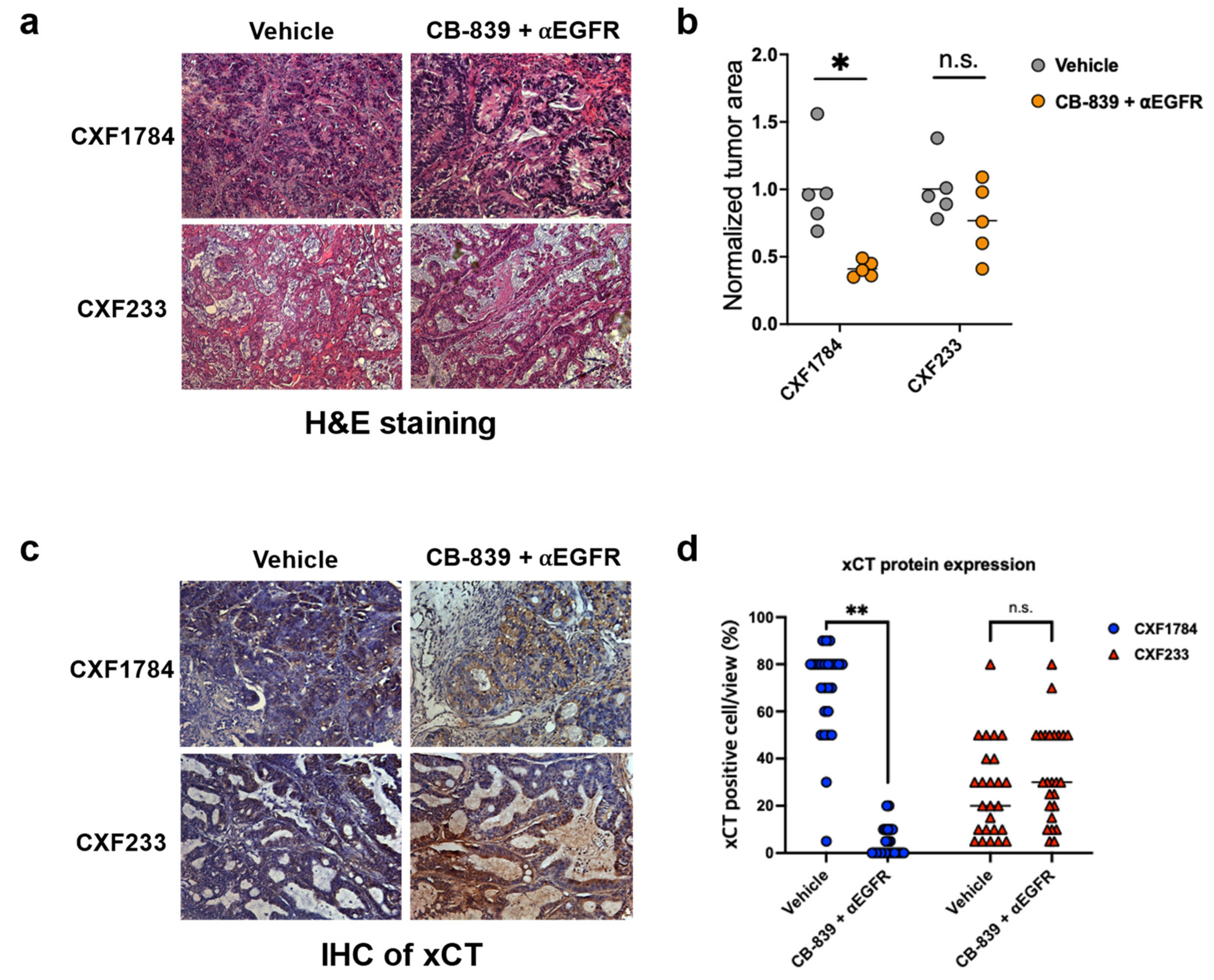
3.4. Histology
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Xie, Y.H.; Chen, Y.X.; Fang, J.Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target. Ther. 2020, 5, 22. [Google Scholar] [CrossRef]
- Cutsem, E.V.; Köhne, C.-H.; Láng, I.; Folprecht, G.; Nowacki, M.P.; Cascinu, S.; Shchepotin, I.; Maurel, J.; Cunningham, D.; Tejpar, S.; et al. Cetuximab Plus Irinotecan, Fluorouracil, and Leucovorin As First-Line Treatment for Metastatic Colorectal Cancer: Updated Analysis of Overall Survival According to Tumor KRAS and BRAF Mutation Status. J. Clin. Oncol. 2011, 29, 2011–2019. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Bondarenko, I.; Hartmann, J.T.; de Braud, F.; Schuch, G.; Zubel, A.; Celik, I.; Schlichting, M.; Koralewski, P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: The OPUS study. Ann. Oncol. 2011, 22, 1535–1546. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef]
- Cohen, A.S.; Geng, L.; Zhao, P.; Fu, A.; Schulte, M.L.; Graves-Deal, R.; Washington, M.K.; Berlin, J.; Coffey, R.J.; Manning, H.C. Combined blockade of EGFR and glutamine metabolism in preclinical models of colorectal cancer. Transl. Oncol. 2020, 13, 100828. [Google Scholar] [CrossRef]
- Koglin, N.; Mueller, A.; Berndt, M.; Schmitt-Willich, H.; Toschi, L.; Stephens, A.W.; Gekeler, V.; Friebe, M.; Dinkelborg, L.M. Specific PET imaging of xC−Transporter activity using a 18F-labeled glutamate derivative reveals a dominant pathway in tumor metabolism. Clin. Cancer Res. 2011, 17, 6000–6011. [Google Scholar] [CrossRef]
- Greenwood, H.E.; McCormick, P.N.; Gendron, T.; Glaser, M.; Pereira, R.; Maddocks, O.D.K.; Sander, K.; Zhang, T.; Koglin, N.; Lythgoe, M.F.; et al. Measurement of Tumor Antioxidant Capacity and Prediction of Chemotherapy Resistance in Preclinical Models of Ovarian Cancer by Positron Emission Tomography. Clin. Cancer Res. 2019, 25, 2471–2482. [Google Scholar] [CrossRef]
- McCormick, P.N.; Greenwood, H.E.; Glaser, M.; Maddocks, O.D.K.; Gendron, T.; Sander, K.; Gowrishankar, G.; Hoehne, A.; Zhang, T.; Shuhendler, A.J.; et al. Assessment of Tumor Redox Status through (S)-4-(3-[18F]fluoropropyl)-L-Glutamic Acid PET Imaging of System xC−activity. Cancer Res. 2019, 79, 853–863. [Google Scholar] [CrossRef]
- Kavanaugh, G.; Williams, J.; Morris, A.S.; Nickels, M.L.; Walker, R.; Koglin, N.; Stephens, A.W.; Washington, M.K.; Geevarghese, S.K.; Liu, Q.; et al. Utility of [18F]FSPG PET to Image Hepatocellular Carcinoma: First Clinical Evaluation in a US Population. Mol. Imaging Biol. 2016, 18, 924–934. [Google Scholar] [CrossRef]
- Paez, R.; Shah, C.; Cords, A.J.; Muterspaugh, A.; Helton, J.E.; Antic, S.; Eisenberg, R.; Chen, H.; Grogan, E.L.; Manning, H.C.; et al. 18F-FSPG PET imaging for the evaluation of indeterminate pulmonary nodules. PLoS ONE 2022, 17, e0265427. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Baek, S.; Choi, C.M.; Ahn, S.H.; Lee, J.W.; Gong, G.; Ryu, J.S.; Oh, S.J.; Bacher-Stier, C.; Fels, L.; Koglin, N.; et al. Exploratory clinical trial of (4S)-4-(3-[18F]fluoropropyl)-L-glutamate for imaging xC-transporter using positron emission tomography in patients with non-small cell lung or breast cancer. Clin. Cancer Res. 2012, 18, 5427–5437. [Google Scholar] [CrossRef]
- Wilson, P.M.; Labonte, M.J.; Lenz, H.J. Molecular markers in the treatment of metastatic colorectal cancer. Cancer J. 2010, 16, 262–272. [Google Scholar] [CrossRef]
- Markowitz, S.D.; Bertagnolli, M.M. Molecular origins of cancer: Molecular basis of colorectal cancer. N. Engl. J. Med. 2009, 361, 2449–2460. [Google Scholar] [CrossRef]
- Thanki, K.; Nicholls, M.E.; Gajjar, A.; Senagore, A.J.; Qiu, S.; Szabo, C.; Hellmich, M.R.; Chao, C. Consensus Molecular Subtypes of Colorectal Cancer and their Clinical Implications. Int. Biol. Biomed. J. 2017, 3, 105–111. [Google Scholar]
- Ten Hoorn, S.; de Back, T.R.; Sommeijer, D.W.; Vermeulen, L. Clinical Value of Consensus Molecular Subtypes in Colorectal Cancer: A Systematic Review and Meta-Analysis. J. Natl. Cancer Inst. 2022, 114, 503–516. [Google Scholar] [CrossRef]
- Herreros-Villanueva, M.; Gomez-Manero, N.; Muniz, P.; Garcia-Giron, C.; Coma del Corral, M.J. PIK3CA mutations in KRAS and BRAF wild type colorectal cancer patients. A study of Spanish population. Mol. Biol. Rep. 2011, 38, 1347–1351. [Google Scholar] [CrossRef]
- Loree, J.M.; Bailey, A.M.; Johnson, A.M.; Yu, Y.; Wu, W.; Bristow, C.A.; Davis, J.S.; Shaw, K.R.; Broaddus, R.; Banks, K.C.; et al. Molecular Landscape of ERBB2/ERBB3 Mutated Colorectal Cancer. J. Natl. Cancer Inst. 2018, 110, 1409–1417. [Google Scholar] [CrossRef]
- Williams, C.S.; Bernard, J.K.; Demory Beckler, M.; Almohazey, D.; Washington, M.K.; Smith, J.J.; Frey, M.R. ERBB4 is over-expressed in human colon cancer and enhances cellular transformation. Carcinogenesis 2015, 36, 710–718. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xia, X.; Huang, P. xCT: A Critical Molecule That Links Cancer Metabolism to Redox Signaling. Mol. Ther. 2020, 28, 2358–2366. [Google Scholar] [CrossRef] [PubMed]
- Tarangelo, A.; Magtanong, L.; Bieging-Rolett, K.T.; Li, Y.; Ye, J.; Attardi, L.D.; Dixon, S.J. p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep. 2018, 22, 569–575. [Google Scholar] [CrossRef]
- Liu, D.S.; Duong, C.P.; Haupt, S.; Montgomery, K.G.; House, C.M.; Azar, W.J.; Pearson, H.B.; Fisher, O.M.; Read, M.; Guerra, G.R.; et al. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat. Commun. 2017, 8, 14844. [Google Scholar] [CrossRef]
- Seib, T.M.; Patel, S.A.; Bridges, R.J. Regulation of the system x−Ccystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia 2011, 59, 1387–1401. [Google Scholar] [CrossRef]
- Nandar, W.; Neely, E.B.; Unger, E.; Connor, J.R. A mutation in the HFE gene is associated with altered brain iron profiles and increased oxidative stress in mice. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Habib, E.; Linher-Melville, K.; Lin, H.X.; Singh, G. Expression of xCT and activity of system xc−are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol. 2015, 5, 33–42. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Tsuchihashi, K.; Ishimoto, T.; Yae, T.; Motohara, T.; Sugihara, E.; Onishi, N.; Masuko, T.; Yoshizawa, K.; Kawashiri, S.; et al. xCT inhibition depletes CD44v-expressing tumor cells that are resistant to EGFR-targeted therapy in head and neck squamous cell carcinoma. Cancer Res. 2013, 73, 1855–1866. [Google Scholar] [CrossRef]
- Tsuchihashi, K.; Okazaki, S.; Ohmura, M.; Ishikawa, M.; Sampetrean, O.; Onishi, N.; Wakimoto, H.; Yoshikawa, M.; Seishima, R.; Iwasaki, Y.; et al. The EGF Receptor Promotes the Malignant Potential of Glioma by Regulating Amino Acid Transport System xc(—). Cancer Res. 2016, 76, 2954–2963. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.R.; Salem, K.; Jeffery, J.; Powers, G.L.; Yan, Y.; Shoghi, K.I.; Mahajan, A.M.; Fowler, A.M. Sex as a Biologic Variable in Preclinical Imaging Research: Initial Observations with 18F-FLT. J. Nucl. Med. 2018, 59, 833–838. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDX ID | CXF233 | CXF1784 | CXF1972 | CXF1753 |
|---|---|---|---|---|
| Patient information | ||||
| Gender | Female | Male | Male | Male |
| Age | 74 | 65 | 62 | 61 |
| Histology | Adenocarcinoma | Adenocarcinoma | Adenocarcinoma | Adenocarcinoma |
| Stage at surgery | TxN2M0 | M1 liver | N/A | T3N1M1 |
| Chemotherapy prior to surgery | No | FolFOX | not known | not known |
| Origin of xenograft | Primary (Colon) | Metastasis (Liver) | Metastasis (Pleura) | Metastasis (Peritoneum) |
| PDX ID | Group | Mouse ID | Tumor Area (mm2) | Normalized Tumor Area * | Alive Tumor Area (%) |
|---|---|---|---|---|---|
| CXF1784 | Vehicle | V1 | 32.75 | 0.69 | 69.20% |
| V2 | 45.82 | 0.96 | 47.39% | ||
| V3 | 46.48 | 0.97 | 43.39% | ||
| V4 | 74.48 | 1.56 | 40.11% | ||
| V5 | 39.37 | 0.82 | 20.15% | ||
| CB-839 + 𝛼EGFR | CB + P1 | 17.01 | 0.36 | 61.39% | |
| CB + P2 | 16.75 | 0.35 | 58.41% | ||
| CB + P3 | 21.74 | 0.45 | 66.66% | ||
| CB + P4 | 23.63 | 0.49 | 71.28% | ||
| CB + P5 | 19.34 | 0.4 | 58.00% | ||
| CXF233 | Vehicle | V6 | 65.82 | 0.89 | 33.28% |
| V7 | 101.79 | 1.38 | 48.53% | ||
| V8 | 69.99 | 0.95 | 55.25% | ||
| V9 | 57.45 | 0.78 | 14.34% | ||
| V10 | 74.95 | 1.01 | 20.69% | ||
| CB-839 + 𝛼EGFR | CB + CTX1 | 72.72 | 0.98 | 33.15% | |
| CB + CTX2 | 30.64 | 0.41 | 23.30% | ||
| CB + CTX3 | 56.56 | 0.76 | 15.47% | ||
| CB + CTX4 | 80.67 | 1.09 | 16.64% | ||
| CB + CTX5 | 44.46 | 0.6 | 21.23% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bae, S.-W.; Wang, J.; Georgiou, D.K.; Wen, X.; Cohen, A.S.; Geng, L.; Tantawy, M.N.; Manning, H.C. Feasibility of [18F]FSPG PET for Early Response Assessment to Combined Blockade of EGFR and Glutamine Metabolism in Wild-Type KRAS Colorectal Cancer. Tomography 2023, 9, 497-508. https://doi.org/10.3390/tomography9020041
Bae S-W, Wang J, Georgiou DK, Wen X, Cohen AS, Geng L, Tantawy MN, Manning HC. Feasibility of [18F]FSPG PET for Early Response Assessment to Combined Blockade of EGFR and Glutamine Metabolism in Wild-Type KRAS Colorectal Cancer. Tomography. 2023; 9(2):497-508. https://doi.org/10.3390/tomography9020041
Chicago/Turabian StyleBae, Seong-Woo, Jianbo Wang, Dimitra K. Georgiou, Xiaoxia Wen, Allison S. Cohen, Ling Geng, Mohammed Noor Tantawy, and H. Charles Manning. 2023. "Feasibility of [18F]FSPG PET for Early Response Assessment to Combined Blockade of EGFR and Glutamine Metabolism in Wild-Type KRAS Colorectal Cancer" Tomography 9, no. 2: 497-508. https://doi.org/10.3390/tomography9020041
APA StyleBae, S.-W., Wang, J., Georgiou, D. K., Wen, X., Cohen, A. S., Geng, L., Tantawy, M. N., & Manning, H. C. (2023). Feasibility of [18F]FSPG PET for Early Response Assessment to Combined Blockade of EGFR and Glutamine Metabolism in Wild-Type KRAS Colorectal Cancer. Tomography, 9(2), 497-508. https://doi.org/10.3390/tomography9020041