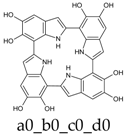
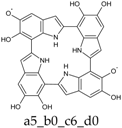
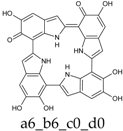
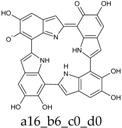
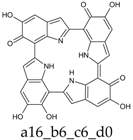
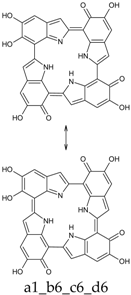
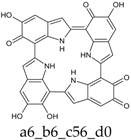
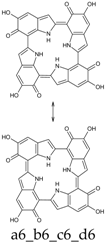
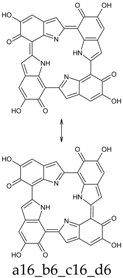
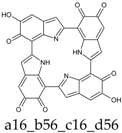
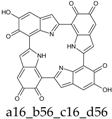
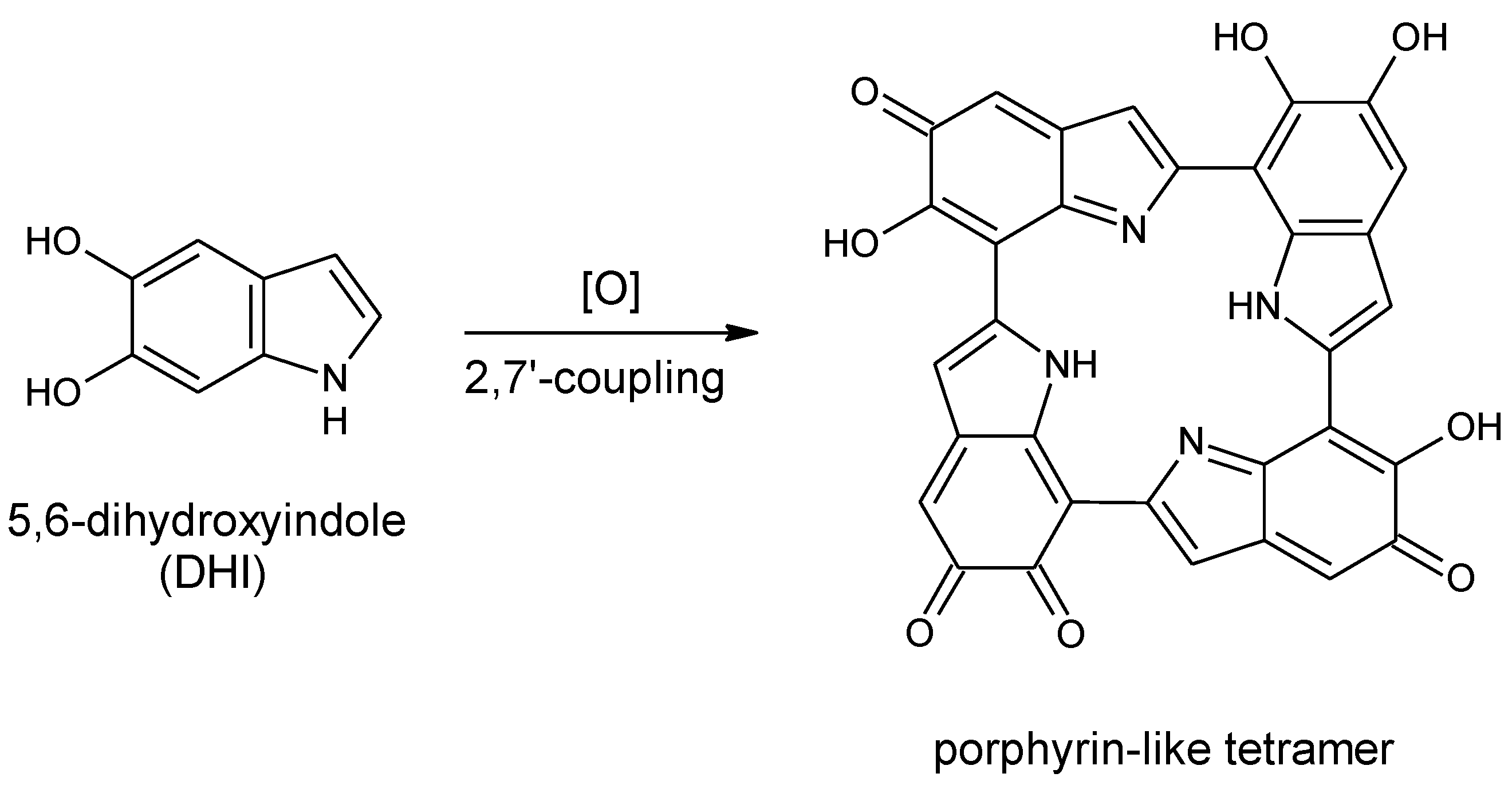
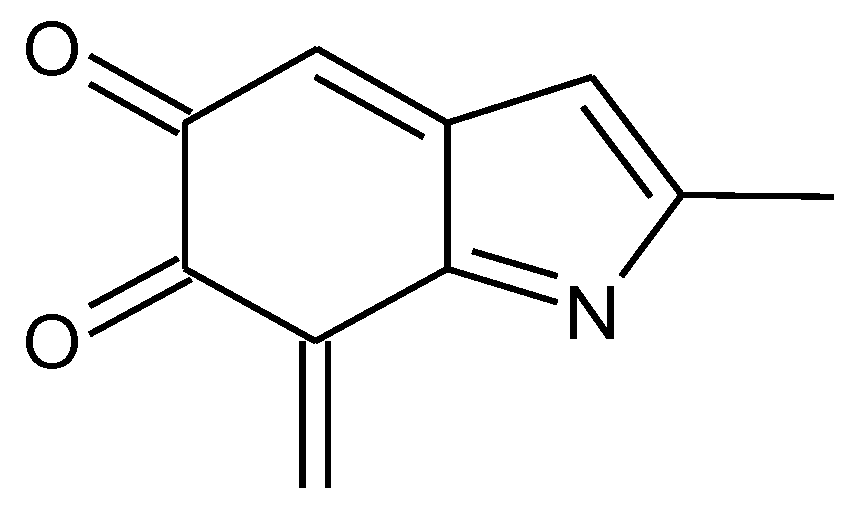
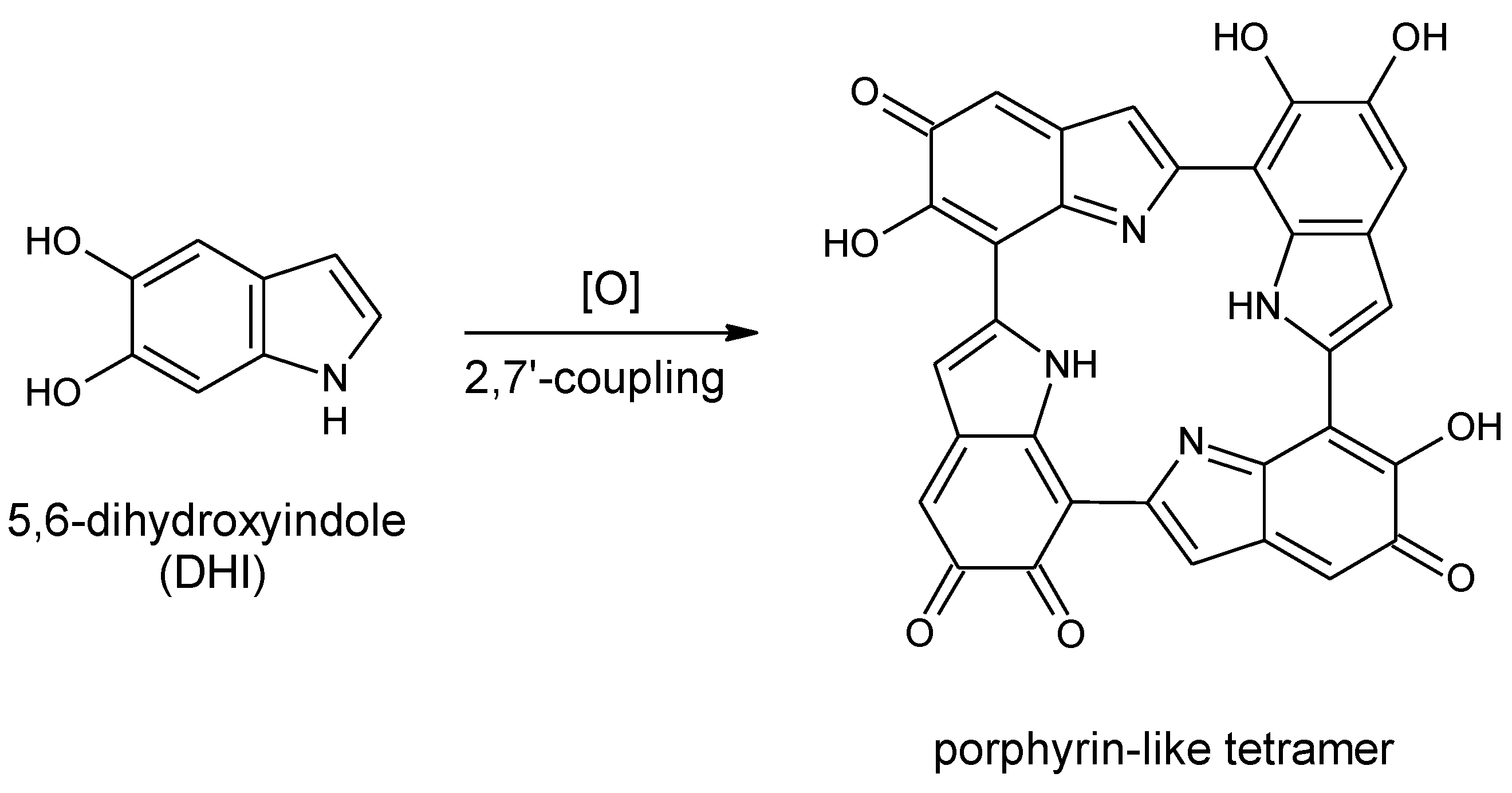
Scheme 1.
Formation of a porphyrin-like tetramer (Kaxiras’s porphyrin, KP) by oxidation of 5,6-dihydroxyindole (DHI).
Scheme 1.
Formation of a porphyrin-like tetramer (Kaxiras’s porphyrin, KP) by oxidation of 5,6-dihydroxyindole (DHI).
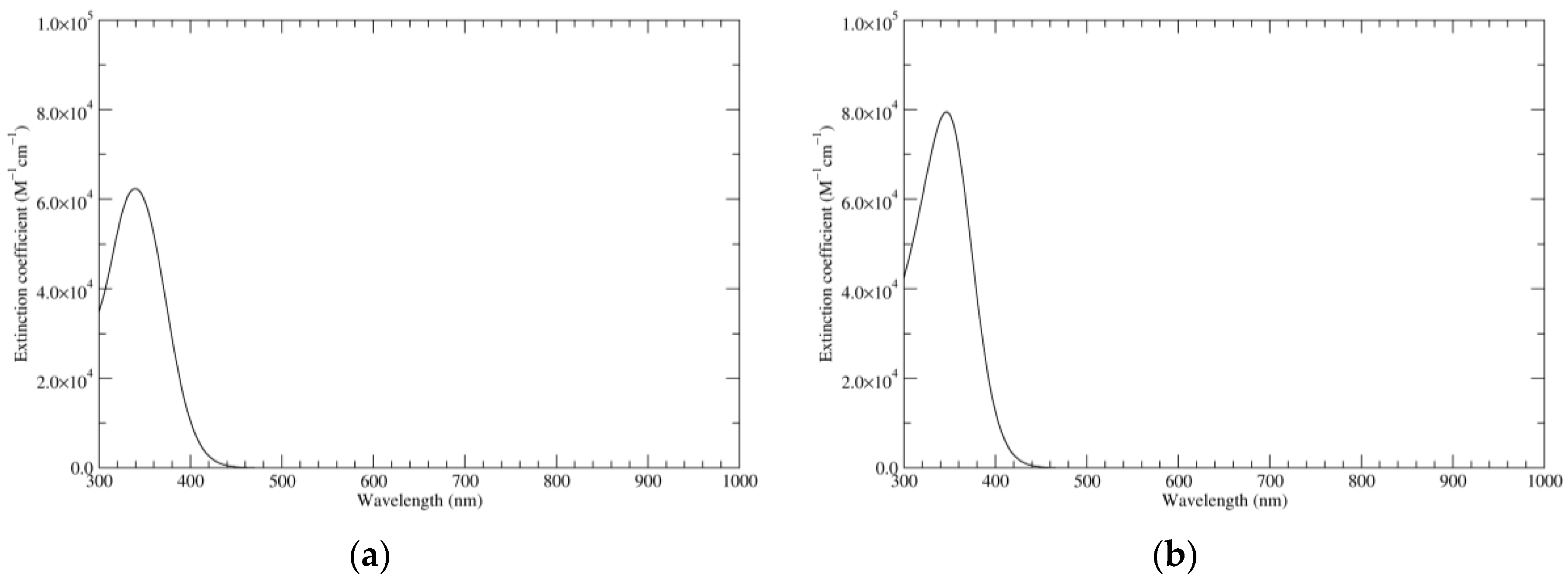
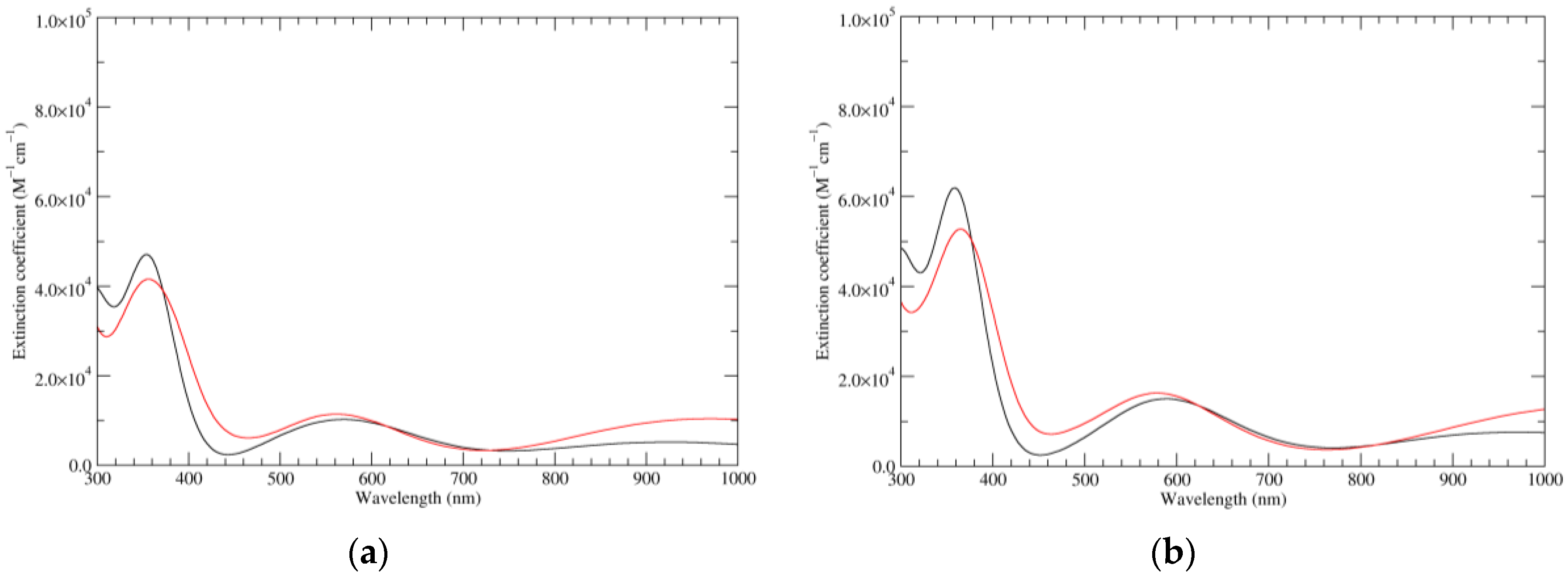
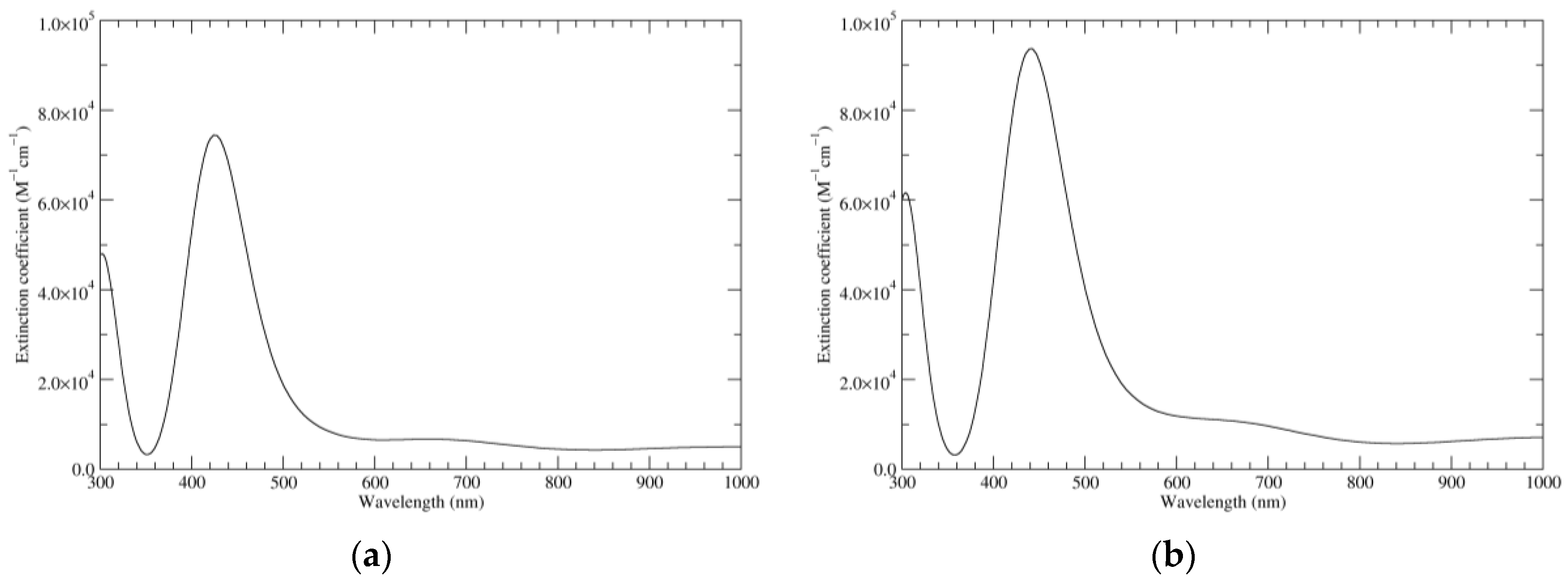
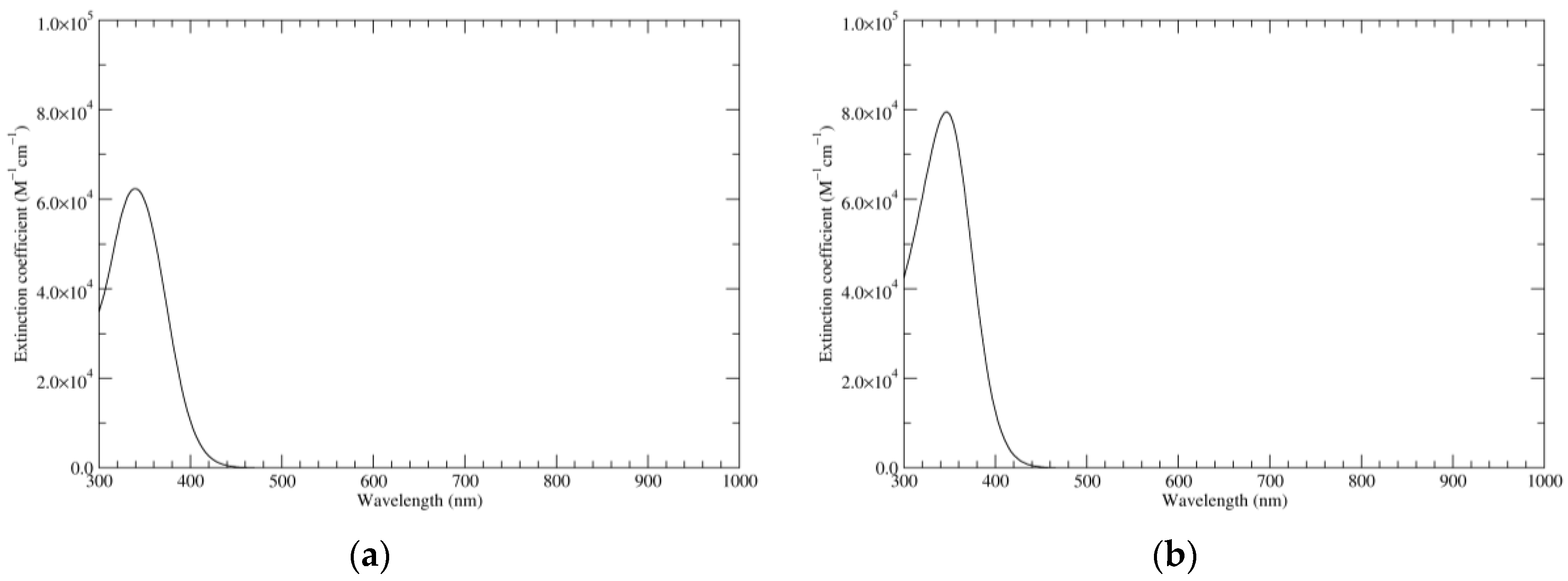
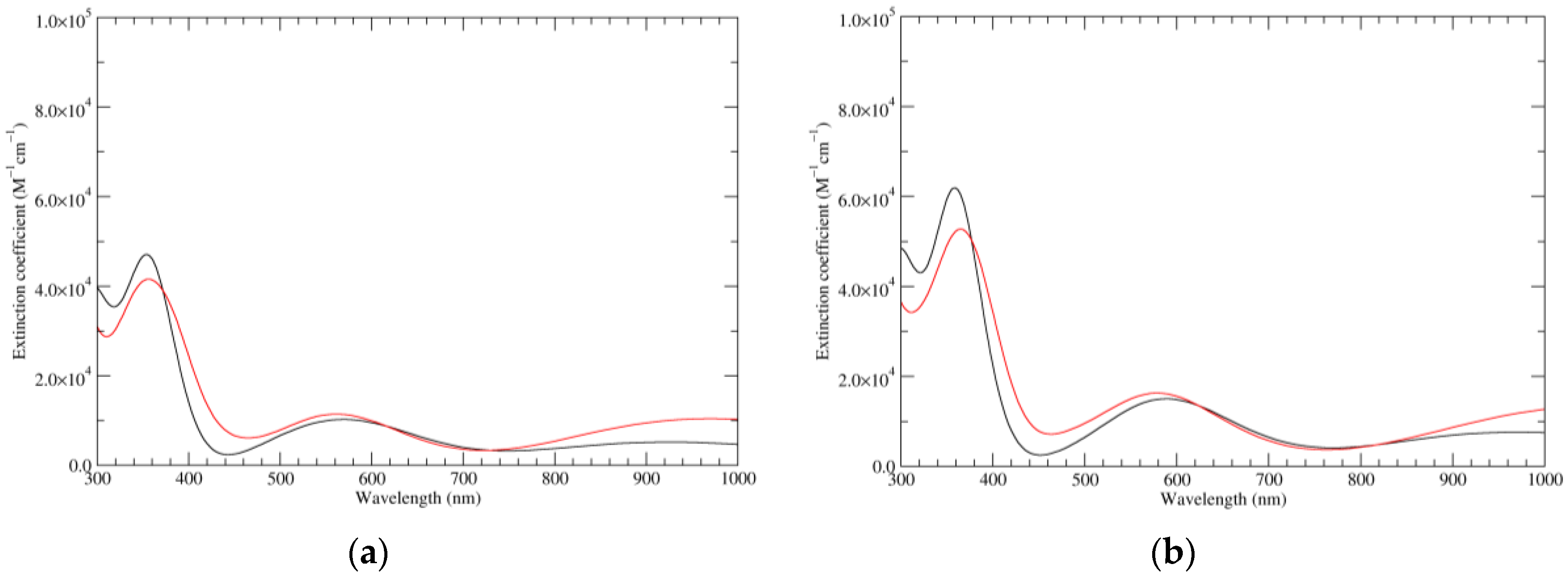
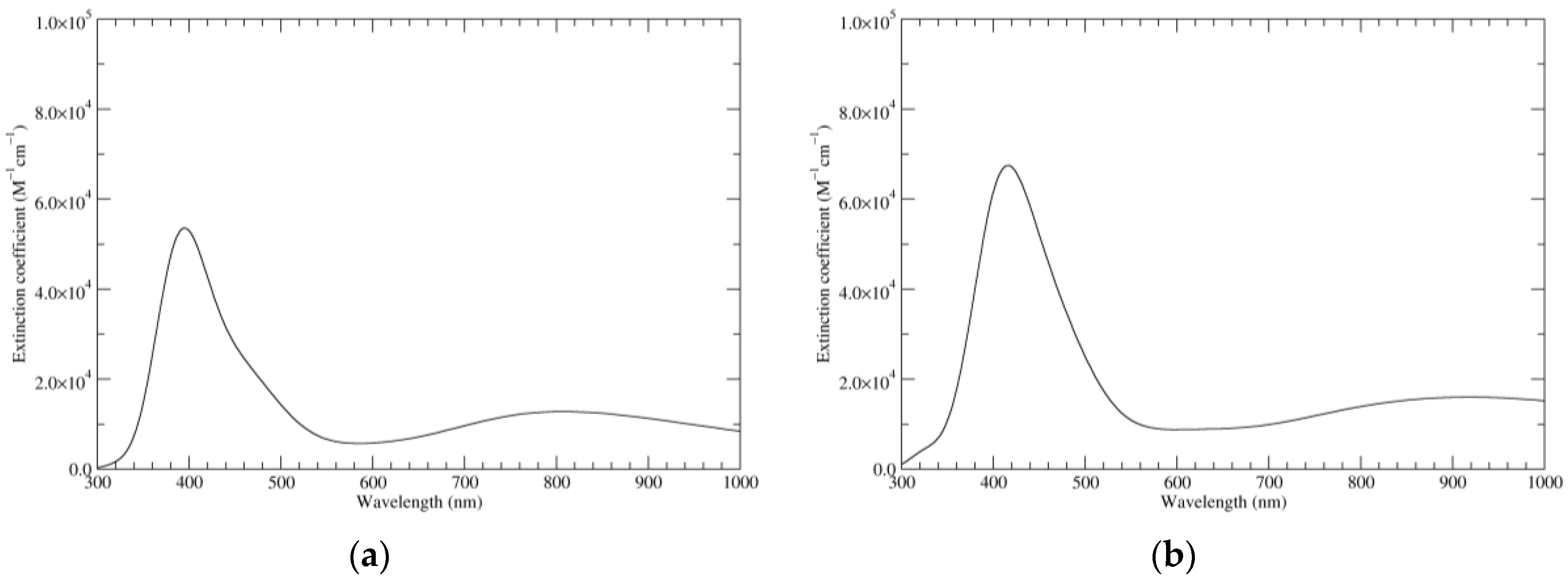
Figure 1.
Computed ultraviolet–visible (UV–Vis) spectra of the most significant tautomers/conformers in the reduced state. (a) Neutral form in vacuo, a0_b0_c0_d0, S4. (b) Neutral form in water, a0_b0_c0_d0, S4.
Figure 1.
Computed ultraviolet–visible (UV–Vis) spectra of the most significant tautomers/conformers in the reduced state. (a) Neutral form in vacuo, a0_b0_c0_d0, S4. (b) Neutral form in water, a0_b0_c0_d0, S4.
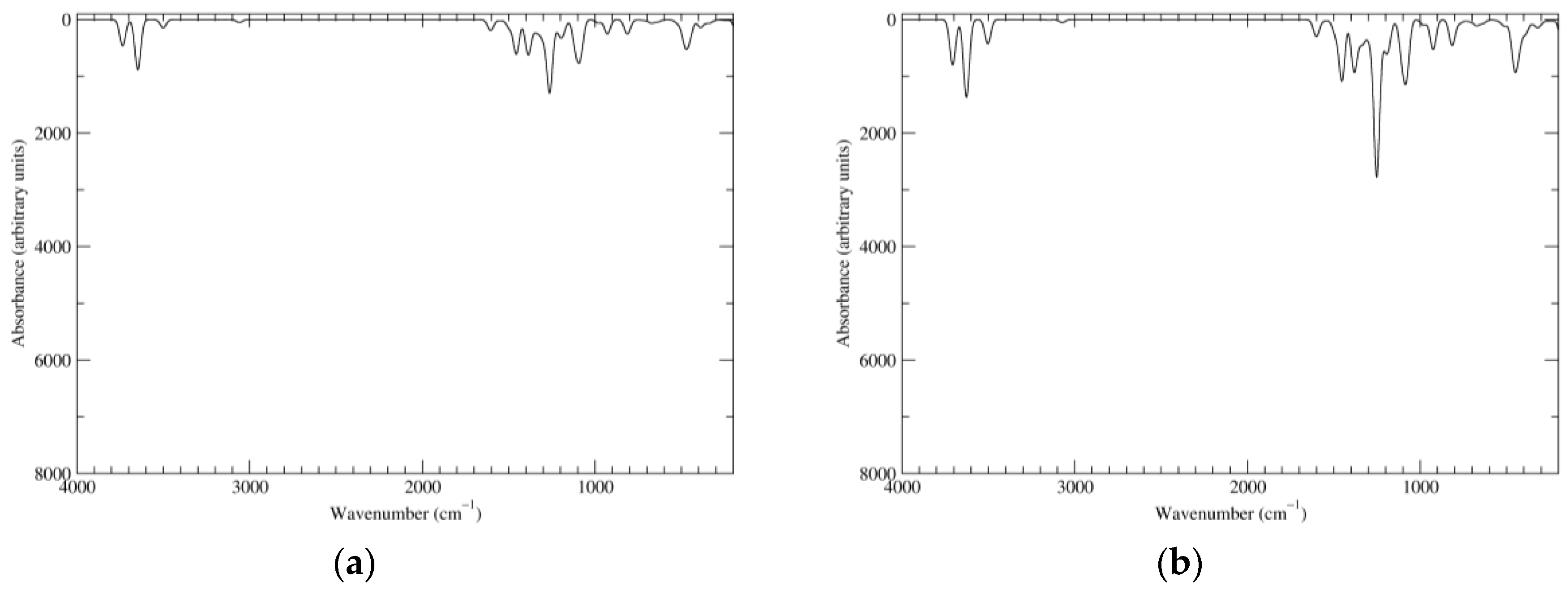
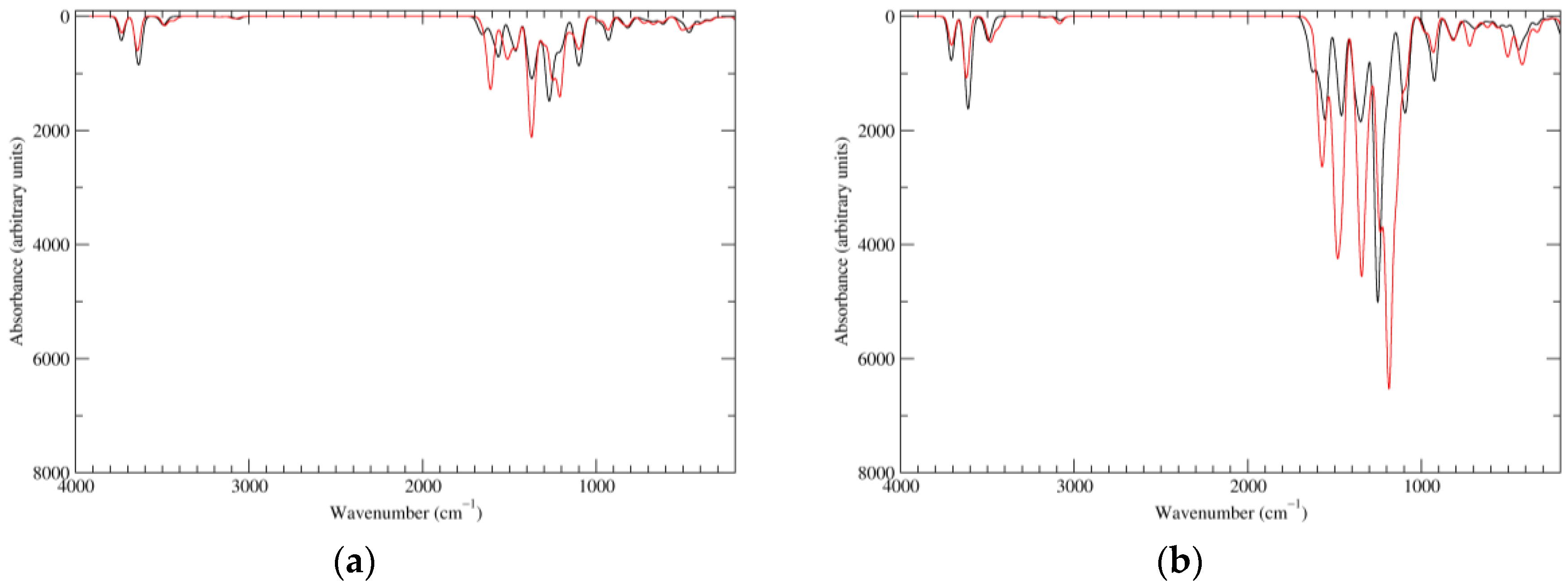
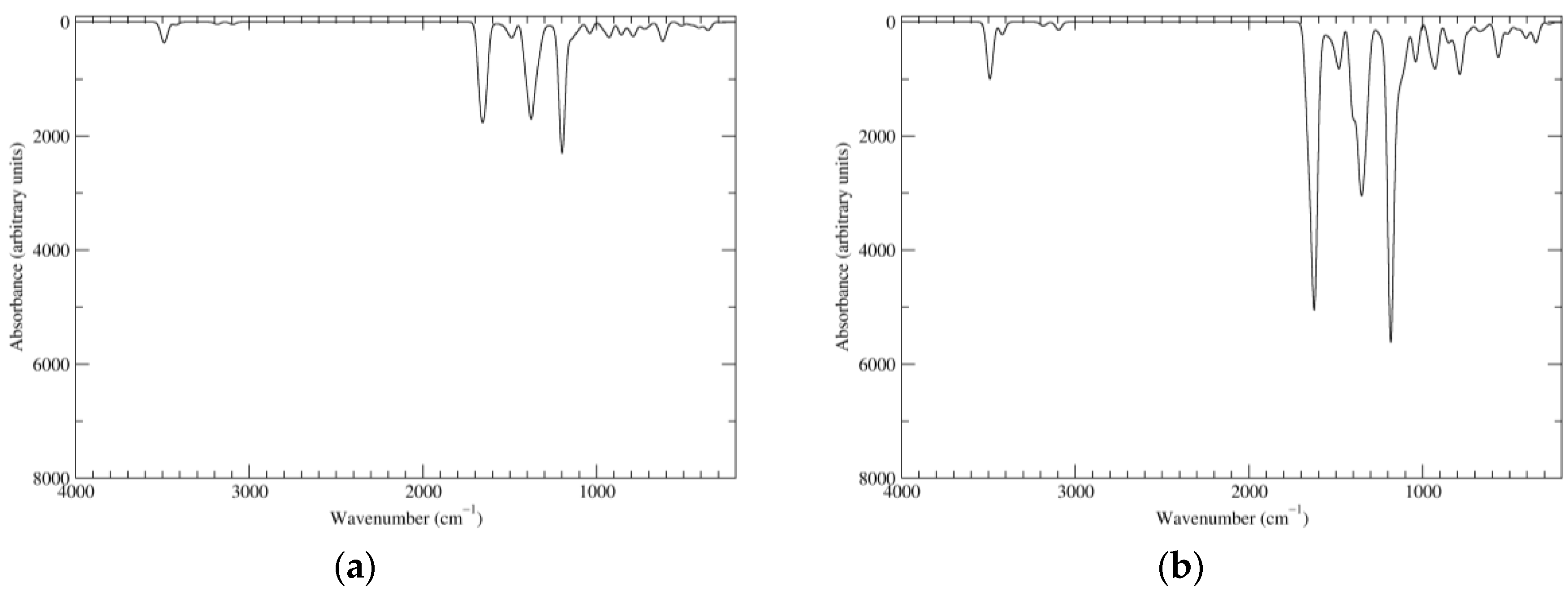
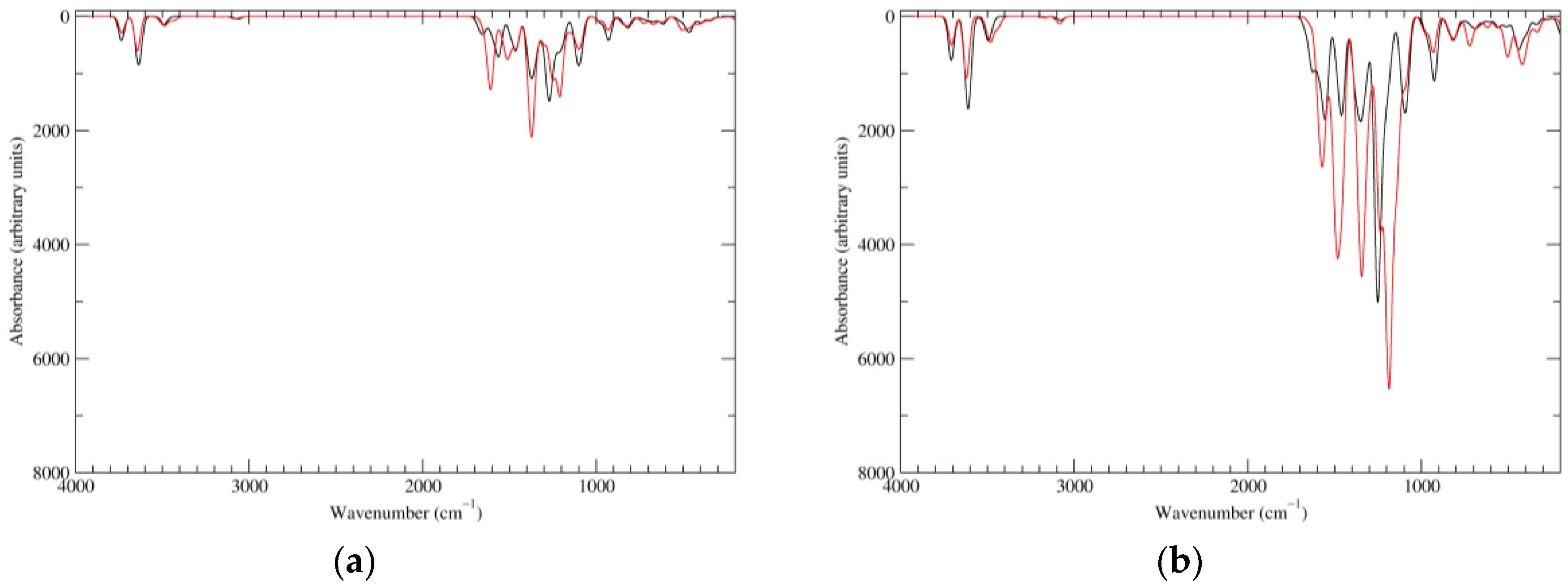
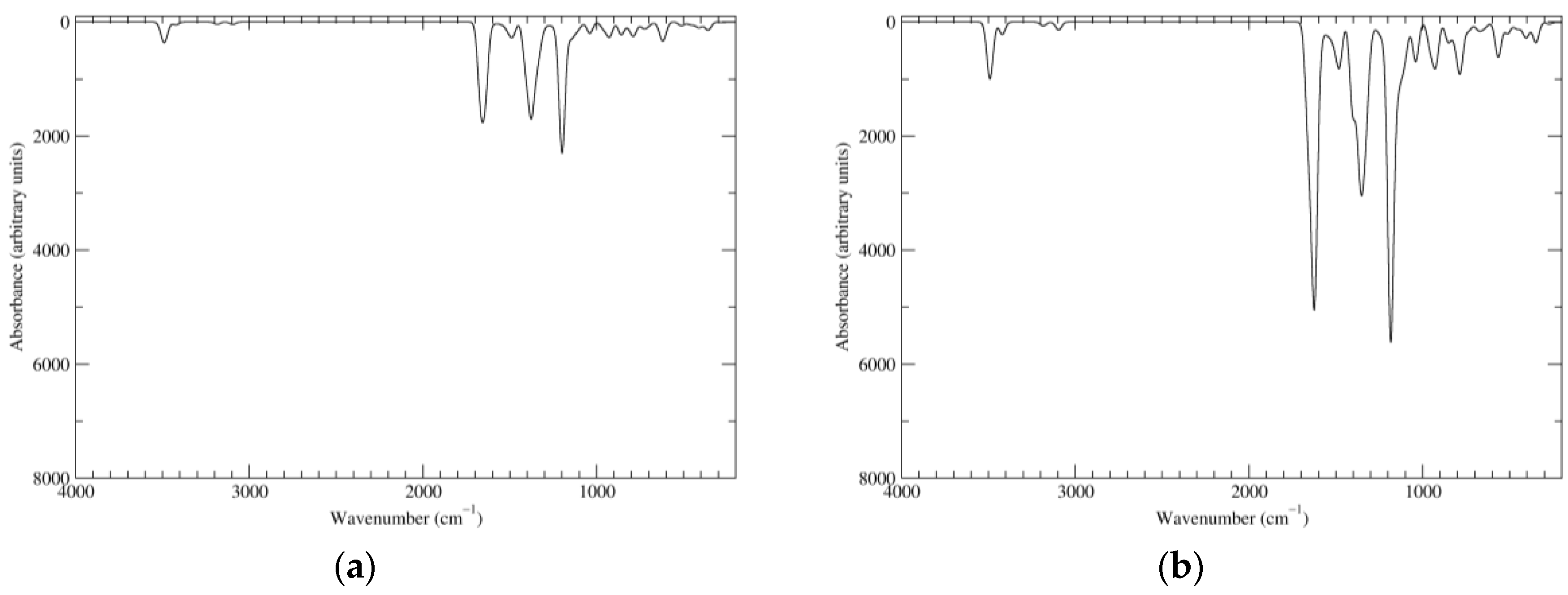
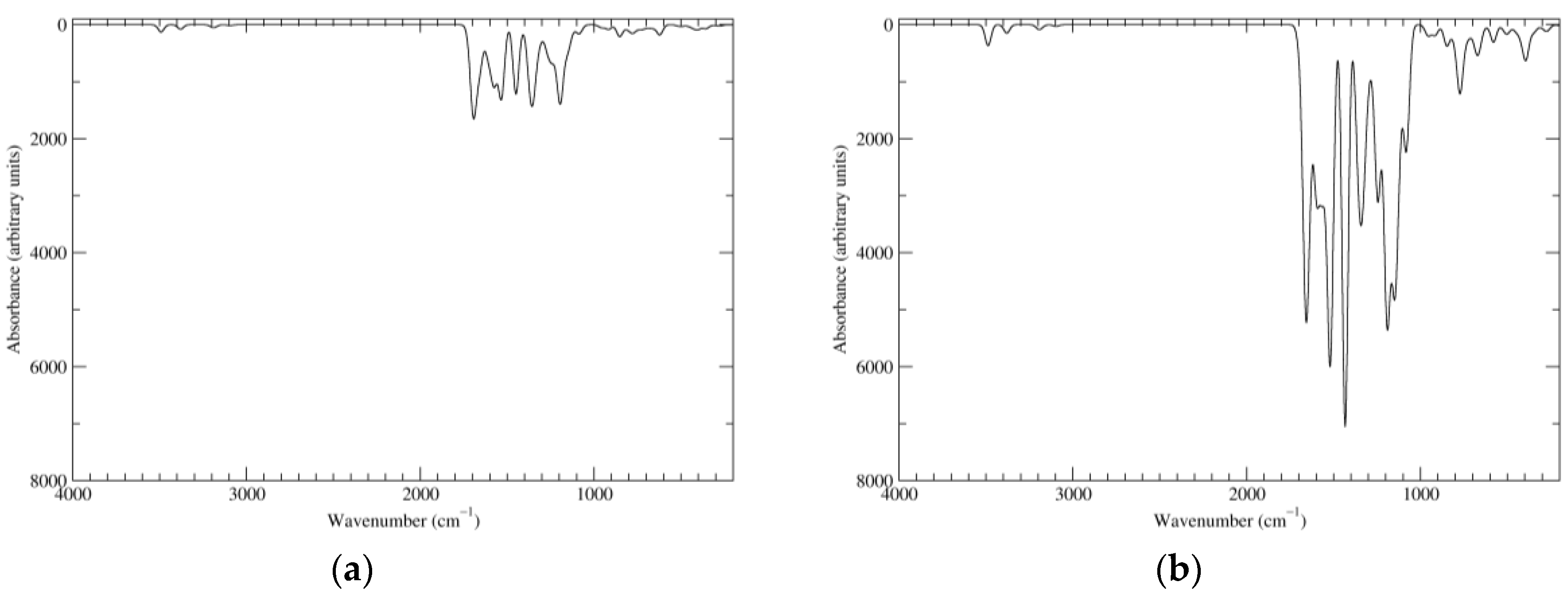
Figure 2.
Computed infrared (IR) spectra of the most significant tautomers/conformers in the reduced state. (a) Neutral form in vacuo, a0_b0_c0_d0, S4. (b) Neutral form in water, a0_b0_c0_d0, S4.
Figure 2.
Computed infrared (IR) spectra of the most significant tautomers/conformers in the reduced state. (a) Neutral form in vacuo, a0_b0_c0_d0, S4. (b) Neutral form in water, a0_b0_c0_d0, S4.
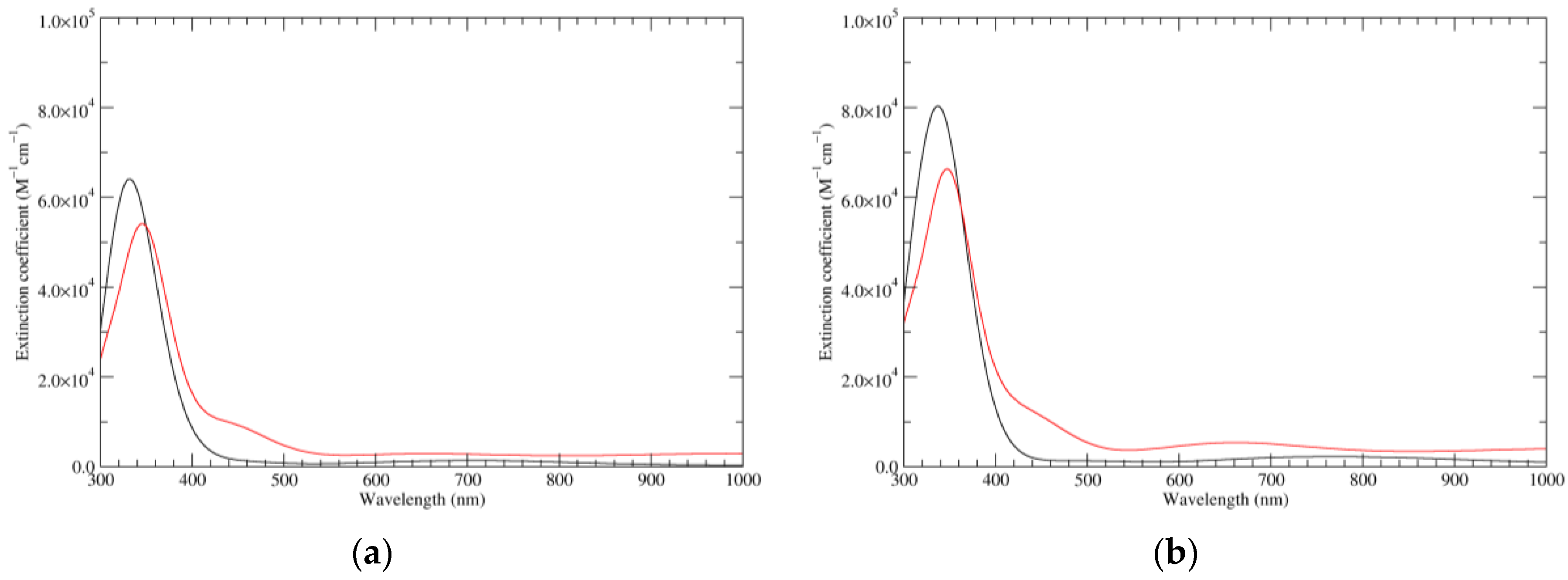
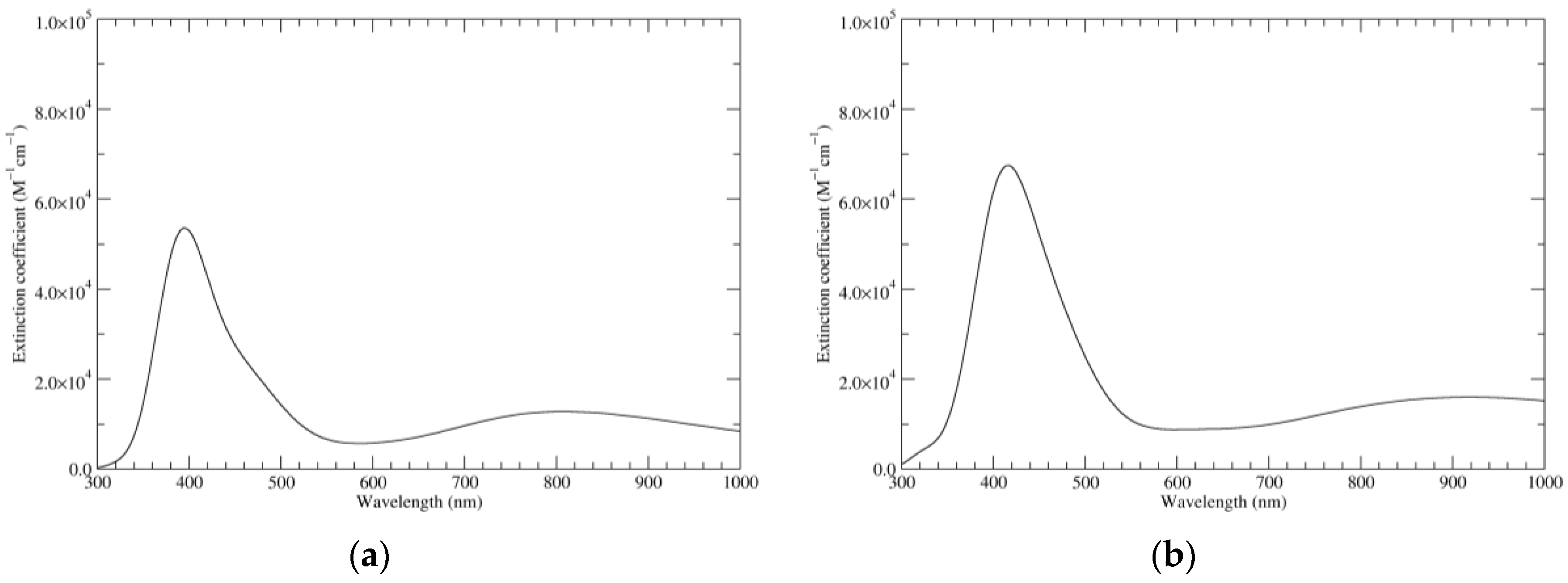
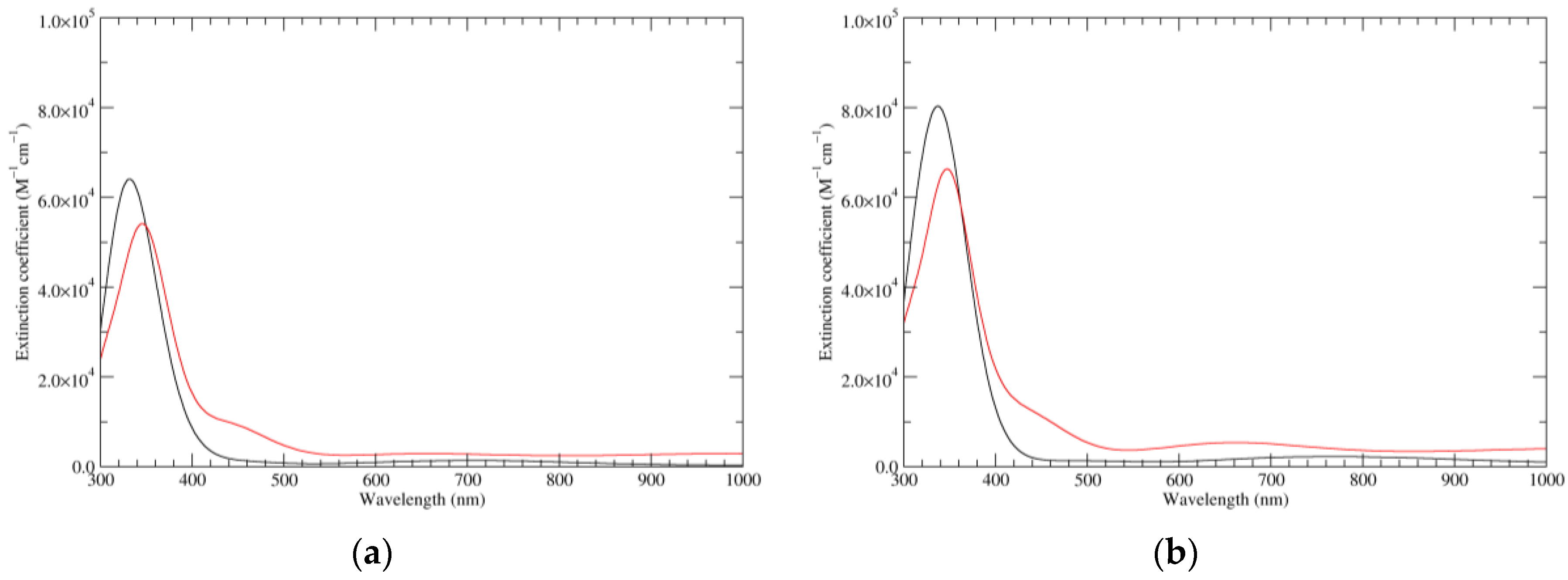
Figure 3.
Computed UV–Vis spectra of the most significant tautomers/conformers in the one-electron oxidation state. (a) Neutral form in vacuo: black line, a5_b0_c0_d0, C1, conf1; red line, a6_b0_c0_d0, C1, conf1. (b) Neutral form in water: black line, a5_b0_c0_d0, C1, conf1; red line, a6_b0_c0_d0 C1, conf1.
Figure 3.
Computed UV–Vis spectra of the most significant tautomers/conformers in the one-electron oxidation state. (a) Neutral form in vacuo: black line, a5_b0_c0_d0, C1, conf1; red line, a6_b0_c0_d0, C1, conf1. (b) Neutral form in water: black line, a5_b0_c0_d0, C1, conf1; red line, a6_b0_c0_d0 C1, conf1.
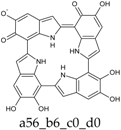
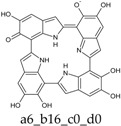
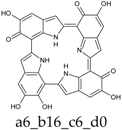
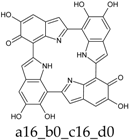
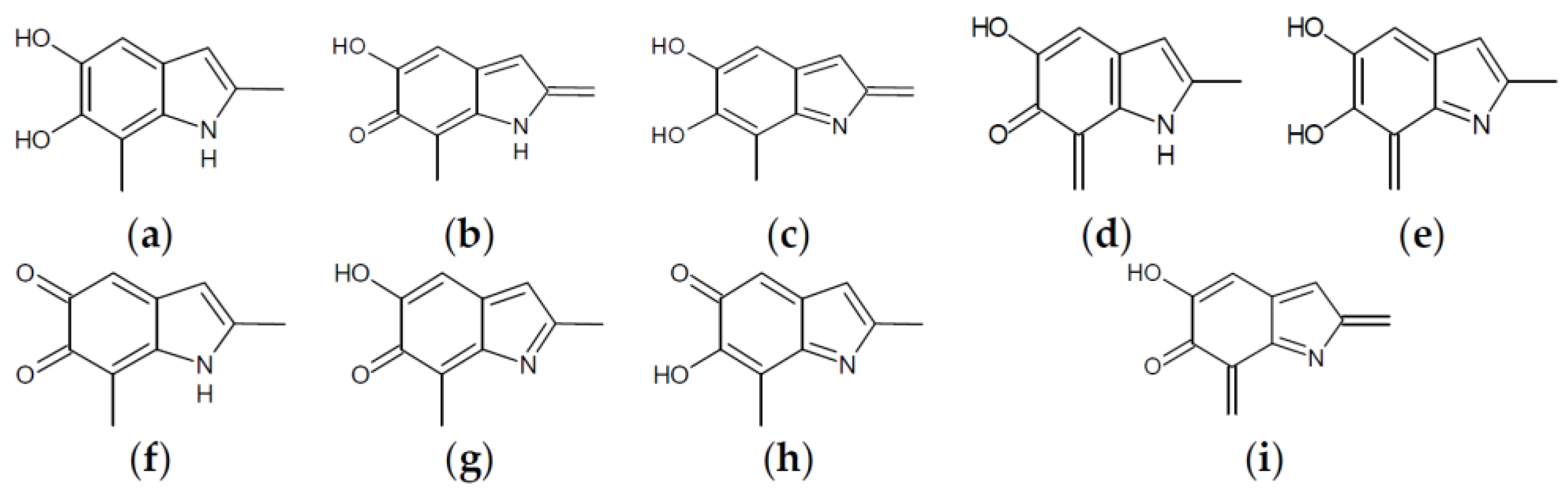
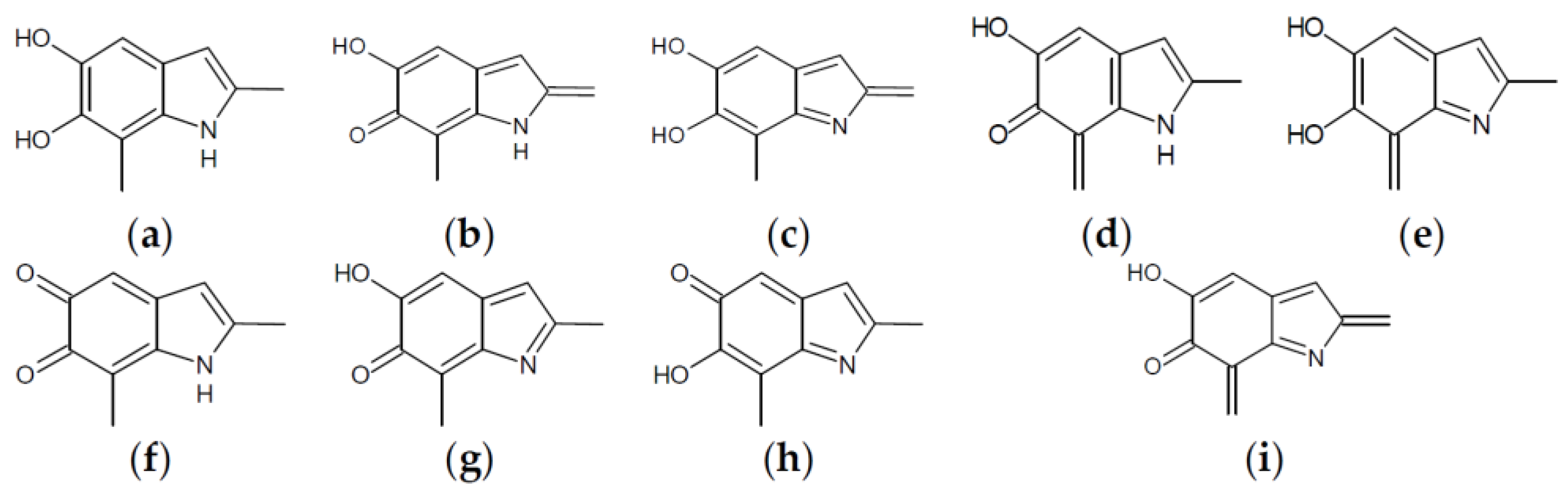
Figure 4.
Building blocks for generation of staring structures of closed-shell tautomers of Kaxiras’s porphyrin. (a) Fully reduced units. (b–e) One-electron oxidized units. (f–i) Two-electron oxidized units.
Figure 4.
Building blocks for generation of staring structures of closed-shell tautomers of Kaxiras’s porphyrin. (a) Fully reduced units. (b–e) One-electron oxidized units. (f–i) Two-electron oxidized units.
Figure 5.
Computed UV–Vis spectra of the most significant tautomers/conformers in the two-electrons oxidation state. (a) Neutral form in vacuo: black line, a16_b0_c0_d0, C1, conf1; red line, a6_b6_c0_d0, C1, conf1. (b) Neutral form in water: black line, a16_b0_c0_d0, C1, conf1; red line, a6_b6_c0_d0, C1, conf1.
Figure 5.
Computed UV–Vis spectra of the most significant tautomers/conformers in the two-electrons oxidation state. (a) Neutral form in vacuo: black line, a16_b0_c0_d0, C1, conf1; red line, a6_b6_c0_d0, C1, conf1. (b) Neutral form in water: black line, a16_b0_c0_d0, C1, conf1; red line, a6_b6_c0_d0, C1, conf1.
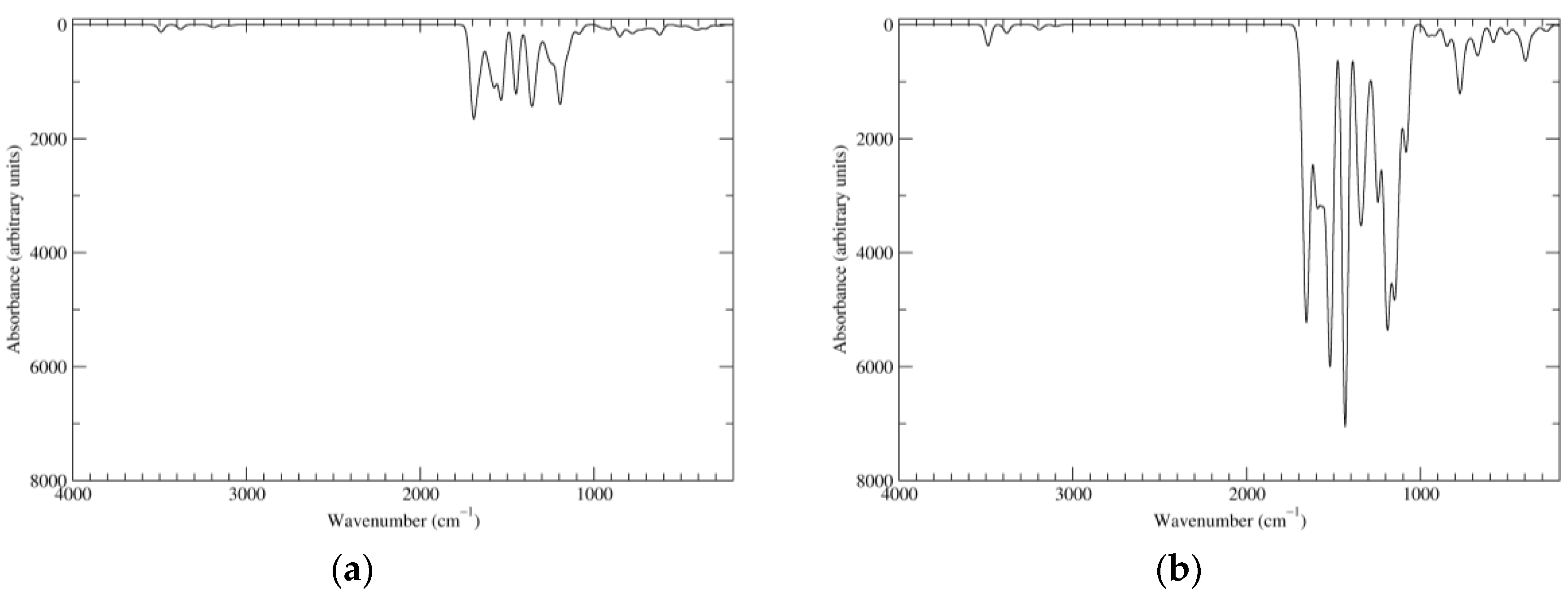
Figure 6.
Computed IR spectra of the most significant tautomers/conformers in the two-electrons oxidation state. (a) Neutral form in vacuo: black line, a16_b0_c0_d0, C1, conf1; red line, a6_b6_c0_d0, C1, conf1. (b) Neutral form in water: black line, a16_b0_c0_d0, C1, conf1; red line, a6_b6_c0_d0, C1, conf1.
Figure 6.
Computed IR spectra of the most significant tautomers/conformers in the two-electrons oxidation state. (a) Neutral form in vacuo: black line, a16_b0_c0_d0, C1, conf1; red line, a6_b6_c0_d0, C1, conf1. (b) Neutral form in water: black line, a16_b0_c0_d0, C1, conf1; red line, a6_b6_c0_d0, C1, conf1.
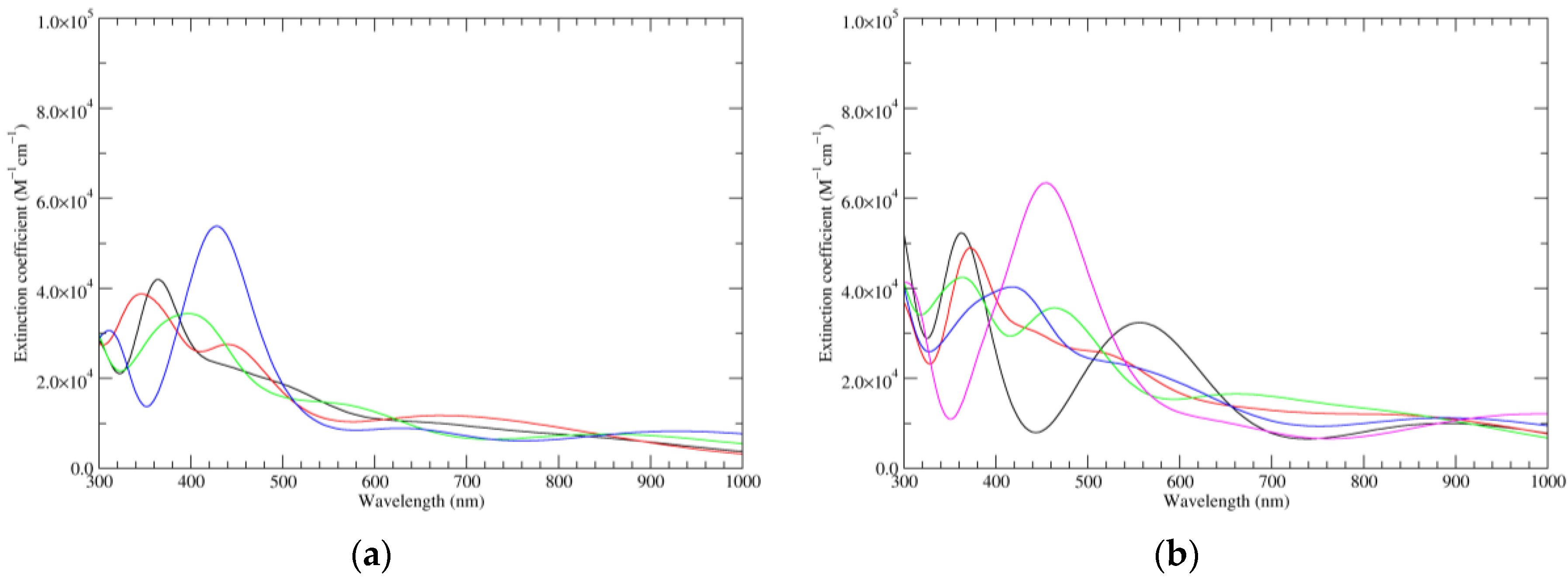
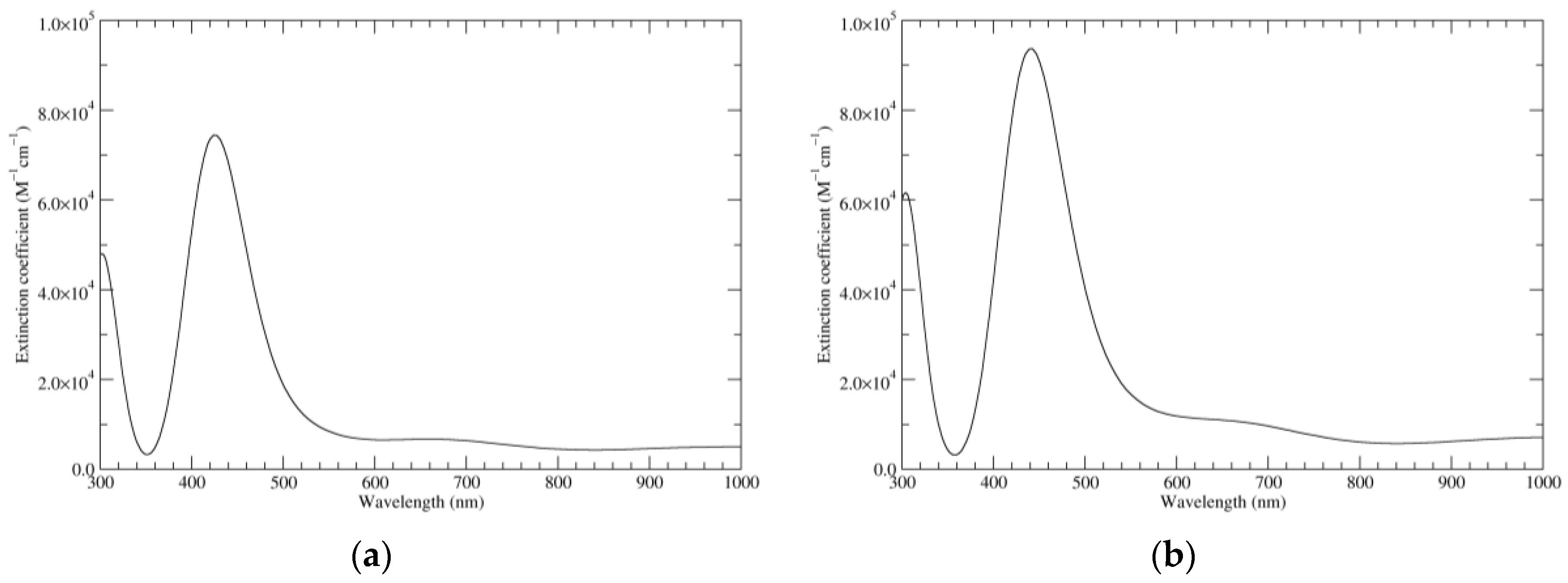
Figure 7.
Computed UV–Vis spectra of the most significant tautomers/conformers in the four-electrons oxidation state. (a) Neutral form in vacuo: black line, a16_b6_c6_d0, C1, conf1; red line, a6_b16_c6_d0, C1, conf1; green line, a6_b6_c16_d0, C1, conf1; blue line, a6_b6_c6_d6, C2, conf1. (b) Neutral form in water: black line, a16_b0_c16_d0, C2, conf1; red line, a16_b6_c6_d0, C1, conf1; green line, a6_b16_c6_d0, C1, conf1; blue line, a6_b6_c16_d0, C1, conf1; magenta line, a6_b6_c6_d6, C2, conf1.
Figure 7.
Computed UV–Vis spectra of the most significant tautomers/conformers in the four-electrons oxidation state. (a) Neutral form in vacuo: black line, a16_b6_c6_d0, C1, conf1; red line, a6_b16_c6_d0, C1, conf1; green line, a6_b6_c16_d0, C1, conf1; blue line, a6_b6_c6_d6, C2, conf1. (b) Neutral form in water: black line, a16_b0_c16_d0, C2, conf1; red line, a16_b6_c6_d0, C1, conf1; green line, a6_b16_c6_d0, C1, conf1; blue line, a6_b6_c16_d0, C1, conf1; magenta line, a6_b6_c6_d6, C2, conf1.
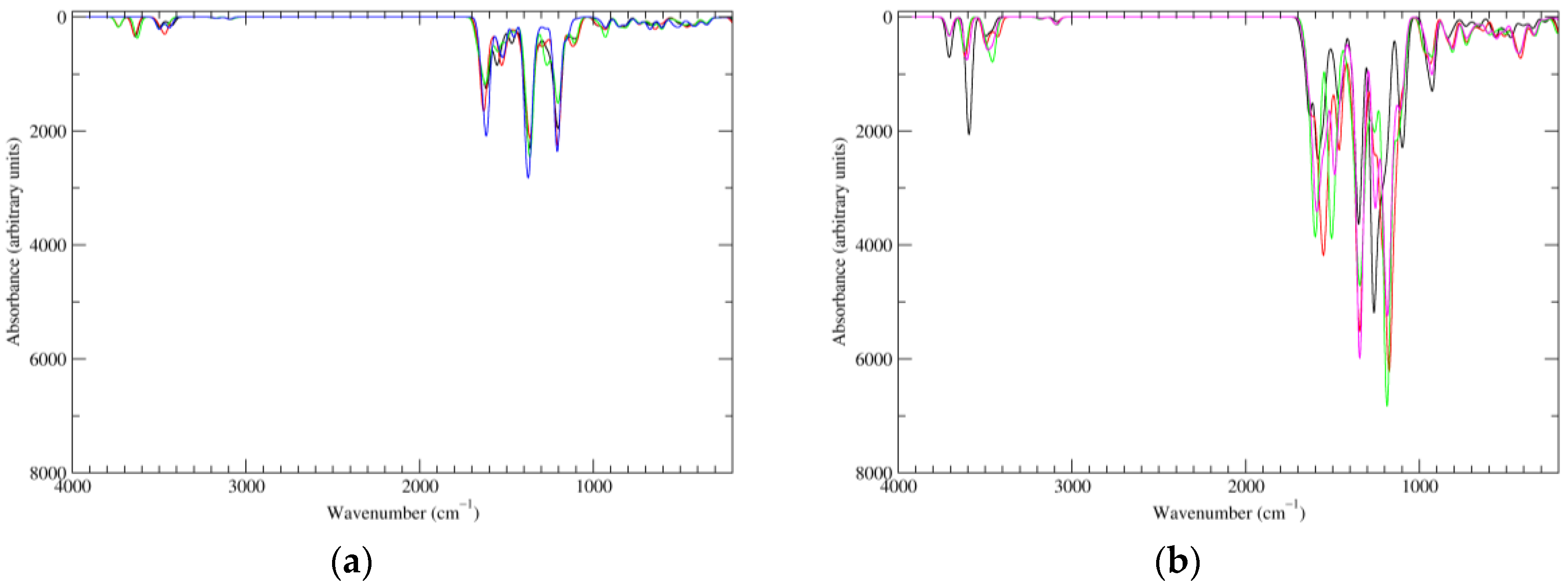
Figure 8.
Computed IR spectra of the most significant tautomers/conformers in the four-electrons oxidation state. (a) Neutral form in vacuo: black line, a16_b6_c6_d0, C1, conf1; red line, a6_b16_c6_d0, C1, conf1; green line, a6_b6_c16_d0, C1, conf1; blue line, a6_b6_c6_d6, C2, conf1. (b) Neutral form in water: black line, a16_b0_c16_d0, C2, conf1; red line, a16_b6_c6_d0, C1, conf1; green line, a6_b16_c6_d0, C1, conf1; blue line, a6_b6_c16_d0, C1, conf1; magenta line, a6_b6_c6_d6, C2, conf1.
Figure 8.
Computed IR spectra of the most significant tautomers/conformers in the four-electrons oxidation state. (a) Neutral form in vacuo: black line, a16_b6_c6_d0, C1, conf1; red line, a6_b16_c6_d0, C1, conf1; green line, a6_b6_c16_d0, C1, conf1; blue line, a6_b6_c6_d6, C2, conf1. (b) Neutral form in water: black line, a16_b0_c16_d0, C2, conf1; red line, a16_b6_c6_d0, C1, conf1; green line, a6_b16_c6_d0, C1, conf1; blue line, a6_b6_c16_d0, C1, conf1; magenta line, a6_b6_c6_d6, C2, conf1.
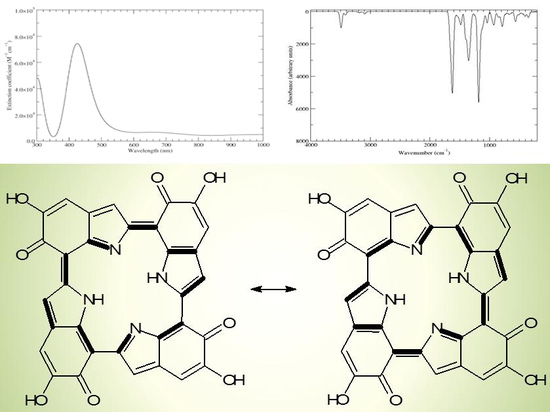
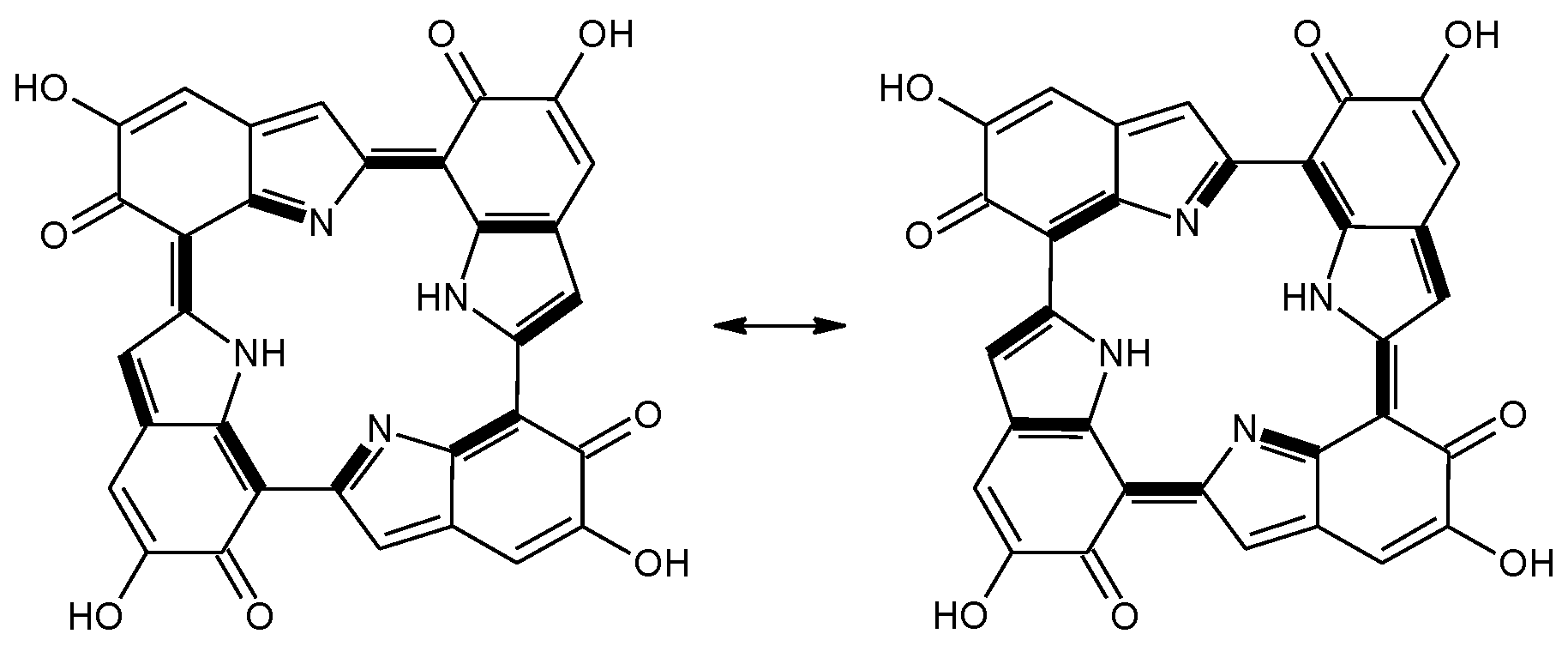
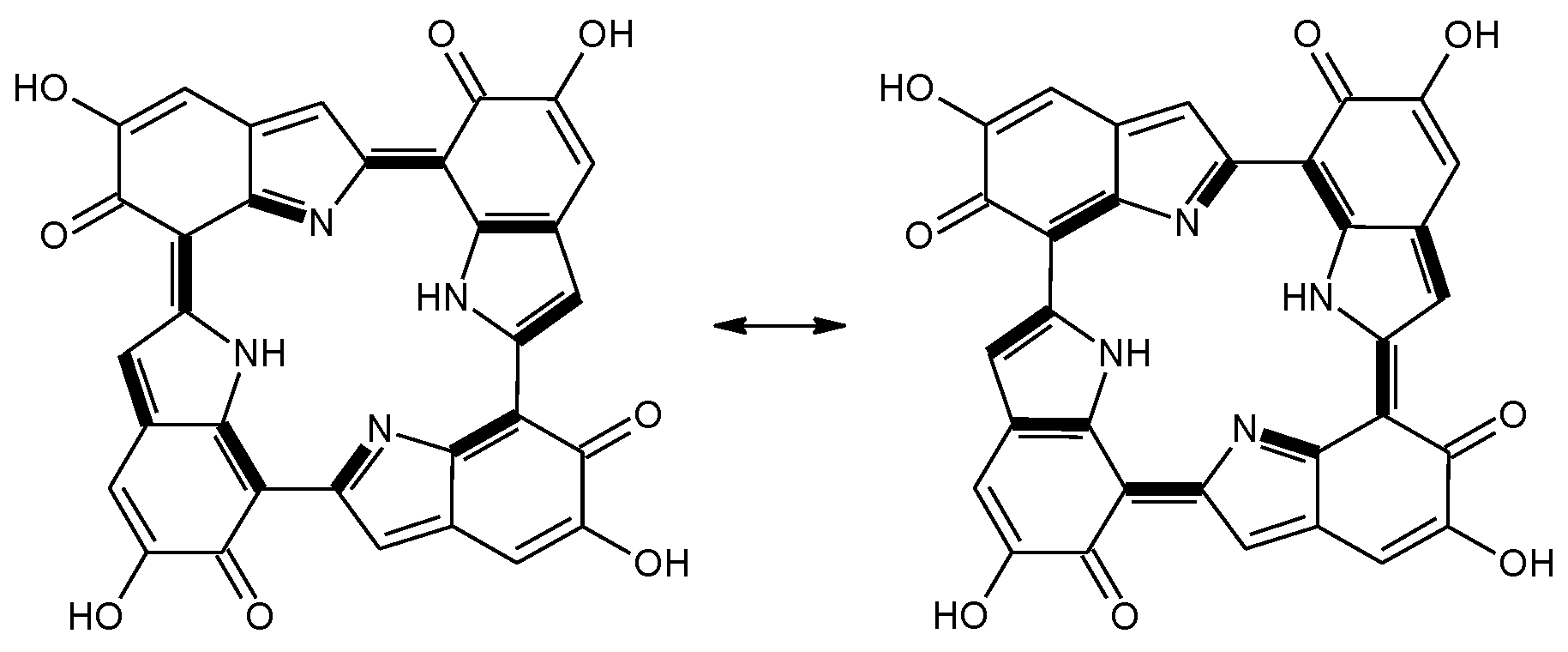
Scheme 2.
a16_b6_c16_d6 as a hybrid between two equivalent contributing structures. The pattern of conjugated double bonds forming an 18-electron aromatic system is highlighted in bold.
Scheme 2.
a16_b6_c16_d6 as a hybrid between two equivalent contributing structures. The pattern of conjugated double bonds forming an 18-electron aromatic system is highlighted in bold.
Figure 9.
Computed UV–Vis spectra of the most significant tautomers/conformers in the six-electron oxidation state. (a) Neutral form in vacuo, a16_b6_c16_d6, C2, conf1. (b) Neutral form in water, a16_b6_c16_d6, C2, conf1.
Figure 9.
Computed UV–Vis spectra of the most significant tautomers/conformers in the six-electron oxidation state. (a) Neutral form in vacuo, a16_b6_c16_d6, C2, conf1. (b) Neutral form in water, a16_b6_c16_d6, C2, conf1.
Figure 10.
Computed IR spectra of the most significant tautomers/conformers in the six-electrons oxidation state. (a) Neutral form in vacuo, a16_b6_c16_d6, C2, conf1. (b) Neutral form in water, a16_b6_c16_d6, C2, conf1.
Figure 10.
Computed IR spectra of the most significant tautomers/conformers in the six-electrons oxidation state. (a) Neutral form in vacuo, a16_b6_c16_d6, C2, conf1. (b) Neutral form in water, a16_b6_c16_d6, C2, conf1.
Figure 11.
Computed UV–Vis spectra of the most significant tautomers/conformers in the eight-electrons oxidation state. (a) Neutral form in vacuo, a16_b56_c16_d56, C2, conf1. (b) Neutral form in water, a16_b56_c16_d56, C2, conf1.
Figure 11.
Computed UV–Vis spectra of the most significant tautomers/conformers in the eight-electrons oxidation state. (a) Neutral form in vacuo, a16_b56_c16_d56, C2, conf1. (b) Neutral form in water, a16_b56_c16_d56, C2, conf1.
Figure 12.
Computed IR spectra of the most significant tautomers/conformers in the eight-electrons oxidation state. (a) Neutral form in vacuo, a16_b56_c16_d56, C2, conf1. (b) Neutral form in water, a16_b56_c16_d56, C2, conf1.
Figure 12.
Computed IR spectra of the most significant tautomers/conformers in the eight-electrons oxidation state. (a) Neutral form in vacuo, a16_b56_c16_d56, C2, conf1. (b) Neutral form in water, a16_b56_c16_d56, C2, conf1.
Figure 13.
A three-electron oxidized building block, which would be needed for the generation of closed-shell tautomers at oxidation levels above KP-8e.
Figure 13.
A three-electron oxidized building block, which would be needed for the generation of closed-shell tautomers at oxidation levels above KP-8e.
Table 1.
KP-Red, neutral form in vacuo.
Table 1.
KP-Red, neutral form in vacuo.
| Tautomer | Conformer a | E (Ha) b | HRRHO (Ha) c | GRRHO (Ha) d |
|---|
![Biomimetics 02 00021 i001]() | S4 | −2050.155247 (0.0) | −2049.641430 (0.0) | −2049.739870 (0.0) |
Table 2.
KP-Red, neutral form in water.
Table 2.
KP-Red, neutral form in water.
| Tautomer | Conformer a | GPCM (Ha) b | HPCM,RRHO (Ha) c | GPCM,RRHO (Ha) d | GSMD (Ha) e | GSMD,RRHO (Ha) f |
|---|
![Biomimetics 02 00021 i002]() | S4 | −2050.191047 (0.0) | −2049.678884 (0.0) | −2049.779144 (0.0) | −2050.220499 (0.0) | −2049.808596 (0.0) |
Table 3.
KP-Red, monoanionic form in water.
Table 4.
KP-1e, neutral form in vacuo.
Table 4.
KP-1e, neutral form in vacuo.
| Tautomer | Conformer a | E (Ha) b | HRRHO (Ha) c | GRRHO (Ha) d |
|---|
![Biomimetics 02 00021 i006]() | C1, conf1 | −2049.532339 (5.5) | −2049.031222 (5.4) | −2049.129769 (5.2) |
![Biomimetics 02 00021 i007]() | C1, conf1 | −2049.541050 (0.0) | −2049.039878 (0.0) | −2049.138067 (0.0) |
Table 5.
KP-1e, neutral form in water.
Table 5.
KP-1e, neutral form in water.
| Tautomer | Conformer a | GPCM (Ha) b | HPCM,RRHO (Ha) c | GPCM,RRHO (Ha) d | GSMD (Ha) e | GSMD,RRHO (Ha) f |
|---|
![Biomimetics 02 00021 i008]() | C1, conf1 | −2049.567367 (4.9) | −2049.067779 (4.8) | −2049.167187 (5.1) | −2049.594711 (4.7) | −2049.194531 (4.9) |
![Biomimetics 02 00021 i009]() | C1, conf1 | −2049.575106 (0.0) | −2049.075489 (0.0) | −2049.175302 (0.0) | −2049.602176 (0.0) | −2049.202372 (0.0) |
Table 6.
KP-1e, monoanionic form in water.
Table 6.
KP-1e, monoanionic form in water.
| Tautomer | Conformer a | GPCM (Ha) b | HPCM,RRHO (Ha) | GPCM,RRHO (Ha) | GSMD (Ha) c | GSMD,RRHO (Ha) d |
|---|
![Biomimetics 02 00021 i010]() | C1, conf1 | −2049.105711 (7.3) | - | - | - | - |
![Biomimetics 02 00021 i011]() | C1, conf1 | −2049.103783 (8.5) | - | - | - | - |
![Biomimetics 02 00021 i012]() | C1, conf1 | −2049.108387 (5.6) | - | - | - | - |
![Biomimetics 02 00021 i013]() | C1, conf1 | −2049.104303 (8.2) | - | - | - | - |
![Biomimetics 02 00021 i014]() | C1, conf1 | −2049.106920 (6.5) | - | - | - | - |
![Biomimetics 02 00021 i015]() | C1, conf1 | −2049.117326 (0.0) | −2048.630807 (0.0) | −2048.728019 (0.0) | −2049.141803 (0.0) | −2048.752496 (0.0) |
Table 7.
KP-2e, neutral form in vacuo.
Table 7.
KP-2e, neutral form in vacuo.
| Tautomer | Conformer a | E (Ha) b | HRRHO (Ha) c | GRRHO (Ha) d |
|---|
![Biomimetics 02 00021 i016]() | C1, conf1 | −2048.922889 (4.2) | −2048.433678 (4.0) | −2048.531488 (3.1) |
![Biomimetics 02 00021 i017]() | C1, conf1 | −2048.929568 (0.0) | −2048.440034 (0.0) | −2048.536382 (0.0) |
Table 8.
KP-2e, neutral form in water.
Table 9.
KP-2e, monoanionic form in water.
Table 10.
KP-4e, neutral form in vacuo.
Table 11.
KP-4e, neutral form in water.
Table 11.
KP-4e, neutral form in water.
| Tautomer | Conformer a | GPCM (Ha) b | HPCM,RRHO (Ha) c | GPCM,RRHO (Ha) d | GSMD (Ha) e | GSMD,RRHO (Ha) f |
|---|
![Biomimetics 02 00021 i032]() | C1, conf1 | −2047.716730 (5.1) | - | - | - | - |
| C2, conf1 | −2047.716589 (5.2) | - | - | - | - |
![Biomimetics 02 00021 i033]() | C1, conf1 | −2047.722869 (1.3) | −2047.260061 (1.3) | −2047.355968 (1.4) | −2047.741845 (1.6) | −2047.374944 (1.5) |
![Biomimetics 02 00021 i034]() | C1, conf1 | −2047.714953 (6.2) | - | - | - | - |
![Biomimetics 02 00021 i035]() | C1, conf1 | −2047.714698 (6.4) | - | - | - | - |
![Biomimetics 02 00021 i036]() | C1, conf1 | −2047.724901 (0.0) | −2047.262199 (0.0) | −2047.358235 (0.0) | −2047.743929 (0.2) | −2047.377263 (0.0) |
![Biomimetics 02 00021 i037]() | C1, conf1 | −2047.722818 (1.3) | −2047.259858 (1.5) | −2047.355768 (1.5) | −2047.742349 (1.2) | −2047.375299 (1.2) |
![Biomimetics 02 00021 i038]() | C1, conf1 | −2047.715313 (6.0) | - | - | - | - |
![Biomimetics 02 00021 i039]() | C1, conf1 | −2047.723719 (0.7) | −2047.260186 (1.3) | −2047.354624 (2.3) | −2047.744321 (0.0) | −2047.375226 (1.3) |
| C2, conf1 | −2047.723546 (0.9) | −2047.260328 (1.2) | −2047.354611 (2.3) | −2047.743800 (0.3) | −2047.374865 (1.5) |
Table 12.
KP-6e, neutral form in vacuo.
Table 12.
KP-6e, neutral form in vacuo.
| Tautomer | Conformer a | E (Ha) b | HRRHO (Ha) c | GRRHO (Ha) d |
|---|
![Biomimetics 02 00021 i040]() | C1, conf1 | −2046.467842 (9.1) | - | - |
![Biomimetics 02 00021 i041]() | C1, conf1 | −2046.482284 (0.0) | - | - |
| C2, conf1 | −2046.482242 (0.0) | −2046.043017 (0.0) | −2046.138676 (0.0) |
Table 13.
KP-6e, neutral form in water.
Table 13.
KP-6e, neutral form in water.
| Tautomer | Conformer a | GPCM (Ha) b | HPCM,RRHO (Ha) c | GPCM,RRHO (Ha) d | GSMD (Ha) e | GSMD,RRHO (Ha) f |
|---|
![Biomimetics 02 00021 i042]() | C1, conf1 | −2046.484846 (7.9) | - | - | - | - |
![Biomimetics 02 00021 i043]() | C1, conf1 | −2046.497644 (−0.1) | - | - | - | - |
| C2, conf1 | −2046.497479 (0.0) | −2046.059837 (0.0) | −2046.154721 (0.0) | −2046.509330 (0.0) | −2046.166572 (0.0) |
Table 14.
Excitations underlying the UV–Vis spectrum of a16_b6_c16_d6, computed at the C2h geometry in vacuo.
Table 14.
Excitations underlying the UV–Vis spectrum of a16_b6_c16_d6, computed at the C2h geometry in vacuo.
| Symmetry | Main CI Contributions (Coefficients) a | λ (nm) (f) b |
|---|
| 1Bu | HOMO−1 → LUMO (−0.12); HOMO → LUMO (0.60); HOMO → LUMO+1 (−0.32) | 1080.6 (0.03) |
| 1Bu | HOMO−4 → LUMO+1 (0.14); HOMO−1 → LUMO (0.16); HOMO−1 → LUMO+1 (−0.15); HOMO → LUMO (0.32); HOMO → LUMO+1 (0.57) | 988.5 (0.06) |
| 1Ag | HOMO−3 → LUMO (0.12); HOMO−2 → LUMO (−0.26); HOMO−2 → LUMO+1 (0.41); HOMO−1 → LUMO+2 (−0.11); HOMO → LUMO+2 (0.49) | 779.9 (0.00) |
| 1Bu | HOMO−2 → LUMO+2 (−0.11); HOMO−1 → LUMO (0.34); HOMO−1 → LUMO+1 (0.59) | 729.8 (0.02) |
| 1Ag | HOMO−2 → LUMO (0.64); HOMO−2 → LUMO+1 (0.22); HOMO → LUMO+2 (0.12) | 714.7 (0.00) |
| 1Ag | HOMO−3 → LUMO (−0.35); HOMO−2 → LUMO+1 (0.46); HOMO−1 → LUMO+2 (−0.14); HOMO → LUMO+2 (−0.35) | 679.8 (0.00) |
| 1Bu | HOMO−4 → LUMO (0.30); HOMO−1 → LUMO (0.51); HOMO−1 → LUMO+1 (−0.31); HOMO → LUMO+1 (−0.20) | 678.2 (0.09) |
| 1Ag | HOMO−3 → LUMO (0.57); HOMO−2 → LUMO+1 (0.20); HOMO → LUMO+2 (−0.34) | 638.0 (0.00) |
| 1Ag | HOMO−3 → LUMO (−0.12); HOMO−3 → LUMO+1 (0.67); HOMO−1 → LUMO+2 (0.14) | 579.1 (0.00) |
| 1Bu | HOMO−5 → LUMO (0.20); HOMO−5 → LUMO+1 (0.17); HOMO−4 → LUMO (0.54); HOMO−3 → LUMO+2 (0.12); HOMO−2 → LUMO+2 (0.24); HOMO−1 → LUMO (−0.21) | 568.8 (0.05) |
| 1Bu | HOMO−5 → LUMO (−0.36); HOMO−5 → LUMO+1 (0.12); HOMO−4 → LUMO+1 (0.49); HOMO−2 → LUMO+2 (0.29); HOMO−1 → LUMO+1 (0.10) | 519.9 (0.04) |
| 1Ag | HOMO−3 → LUMO+1 (−0.16); HOMO−2 → LUMO+1 (0.15);
HOMO−1 → LUMO+2 (0.66) | 506.4 (0.00) |
| 1Bu | HOMO−5 → LUMO+1 (−0.36); HOMO−4 → LUMO (−0.15);
HOMO−4 → LUMO+1 (−0.19); HOMO−2 → LUMO+2 (0.54) | 487.2 (0.10) |
| 1Bu | HOMO−5 → LUMO (−0.42); HOMO−4 → LUMO+1 (−0.20);
HOMO−3 → LUMO+2 (0.51); HOMO−1 → LUMO (−0.10) | 458.4 (0.06) |
| 1Bu | HOMO−5 → LUMO (0.30); HOMO−5 → LUMO+1 (0.35); HOMO−4 → LUMO (−0.27); HOMO−4 → LUMO+1 (0.12); HOMO−3 → LUMO+2 (0.36);
HOMO−2 → LUMO+2 (0.11); HOMO−1 → LUMO (0.15); HOMO → LUMO+1 (−0.13);
HOMO → LUMO+4 (0.13) | 430.7 (0.56) |
| 1Ag | HOMO−5 → LUMO+2 (0.12); HOMO−4 → LUMO+2 (0.68); HOMO−3 → LUMO (−0.11) | 418.5 (0.00) |
| 1Bu | HOMO−5 → LUMO (−0.19); HOMO−5 → LUMO+1 (0.43); HOMO−4 → LUMO+1 (−0.35); HOMO−3 → LUMO+2 (−0.26); HOMO−2 → LUMO+2 (0.15);
HOMO → LUMO (0.16); HOMO → LUMO+4 (0.11); HOMO → LUMO+5 (−0.12) | 418.0 (0.79) |
| 1Bg | HOMO-10 → LUMO (0.24); HOMO-10 → LUMO+1 (0.20); HOMO-9 → LUMO+2 (−0.25); HOMO-8 → LUMO+2 (−0.10); HOMO-6 → LUMO (0.54);
HOMO-6 → LUMO+1 (0.15) | 407.1 (0.00) |
| 1Au | HOMO-10 → LUMO+2 (−0.12); HOMO-9 → LUMO (0.49); HOMO-9 → LUMO+1 (0.14); HOMO-8 → LUMO (0.23); HOMO-8 → LUMO+1 (0.27); HOMO-6 → LUMO+2 (−0.28) | 404.4 (0.00) |
Table 15.
KP-8e, neutral form in vacuo.
Table 15.
KP-8e, neutral form in vacuo.
| Tautomer | Conformer a | E (Ha) b | HRRHO (Ha) c | GRRHO (Ha) d |
|---|
![Biomimetics 02 00021 i044]() | C1, conf1 | −2045.192843 (−0.1) | - | - |
| C1, conf2 | −2045.192633 (0.0) | - | - |
| C2, conf1 | −2045.192633 (0.0) | −2044.778733 (0.0) | −2044.873783 (0.0) |
Table 16.
KP-8e, neutral form in water.
Table 17.
HOMO and LUMO energies, and corresponding HOMO–LUMO gaps, computed at the PBE0/6-311++G(2d,2p)//BE0/6-31+G(d,p) level in vacuo and in water.
Table 17.
HOMO and LUMO energies, and corresponding HOMO–LUMO gaps, computed at the PBE0/6-311++G(2d,2p)//BE0/6-31+G(d,p) level in vacuo and in water.
| Oxidation State | Tautomer | Conformer | EHOMO (eV) | ELUMO (eV) | Gap (eV) | EHOMO (eV) | ELUMO (eV) | Gap (eV) |
|---|
| In Vacuo | In Water |
|---|
| KP-Red | a0_b0_c0_d0 | S4 | −5.074 | −1.308 | 3.767 | −5.363 | −1.499 | 3.864 |
| KP-1e | a5_b0_c0_d0 | C1, conf1 | −5.341 | −1.556 | 3.784 | −5.465 | −1.644 | 3.822 |
| a6_b0_c0_d0 | C1, conf1 | −5.073 | −1.512 | 3.561 | −5.314 | −1.609 | 3.705 |
| KP-2e | a16_b0_c0_d0 | C1, conf1 | −5.269 | −3.449 | 1.821 | −5.480 | −3.696 | 1.783 |
| a6_b6_c0_d0 | C1, conf1 | −5.272 | −3.708 | 1.565 | −5.329 | −3.834 | 1.495 |
| KP-4e | a16_b0_c16_d0 | C2, conf1 | - | - | - | −5.603 | −3.748 | 1.855 |
| a16_b6_c6_d0 | C1, conf1 | −5.506 | −4.114 | 1.392 | −5.443 | −4.086 | 1.357 |
| a6_b16_c6_d0 | C1, conf1 | −5.479 | −4.084 | 1.395 | −5.443 | −4.057 | 1.386 |
| a6_b6_c16_d0 | C1, conf1 | −5.451 | −4.053 | 1.398 | −5.418 | −4.020 | 1.398 |
| a6_b6_c6_d6 | C2, conf1 | −5.479 | −4.371 | 1.108 | −5.280 | −4.145 | 1.135 |
| KP-6e | a16_b6_c16_d6 | C2, conf1 | −5.978 | −4.280 | 1.698 | −5.890 | −4.171 | 1.719 |
| KP-8e | a16_b56_c16_d56 | C2, conf1 | −6.393 | −4.787 | 1.605 | −6.132 | −4.558 | 1.574 |
Table 18.
Reaction free energies computed for several disproportionation processes involving KP.
Table 18.
Reaction free energies computed for several disproportionation processes involving KP.
| Reaction | ∆GRRHO (kcal mol−1)
(In Vacuo) a | ∆GSMD,RRHO (kcal mol−1)
(In Water) b |
|---|
| 2 KP-1e ⇌ KP-Red + KP-2e | −0.1 (−0.04) | 1.6 (0.8) |
| 2 KP-2e ⇌ KP-Red + KP-4e | −0.1 (−0.05) | 0.8 (0.4) |
| 2 KP-4e ⇌ KP-2e + KP-6e | −5.6 (−2.8) | −3.5 (−1.8) |
| 2 KP-6e ⇌ KP-4e + KP-8e | 44.3 (22.1) | 24.8 (12.4) |
| 6 KP-1e ⇌ 5 KP-Red + KP-6e | −6.3 (−1.0) | 2.9 (0.5) |
| 3 KP-2e ⇌ 2 KP-Red + KP-6e | −5.8 (−1.9) | −1.9 (−0.6) |
| 3 KP-4e ⇌ KP-Red + 2 KP-6e | −11.3 (−3.8) | −6.2 (−2.1) |
| 4 KP-6e ⇌ KP-Red + 3 KP-8e | 121.4 (30.4) | 68.2 (17.1) |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}