
Genetic and Epigenetic Interconnections Between Atopic Dermatitis, Allergic Rhinitis, and Rhinitis with Nasal Polyps


 and
and
Abstract

1. Introduction
2. The Genetics and Epigenetics of Atopic Dermatitis (AD)
2.1. Epigenetic Patterns in AD Pathogenesis
2.2. MicroRNAs and Immune Modulation
2.3. Linking Genetics, Epigenetics, and the Environment in AD
3. The Genetics and Epigenetics of Allergic Rhinitis (AR)
3.1. Genetics of AR
3.2. Epigenetics in AR
3.2.1. DNA Methylation in AR
3.2.2. Histone Deacetylation in AR
3.2.3. MicroRNAs in AR
4. Epigenetics in Chronic Sinusitis with Nasal Polyps (CRSwNP)
4.1. DNA Methylation in CRSwNP
4.2. Histone Modification in CRSwNP
4.3. MicroRNAs in CRSwNP
4.4. Matrix Metalloproteinases in CRSwNP
5. Links Between Atopic Dermatitis, Rhinitis, and CRSwNP
5.1. The Connection Between Periostin, IL-13, and IL-4 in AD, AR, and CRSwNP
5.2. The Connection of Metalloproteinases and microRNAS in AD, AR, and CRSwNP
5.3. Links Between Metalloproteinases, the Inflammasome, and Staphylococcus in AD, AR, and CRSwNP
5.3.1. Genetic and Epigenetic Modifications Influencing Inflammasome Activity
5.3.2. Role of Staphylococcus aureus in Modulating Inflammasomes and Metalloproteinases
6. Links Between AD and CRSwNP
6.1. Immunological Overlap and Pathophysiology
6.2. Shared Comorbidities and Risk Factors
6.3. Therapeutic Advances: Biologic Agents and Integrated Management
6.4. Quality of Life and Unmet Needs
7. Conclusions and Key Novel Findings
Author Contributions
Funding
Conflicts of Interest
References
- Lee, H.H.; Patel, K.R.; Singam, V.; Rastogi, S.; Silverberg, J.I. A systematic review and meta-analysis of the prevalence and phenotype of adult-onset atopic dermatitis. J. Am. Acad. Dermatol. 2019, 80, 1526–1532.e1527. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, M.; Vâță, D.; Alexa, A.I.; Trandafir, L.; Patrașcu, A.I.; Hâncu, M.F.; Gheucă-Solovăstru, L. Atopic Dermatitis-Beyond the Skin. Diagnostics 2021, 11, 1553. [Google Scholar] [CrossRef] [PubMed]
- Watson, W.; Kapur, S. Atopic dermatitis. Allergy Asthma Clin. Immunol. 2011, 7 (Suppl. S1), S4. [Google Scholar] [CrossRef] [PubMed]
- Fadadu, R.P.; Abuabara, K.; Balmes, J.R.; Hanifin, J.M.; Wei, M.L. Air Pollution and Atopic Dermatitis, from Molecular Mechanisms to Population-Level Evidence: A Review. Int. J. Environ. Res. Public Health 2023, 20, 2526. [Google Scholar] [CrossRef]
- Grafanaki, K.; Bania, A.; Kaliatsi, E.G.; Vryzaki, E.; Vasilopoulos, Y.; Georgiou, S. The Imprint of Exposome on the Development of Atopic Dermatitis across the Lifespan: A Narrative Review. J. Clin. Med. 2023, 12, 2180. [Google Scholar] [CrossRef]
- Spergel, J.M. From atopic dermatitis to asthma: The atopic march. Ann. Allergy Asthma Immunol. 2010, 105, 99–117. [Google Scholar] [CrossRef]
- Bantz, S.K.; Zhu, Z.; Zheng, T. The Atopic March: Progression from Atopic Dermatitis to Allergic Rhinitis and Asthma. J. Clin. Cell. Immunol. 2014, 5, 202. [Google Scholar] [CrossRef]
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic dermatitis. Nat. Rev. Dis. Primers 2018, 4, 1. [Google Scholar] [CrossRef]
- Drislane, C.; Irvine, A.D. The role of filaggrin in atopic dermatitis and allergic disease. Ann. Allergy Asthma Immunol. 2020, 124, 36–43. [Google Scholar] [CrossRef]
- Weidinger, S.; O’Sullivan, M.; Illig, T.; Baurecht, H.; Depner, M.; Rodriguez, E.; Ruether, A.; Klopp, N.; Vogelberg, C.; Weiland, S.K.; et al. Filaggrin mutations, atopic eczema, hay fever, and asthma in children. J. Allergy Clin. Immunol. 2008, 121, 1203–1209.e1201. [Google Scholar] [CrossRef]
- Moosbrugger-Martinz, V.; Leprince, C.; Méchin, M.C.; Simon, M.; Blunder, S.; Gruber, R.; Dubrac, S. Revisiting the Roles of Filaggrin in Atopic Dermatitis. Int. J. Mol. Sci. 2022, 23, 5318. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Vandeplas, G.; Huynh, T.M.T.; Joish, V.N.; Mannent, L.; Tomassen, P.; Van Zele, T.; Cardell, L.O.; Arebro, J.; Olze, H.; et al. The Global Allergy and Asthma European Network (GALEN rhinosinusitis cohort: A large European cross-sectional study of chronic rhinosinusitis patients with and without nasal polyps. Rhinology 2019, 57, 32–42. [Google Scholar] [CrossRef] [PubMed]
- González-Mendoza, T.; Bedolla-Barajas, M.; Bedolla-Pulido, T.R.; Morales-Romero, J.; Pulido-Guillén, N.A.; Lerma-Partida, S.; Meza-López, C. The prevalence of allergic rhinitis and atopic dermatitis in late adolescents differs according to their gender. Rev. Alerg. Mex. 2019, 66, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, A.; Thomsen, S.F.; Lacour, J.P.; Jaumont, X.; Lazarewicz, S. Targeting immunoglobulin E in atopic dermatitis: A review of the existing evidence. World Allergy Organ. J. 2021, 14, 100519. [Google Scholar] [CrossRef]
- Simpson, E.L.; De Benedetto, A.; Boguniewicz, M.; Ong, P.Y.; Lussier, S.; Villarreal, M.; Schneider, L.C.; Paller, A.S.; Guttman-Yassky, E.; Hanifin, J.M.; et al. Phenotypic and Endotypic Determinants of Atopic Dermatitis Severity From the Atopic Dermatitis Research Network (ADRN) Registry. J. Allergy Clin. Immunol. Pract. 2023, 11, 2504–2515. [Google Scholar] [CrossRef]
- Tsuge, M.; Ikeda, M.; Matsumoto, N.; Yorifuji, T.; Tsukahara, H. Current Insights into Atopic March. Children 2021, 8, 1067. [Google Scholar] [CrossRef]
- Paller, A.S.; Spergel, J.M.; Mina-Osorio, P.; Irvine, A.D. The atopic march and atopic multimorbidity: Many trajectories, many pathways. J. Allergy Clin. Immunol. 2019, 143, 46–55. [Google Scholar] [CrossRef]
- Shankar, A.; McAlees, J.W.; Lewkowich, I.P. Modulation of IL-4/IL-13 cytokine signaling in the context of allergic disease. J. Allergy Clin. Immunol. 2022, 150, 266–276. [Google Scholar] [CrossRef]
- David, M.; Ford, D.; Bertoglio, J.; Maizel, A.L.; Pierre, J. Induction of the IL-13 receptor alpha2-chain by IL-4 and IL-13 in human keratinocytes: Involvement of STAT6, ERK and p38 MAPK pathways. Oncogene 2001, 20, 6660–6668. [Google Scholar] [CrossRef]
- Tian, J.; Zhang, D.; Yang, Y.; Huang, Y.; Wang, L.; Yao, X.; Lu, Q. Global epidemiology of atopic dermatitis: A comprehensive systematic analysis and modelling study. Br. J. Dermatol. 2023, 190, 55–61. [Google Scholar] [CrossRef]
- Grafanaki, K.; Antonatos, C.; Maniatis, A.; Petropoulou, A.; Vryzaki, E.; Vasilopoulos, Y.; Georgiou, S.; Gregoriou, S. Intrinsic Effects of Exposome in Atopic Dermatitis: Genomics, Epigenomics and Regulatory Layers. J. Clin. Med. 2023, 12, 4000. [Google Scholar] [CrossRef] [PubMed]
- Potaczek, D.P.; Alashkar Alhamwe, B.; Miethe, S.; Garn, H. Epigenetic Mechanisms in Allergy Development and Prevention. Handb. Exp. Pharmacol. 2022, 268, 331–357. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.D.; de Guzman Strong, C. Current understanding of epigenetics in atopic dermatitis. Exp. Dermatol. 2021, 30, 1150–1155. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, E.; Baurecht, H.; Wahn, A.F.; Kretschmer, A.; Hotze, M.; Zeilinger, S.; Klopp, N.; Illig, T.; Schramm, K.; Prokisch, H.; et al. An integrated epigenetic and transcriptomic analysis reveals distinct tissue-specific patterns of DNA methylation associated with atopic dermatitis. J. Investig. Dermatol. 2014, 134, 1873–1883. [Google Scholar] [CrossRef]
- Acevedo, N.; Benfeitas, R.; Katayama, S.; Bruhn, S.; Andersson, A.; Wikberg, G.; Lundeberg, L.; Lindvall, J.M.; Greco, D.; Kere, J.; et al. Epigenetic alterations in skin homing CD4+CLA+ T cells of atopic dermatitis patients. Sci. Rep. 2020, 10, 18020. [Google Scholar] [CrossRef]
- Facheris, P.; Jeffery, J.; Del Duca, E.; Guttman-Yassky, E. The translational revolution in atopic dermatitis: The paradigm shift from pathogenesis to treatment. Cell. Mol. Immunol. 2023, 20, 448–474. [Google Scholar] [CrossRef]
- Martin, M.J.; Estravís, M.; García-Sánchez, A.; Dávila, I.; Isidoro-García, M.; Sanz, C. Genetics and Epigenetics of Atopic Dermatitis: An Updated Systematic Review. Genes 2020, 11, 442. [Google Scholar] [CrossRef]
- Chen, L.; Qi, X.; Wang, J.; Yin, J.; Sun, P.; Sun, Y.; Wu, Y.; Zhang, L.; Gao, X. Identification of novel candidate genes and predicted miRNAs in atopic dermatitis patients by bioinformatic methods. Sci. Rep. 2022, 12, 22067. [Google Scholar] [CrossRef]
- Eapen, A.A.; Parameswaran, S.; Forney, C.; Edsall, L.E.; Miller, D.; Donmez, O.; Dunn, K.; Lu, X.; Granitto, M.; Rowden, H.; et al. Epigenetic and transcriptional dysregulation in CD4+ T cells in patients with atopic dermatitis. PLoS Genet. 2022, 18, e1009973. [Google Scholar] [CrossRef]
- Fokkens, W.J.; Lund, V.J.; Hopkins, C.; Hellings, P.W.; Kern, R.; Reitsma, S.; Toppila-Salmi, S.; Bernal-Sprekelsen, M.; Mullol, J.; Alobid, I.; et al. European Position Paper on Rhinosinusitis and Nasal Polyps 2020. Rhinology 2020, 58, 1–464. [Google Scholar] [CrossRef]
- Liu, T.; Sun, Y.; Bai, W. The Role of Epigenetics in the Chronic Sinusitis with Nasal Polyp. Curr. Allergy Asthma Rep. 2020, 21, 1. [Google Scholar] [CrossRef] [PubMed]
- Fokkens, W.J. EPOS2020: A major step forward. Rhinology 2020, 58, 1. [Google Scholar] [CrossRef] [PubMed]
- Johansson, L.; Akerlund, A.; Holmberg, K.; Melén, I.; Bende, M. Prevalence of nasal polyps in adults: The Skövde population-based study. Ann. Otol. Rhinol. Laryngol. 2003, 112, 625–629. [Google Scholar] [CrossRef] [PubMed]
- Ogulur, I.; Mitamura, Y.; Yazici, D.; Pat, Y.; Ardicli, S.; Li, M.; D’Avino, P.; Beha, C.; Babayev, H.; Zhao, B.; et al. Type 2 immunity in allergic diseases. Cell. Mol. Immunol. 2025, 22, 211–242. [Google Scholar] [CrossRef]
- Bélanger, É.; Madore, A.M.; Boucher-Lafleur, A.M.; Simon, M.M.; Kwan, T.; Pastinen, T.; Laprise, C. Eosinophil microRNAs Play a Regulatory Role in Allergic Diseases Included in the Atopic March. Int. J. Mol. Sci. 2020, 21, 9011. [Google Scholar] [CrossRef]
- Kaminsky, Z.A.; Tang, T.; Wang, S.C.; Ptak, C.; Oh, G.H.; Wong, A.H.; Feldcamp, L.A.; Virtanen, C.; Halfvarson, J.; Tysk, C.; et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet. 2009, 41, 240–245. [Google Scholar] [CrossRef]
- Hannon, E.; Knox, O.; Sugden, K.; Burrage, J.; Wong, C.C.Y.; Belsky, D.W.; Corcoran, D.L.; Arseneault, L.; Moffitt, T.E.; Caspi, A.; et al. Characterizing genetic and environmental influences on variable DNA methylation using monozygotic and dizygotic twins. PLoS Genet. 2018, 14, e1007544. [Google Scholar] [CrossRef]
- Li, J.; Qiu, C.Y.; Tao, Y.J.; Cheng, L. Epigenetic modifications in chronic rhinosinusitis with and without nasal polyps. Front. Genet. 2022, 13, 1089647. [Google Scholar] [CrossRef]
- Løset, M.; Brown, S.J.; Saunes, M.; Hveem, K. Genetics of Atopic Dermatitis: From DNA Sequence to Clinical Relevance. Dermatology 2019, 235, 355–364. [Google Scholar] [CrossRef]
- Liang, Y.; Chang, C.; Lu, Q. The Genetics and Epigenetics of Atopic Dermatitis-Filaggrin and Other Polymorphisms. Clin. Rev. Allergy Immunol. 2016, 51, 315–328. [Google Scholar] [CrossRef]
- Mu, Z.; Zhang, J. The Role of Genetics, the Environment, and Epigenetics in Atopic Dermatitis. Adv. Exp. Med. Biol. 2020, 1253, 107–140. [Google Scholar] [CrossRef] [PubMed]
- Nedoszytko, B.; Reszka, E.; Gutowska-Owsiak, D.; Trzeciak, M.; Lange, M.; Jarczak, J.; Niedoszytko, M.; Jablonska, E.; Romantowski, J.; Strapagiel, D.; et al. Genetic and Epigenetic Aspects of Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 6484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, H.C.; Feng, C.; Yan, M. Analysis of the Association of Polymorphisms rs5743708 in TLR2 and rs4986790 in TLR4 with Atopic Dermatitis Risk. Immunol. Investig. 2019, 48, 169–180. [Google Scholar] [CrossRef]
- Trzeciak, M.; Wesserling, M.; Bandurski, T.; Glen, J.; Nowicki, R.; Pawelczyk, T. Association of a Single Nucleotide Polymorphism in a Late Cornified Envelope-like Proline-rich 1 Gene (LELP1) with Atopic Dermatitis. Acta Derm. Venereol. 2016, 96, 459–463. [Google Scholar] [CrossRef]
- Brown, S.J.; Elias, M.S.; Bradley, M. Genetics in Atopic Dermatitis: Historical Perspective and Future Prospects. Acta Derm. Venereol. 2020, 100, adv00163. [Google Scholar] [CrossRef]
- Stemmler, S.; Hoffjan, S. Trying to understand the genetics of atopic dermatitis. Mol. Cell. Probes 2016, 30, 374–385. [Google Scholar] [CrossRef]
- Bonamonte, D.; Filoni, A.; Vestita, M.; Romita, P.; Foti, C.; Angelini, G. The Role of the Environmental Risk Factors in the Pathogenesis and Clinical Outcome of Atopic Dermatitis. BioMed Res. Int. 2019, 2019, 2450605. [Google Scholar] [CrossRef]
- Hoffjan, S.; Stemmler, S. Unravelling the complex genetic background of atopic dermatitis: From genetic association results towards novel therapeutic strategies. Arch. Dermatol. Res. 2015, 307, 659–670. [Google Scholar] [CrossRef]
- Tsuji, G.; Hashimoto-Hachiya, A.; Kiyomatsu-Oda, M.; Takemura, M.; Ohno, F.; Ito, T.; Morino-Koga, S.; Mitoma, C.; Nakahara, T.; Uchi, H.; et al. Aryl hydrocarbon receptor activation restores filaggrin expression via OVOL1 in atopic dermatitis. Cell Death Dis. 2017, 8, e2931. [Google Scholar] [CrossRef]
- Zablotna, M.; Sobjanek, M.; Glen, J.; Niedoszytko, M.; Wilkowska, A.; Roszkiewicz, J.; Nedoszytko, B. Association between the -1154 G/A promoter polymorphism of the vascular endothelial growth factor gene and atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2010, 24, 91–92. [Google Scholar] [CrossRef]
- Trzeciak, M.; Gleń, J.; Roszkiewicz, J.; Nedoszytko, B. Association of single nucleotide polymorphism of interleukin-18 with atopic dermatitis. J. Eur. Acad. Dermatol. Venereol. 2010, 24, 78–79. [Google Scholar] [CrossRef] [PubMed]
- Trzeciak, M.; Gleń, J.; Bandurski, T.; Sokołowska-Wojdyło, M.; Wilkowska, A.; Roszkiewicz, J. Relationship between serum levels of interleukin-18, IgE and disease severity in patients with atopic dermatitis. Clin. Exp. Dermatol. 2011, 36, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Nedoszytko, B.; Niedoszytko, M.; Lange, M.; van Doormaal, J.; Gleń, J.; Zabłotna, M.; Renke, J.; Vales, A.; Buljubasic, F.; Jassem, E.; et al. Interleukin-13 promoter gene polymorphism -1112C/T is associated with the systemic form of mastocytosis. Allergy 2009, 64, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Wilkowska, A.; Gleń, J.; Zabłotna, M.; Trzeciak, M.; Ryduchowska, M.; Sobjanek, M.; Nedoszytko, B.; Nowicki, R.; Sokołowska-Wojdyło, M. The association of GM-CSF-677A/C promoter gene polymorphism with the occurrence and severity of atopic dermatitis in a Polish population. Int. J. Dermatol. 2014, 53, e172–e174. [Google Scholar] [CrossRef]
- Totté, J.E.E.; van der Feltz, W.T.; Hennekam, M.; van Belkum, A.; van Zuuren, E.J.; Pasmans, S.G.M.A. Prevalence and odds of Staphylococcus aureus carriage in atopic dermatitis: A systematic review and meta-analysis. Br. J. Dermatol. 2016, 175, 687–695. [Google Scholar] [CrossRef]
- Rebane, A.; Akdis, C.A. MicroRNAs: Essential players in the regulation of inflammation. J. Allergy Clin. Immunol. 2013, 132, 15–26. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, Y.; Zhang, H.; Hu, L.; Liu, J.; Wang, L.; Wang, T.; Zhang, H.; Cong, L.; Wang, Q. Pathogenesis of allergic diseases and implications for therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 138. [Google Scholar] [CrossRef]
- Yazd, N.K.K.; Patel, R.R.; Dellavalle, R.P.; Dunnick, C.A. Genetic Risk Factors for Development of Atopic Dermatitis: A Systematic Review. Curr. Dermatol. Rep. 2017, 6, 297–308. [Google Scholar] [CrossRef]
- Bin, L.; Leung, D.Y. Genetic and epigenetic studies of atopic dermatitis. Allergy Asthma Clin. Immunol. 2016, 12, 52. [Google Scholar] [CrossRef]
- Furue, K.; Ito, T.; Tsuji, G.; Ulzii, D.; Vu, Y.H.; Kido-Nakahara, M.; Nakahara, T.; Furue, M. The IL-13-OVOL1-FLG axis in atopic dermatitis. Immunology 2019, 158, 281–286. [Google Scholar] [CrossRef]
- Dong, S.; Li, D.; Shi, D. Skin barrier-inflammatory pathway is a driver of the psoriasis-atopic dermatitis transition. Front. Med. 2024, 11, 1335551. [Google Scholar] [CrossRef]
- Paternoster, L.; Standl, M.; Chen, C.M.; Ramasamy, A.; Bønnelykke, K.; Duijts, L.; Ferreira, M.A.; Alves, A.C.; Thyssen, J.P.; Albrecht, E.; et al. Meta-analysis of genome-wide association studies identifies three new risk loci for atopic dermatitis. Nat. Genet. 2011, 44, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.D.; Xiao, F.L.; Li, Y.; Zhou, W.M.; Tang, H.Y.; Tang, X.F.; Zhang, H.; Schaarschmidt, H.; Zuo, X.B.; Foelster-Holst, R.; et al. Genome-wide association study identifies two new susceptibility loci for atopic dermatitis in the Chinese Han population. Nat. Genet. 2011, 43, 690–694. [Google Scholar] [CrossRef]
- Weidinger, S.; Willis-Owen, S.A.; Kamatani, Y.; Baurecht, H.; Morar, N.; Liang, L.; Edser, P.; Street, T.; Rodriguez, E.; O’Regan, G.M.; et al. A genome-wide association study of atopic dermatitis identifies loci with overlapping effects on asthma and psoriasis. Hum. Mol. Genet. 2013, 22, 4841–4856. [Google Scholar] [CrossRef]
- Hirota, T.; Takahashi, A.; Kubo, M.; Tsunoda, T.; Tomita, K.; Sakashita, M.; Yamada, T.; Fujieda, S.; Tanaka, S.; Doi, S.; et al. Genome-wide association study identifies eight new susceptibility loci for atopic dermatitis in the Japanese population. Nat. Genet. 2012, 44, 1222–1226. [Google Scholar] [CrossRef]
- Paternoster, L.; Standl, M.; Waage, J.; Baurecht, H.; Hotze, M.; Strachan, D.P.; Curtin, J.A.; Bønnelykke, K.; Tian, C.; Takahashi, A.; et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat. Genet. 2015, 47, 1449–1456. [Google Scholar] [CrossRef]
- Esparza-Gordillo, J.; Schaarschmidt, H.; Liang, L.; Cookson, W.; Bauerfeind, A.; Lee-Kirsch, M.A.; Nemat, K.; Henderson, J.; Paternoster, L.; Harper, J.I.; et al. A functional IL-6 receptor (IL6R) variant is a risk factor for persistent atopic dermatitis. J. Allergy Clin. Immunol. 2013, 132, 371–377. [Google Scholar] [CrossRef]
- Antonatos, C.; Mitsoudi, D.; Pontikas, A.; Akritidis, A.; Xiropotamos, P.; Georgakilas, G.K.; Georgiou, S.; Tsiogka, A.; Gregoriou, S.; Grafanaki, K.; et al. Transcriptome-wide analyses delineate the genetic architecture of expression variation in atopic dermatitis. Hum. Genet. Genom. Adv. 2025, 6, 100422. [Google Scholar] [CrossRef]
- Purwar, R.; Kraus, M.; Werfel, T.; Wittmann, M. Modulation of keratinocyte-derived MMP-9 by IL-13: A possible role for the pathogenesis of epidermal inflammation. J. Investig. Dermatol. 2008, 128, 59–66. [Google Scholar] [CrossRef]
- Kobayashi, T. Is matrix metalloproteinase-9 a culprit involved in dermatitis? Increased expression of gelatinolytic activity in allergic contact dermatitis. Contact Dermat. 2011, 64, 171–172. [Google Scholar] [CrossRef]
- Ziyab, A.H.; Karmaus, W.; Holloway, J.W.; Zhang, H.; Ewart, S.; Arshad, S.H. DNA methylation of the filaggrin gene adds to the risk of eczema associated with loss-of-function variants. J. Eur. Acad. Dermatol. Venereol. 2013, 27, e420–e423. [Google Scholar] [CrossRef]
- Boorgula, M.P.; Taub, M.A.; Rafaels, N.; Daya, M.; Campbell, M.; Chavan, S.; Shetty, A.; Cheadle, C.; Barkataki, S.; Fan, J.; et al. Replicated methylation changes associated with eczema herpeticum and allergic response. Clin. Epigenet. 2019, 11, 122. [Google Scholar] [CrossRef]
- Luo, Y.; Zhou, B.; Zhao, M.; Tang, J.; Lu, Q. Promoter demethylation contributes to TSLP overexpression in skin lesions of patients with atopic dermatitis. Clin. Exp. Dermatol. 2014, 39, 48–53. [Google Scholar] [CrossRef]
- Harb, H.; Renz, H. Update on epigenetics in allergic disease. J. Allergy Clin. Immunol. 2015, 135, 15–24. [Google Scholar] [CrossRef]
- Botchkarev, V.A.; Gdula, M.R.; Mardaryev, A.N.; Sharov, A.A.; Fessing, M.Y. Epigenetic regulation of gene expression in keratinocytes. J. Investig. Dermatol. 2012, 132, 2505–2521. [Google Scholar] [CrossRef]
- Potaczek, D.P.; Harb, H.; Michel, S.; Alhamwe, B.A.; Renz, H.; Tost, J. Epigenetics and allergy: From basic mechanisms to clinical applications. Epigenomics 2017, 9, 539–571. [Google Scholar] [CrossRef]
- Vaher, H.; Runnel, T.; Urgard, E.; Aab, A.; Carreras Badosa, G.; Maslovskaja, J.; Abram, K.; Raam, L.; Kaldvee, B.; Annilo, T.; et al. miR-10a-5p is increased in atopic dermatitis and has capacity to inhibit keratinocyte proliferation. Allergy 2019, 74, 2146–2156. [Google Scholar] [CrossRef]
- Lee, A.Y. The Role of MicroRNAs in Epidermal Barrier. Int. J. Mol. Sci. 2020, 21, 5781. [Google Scholar] [CrossRef]
- Khosrojerdi, M.; Azad, F.J.; Yadegari, Y.; Ahanchian, H.; Azimian, A. The role of microRNAs in atopic dermatitis. Non-Coding RNA Res. 2024, 9, 1033–1039. [Google Scholar] [CrossRef]
- Sonkoly, E.; Janson, P.; Majuri, M.L.; Savinko, T.; Fyhrquist, N.; Eidsmo, L.; Xu, N.; Meisgen, F.; Wei, T.; Bradley, M.; et al. MiR-155 is overexpressed in patients with atopic dermatitis and modulates T-cell proliferative responses by targeting cytotoxic T lymphocyte-associated antigen 4. J. Allergy Clin. Immunol. 2010, 126, 581–589.e20. [Google Scholar] [CrossRef]
- Quinn, S.R.; Mangan, N.E.; Caffrey, B.E.; Gantier, M.P.; Williams, B.R.; Hertzog, P.J.; McCoy, C.E.; O’Neill, L.A. The role of Ets2 transcription factor in the induction of microRNA-155 (miR-155) by lipopolysaccharide and its targeting by interleukin-10. J. Biol. Chem. 2014, 289, 4316–4325. [Google Scholar] [CrossRef]
- Jia, H.Z.; Liu, S.L.; Zou, Y.F.; Chen, X.F.; Yu, L.; Wan, J.; Zhang, H.Y.; Chen, Q.; Xiong, Y.; Yu, B.; et al. MicroRNA-223 is involved in the pathogenesis of atopic dermatitis by affecting histamine-N-methyltransferase. Cell. Mol. Biol. 2018, 64, 103–107. [Google Scholar] [CrossRef]
- Boldin, M.P.; Baltimore, D. MicroRNAs, new effectors and regulators of NF-κB. Immunol. Rev. 2012, 246, 205–220. [Google Scholar] [CrossRef]
- Yang, Z.; Zeng, B.; Wang, C.; Wang, H.; Huang, P.; Pan, Y. MicroRNA-124 alleviates chronic skin inflammation in atopic eczema via suppressing innate immune responses in keratinocytes. Cell. Immunol. 2017, 319, 53–60. [Google Scholar] [CrossRef]
- Liew, W.C.; Sundaram, G.M.; Quah, S.; Lum, G.G.; Tan, J.S.L.; Ramalingam, R.; Common, J.E.A.; Tang, M.B.Y.; Lane, E.B.; Thng, S.T.G.; et al. Belinostat resolves skin barrier defects in atopic dermatitis by targeting the dysregulated miR-335:SOX6 axis. J. Allergy Clin. Immunol. 2020, 146, 606–620.e612. [Google Scholar] [CrossRef]
- Zu, Y.; Chen, X.F.; Li, Q.; Zhang, S.T. CYT387, a Novel JAK2 Inhibitor, Suppresses IL-13-Induced Epidermal Barrier Dysfunction Via miR-143 Targeting IL-13Rα1 and STAT3. Biochem. Genet. 2021, 59, 531–546. [Google Scholar] [CrossRef]
- Rebane, A.; Runnel, T.; Aab, A.; Maslovskaja, J.; Rückert, B.; Zimmermann, M.; Plaas, M.; Kärner, J.; Treis, A.; Pihlap, M.; et al. MicroRNA-146a alleviates chronic skin inflammation in atopic dermatitis through suppression of innate immune responses in keratinocytes. J. Allergy Clin. Immunol. 2014, 134, 836–847.e811. [Google Scholar] [CrossRef]
- Choi, B.Y.; Han, M.; Kwak, J.W.; Kim, T.H. Genetics and Epigenetics in Allergic Rhinitis. Genes 2021, 12, 2004. [Google Scholar] [CrossRef]
- Andiappan, A.K.; de Wang, Y.; Anantharaman, R.; Parate, P.N.; Suri, B.K.; Low, H.Q.; Li, Y.; Zhao, W.; Castagnoli, P.; Liu, J.; et al. Genome-wide association study for atopy and allergic rhinitis in a Singapore Chinese population. PLoS ONE 2011, 6, e19719. [Google Scholar] [CrossRef]
- Campbell, A.; Chanal, I.; Czarlewski, W.; Michel, F.B.; Bousquet, J. Reduction of soluble ICAM-1 levels in nasal secretion by H1-blockers in seasonal allergic rhinitis. Allergy 1997, 52, 1022–1025. [Google Scholar] [CrossRef]
- Jiang, F.; Yan, A. IL-4 rs2243250 polymorphism associated with susceptibility to allergic rhinitis: A meta-analysis. Biosci. Rep. 2021, 41, BSR20210522. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, Y. Association Between Vitamin D Receptor Gene Polymorphism rs2228570 and Allergic Rhinitis. Pharmgenom. Pers. Med. 2020, 13, 327–335. [Google Scholar] [CrossRef]
- Wei, X.; Zhang, Y.; Fu, Z.; Zhang, L. The association between polymorphisms in the MRPL4 and TNF-α genes and susceptibility to allergic rhinitis. PLoS ONE 2013, 8, e57981. [Google Scholar] [CrossRef]
- León, B.; Ballesteros-Tato, A.; Browning, J.L.; Dunn, R.; Randall, T.D.; Lund, F.E. Regulation of T(H)2 development by CXCR5+ dendritic cells and lymphotoxin-expressing B cells. Nat. Immunol. 2012, 13, 681–690. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Shinnakasu, R.; Inoue, T.; Kometani, K.; Moriyama, S.; Adachi, Y.; Nakayama, M.; Takahashi, Y.; Fukuyama, H.; Okada, T.; Kurosaki, T. Regulated selection of germinal-center cells into the memory B cell compartment. Nat. Immunol. 2016, 17, 861–869. [Google Scholar] [CrossRef]
- Roychoudhuri, R.; Clever, D.; Li, P.; Wakabayashi, Y.; Quinn, K.M.; Klebanoff, C.A.; Ji, Y.; Sukumar, M.; Eil, R.L.; Yu, Z.; et al. BACH2 regulates CD8+ T cell differentiation by controlling access of AP-1 factors to enhancers. Nat. Immunol. 2016, 17, 851–860. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef]
- Kassmeier, M.D.; Mondal, K.; Palmer, V.L.; Raval, P.; Kumar, S.; Perry, G.A.; Anderson, D.K.; Ciborowski, P.; Jackson, S.; Xiong, Y.; et al. VprBP binds full-length RAG1 and is required for B-cell development and V(D)J recombination fidelity. EMBO J. 2012, 31, 945–958. [Google Scholar] [CrossRef]
- Hamblet, C.E.; Makowski, S.L.; Tritapoe, J.M.; Pomerantz, J.L. NK Cell Maturation and Cytotoxicity Are Controlled by the Intramembrane Aspartyl Protease SPPL3. J. Immunol. 2016, 196, 2614–2626. [Google Scholar] [CrossRef]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef]
- Gatsounia, A.; Schinas, G.; Danielides, G.; Grafanaki, K.; Mastronikolis, N.; Stathopoulos, C.; Lygeros, S. Epigenetic Mechanisms in CRSwNP: The Role of MicroRNAs as Potential Biomarkers and Therapeutic Targets. Curr. Issues Mol. Biol. 2025, 47, 114. [Google Scholar] [CrossRef]
- Nestor, C.E.; Barrenäs, F.; Wang, H.; Lentini, A.; Zhang, H.; Bruhn, S.; Jörnsten, R.; Langston, M.A.; Rogers, G.; Gustafsson, M.; et al. DNA methylation changes separate allergic patients from healthy controls and may reflect altered CD4+ T-cell population structure. PLoS Genet. 2014, 10, e1004059. [Google Scholar] [CrossRef]
- Swamy, R.S.; Reshamwala, N.; Hunter, T.; Vissamsetti, S.; Santos, C.B.; Baroody, F.M.; Hwang, P.H.; Hoyte, E.G.; Garcia, M.A.; Nadeau, K.C. Epigenetic modifications and improved regulatory T-cell function in subjects undergoing dual sublingual immunotherapy. J. Allergy Clin. Immunol. 2012, 130, 215–224.e217. [Google Scholar] [CrossRef]
- Bayrak Degirmenci, P.; Aksun, S.; Altin, Z.; Bilgir, F.; Arslan, I.B.; Colak, H.; Ural, B.; Solakoglu Kahraman, D.; Diniz, G.; Ozdemir, B.; et al. Allergic Rhinitis and Its Relationship with IL-10, IL-17, TGF-β, IFN-γ, IL 22, and IL-35. Dis. Markers 2018, 2018, 9131432. [Google Scholar] [CrossRef]
- Li, J.Y.; Zhang, Y.; Lin, X.P.; Ruan, Y.; Wang, Y.; Wang, C.S.; Zhang, L. Association between DNA hypomethylation at IL13 gene and allergic rhinitis in house dust mite-sensitized subjects. Clin. Exp. Allergy 2016, 46, 298–307. [Google Scholar] [CrossRef]
- Sarnowski, C.; Laprise, C.; Malerba, G.; Moffatt, M.F.; Dizier, M.H.; Morin, A.; Vincent, Q.B.; Rohde, K.; Esparza-Gordillo, J.; Margaritte-Jeannin, P.; et al. DNA methylation within melatonin receptor 1A (MTNR1A) mediates paternally transmitted genetic variant effect on asthma plus rhinitis. J. Allergy Clin. Immunol. 2016, 138, 748–753. [Google Scholar] [CrossRef]
- Alaskhar Alhamwe, B.; Khalaila, R.; Wolf, J.; von Bülow, V.; Harb, H.; Alhamdan, F.; Hii, C.S.; Prescott, S.L.; Ferrante, A.; Renz, H.; et al. Histone modifications and their role in epigenetics of atopy and allergic diseases. Allergy Asthma Clin. Immunol. 2018, 14, 39. [Google Scholar] [CrossRef]
- Fields, P.E.; Kim, S.T.; Flavell, R.A. Cutting edge: Changes in histone acetylation at the IL-4 and IFN-gamma loci accompany Th1/Th2 differentiation. J. Immunol. 2002, 169, 647–650. [Google Scholar] [CrossRef]
- Jiang, J.; Liu, J.Q.; Li, J.; Li, M.; Chen, H.B.; Yan, H.; Mo, L.H.; Qiu, S.Q.; Liu, Z.G.; Yang, P.C. Trek1 contributes to maintaining nasal epithelial barrier integrity. Sci. Rep. 2015, 5, 9191. [Google Scholar] [CrossRef]
- Wang, Y.; Lv, L.; Zang, H.; Gao, Z.; Zhang, F.; Wang, X.; Zhou, X. Regulation of Trek1 expression in nasal mucosa with allergic rhinitis by specific immunotherapy. Cell Biochem. Funct. 2015, 33, 23–28. [Google Scholar] [CrossRef]
- Cho, J.S.; Kang, J.H.; Han, I.H.; Um, J.Y.; Park, I.H.; Lee, S.H.; Lee, H.M. Antiallergic Effects of Trichostatin A in a Murine Model of Allergic Rhinitis. Clin. Exp. Otorhinolaryngol. 2015, 8, 243–249. [Google Scholar] [CrossRef]
- Chen, R.F.; Huang, H.C.; Ou, C.Y.; Hsu, T.Y.; Chuang, H.; Chang, J.C.; Wang, L.; Kuo, H.C.; Yang, K.D. MicroRNA-21 expression in neonatal blood associated with antenatal immunoglobulin E production and development of allergic rhinitis. Clin. Exp. Allergy 2010, 40, 1482–1490. [Google Scholar] [CrossRef]
- Liu, W.; Zeng, Q.; Luo, R. Correlation between Serum Osteopontin and miR-181a Levels in Allergic Rhinitis Children. Mediators Inflamm. 2016, 2016, 9471215. [Google Scholar] [CrossRef]
- Korde, A.; Ahangari, F.; Haslip, M.; Zhang, X.; Liu, Q.; Cohn, L.; Gomez, J.L.; Chupp, G.; Pober, J.S.; Gonzalez, A.; et al. An endothelial microRNA-1-regulated network controls eosinophil trafficking in asthma and chronic rhinosinusitis. J. Allergy Clin. Immunol. 2020, 145, 550–562. [Google Scholar] [CrossRef]
- Fan, Y.; Tang, Z.; Sun, J.; Zhao, X.; Li, Z.; Zheng, Y.; Zeng, X.; Feng, J. MicroRNA-29a promotes the proliferation of human nasal epithelial cells and inhibits their apoptosis and promotes the development of allergic rhinitis by down-regulating FOS expression. PLoS ONE 2021, 16, e0255480. [Google Scholar] [CrossRef]
- Yamada, Y.; Kosaka, K.; Miyazawa, T.; Kurata-Miura, K.; Yoshida, T. miR-142-3p enhances FcεRI-mediated degranulation in mast cells. Biochem. Biophys. Res. Commun. 2014, 443, 980–986. [Google Scholar] [CrossRef]
- Mayoral, R.J.; Deho, L.; Rusca, N.; Bartonicek, N.; Saini, H.K.; Enright, A.J.; Monticelli, S. MiR-221 influences effector functions and actin cytoskeleton in mast cells. PLoS ONE 2011, 6, e26133. [Google Scholar] [CrossRef]
- Panganiban, R.P.; Wang, Y.; Howrylak, J.; Chinchilli, V.M.; Craig, T.J.; August, A.; Ishmael, F.T. Circulating microRNAs as biomarkers in patients with allergic rhinitis and asthma. J. Allergy Clin. Immunol. 2016, 137, 1423–1432. [Google Scholar] [CrossRef]
- Suojalehto, H.; Lindström, I.; Majuri, M.L.; Mitts, C.; Karjalainen, J.; Wolff, H.; Alenius, H. Altered microRNA expression of nasal mucosa in long-term asthma and allergic rhinitis. Int. Arch. Allergy Immunol. 2014, 163, 168–178. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, D.K.; Yu, M.S.; Cha, M.J.; Yu, S.L.; Kang, J. Role of epigenetics in the pathogenesis of chronic rhinosinusitis with nasal polyps. Mol. Med. Rep. 2018, 17, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Vickery, T.W.; Ramakrishnan, V.R.; Suh, J.D. The Role of Staphylococcus aureus in Patients with Chronic Sinusitis and Nasal Polyposis. Curr. Allergy Asthma Rep. 2019, 19, 21. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, L.; Li, S.; Wang, K.; Ke, R.; Shi, W.; Wang, J.; Yan, X.; Zhang, Q.; Wang, Q.; et al. Activation of AMPK inhibits TGF-β1-induced airway smooth muscle cells proliferation and its potential mechanisms. Sci. Rep. 2018, 8, 3624. [Google Scholar] [CrossRef]
- Lu, Y.; Li, Z.; Xie, B.; Song, Y.; Ye, X.; Liu, P. hsa-miR-20a-5p attenuates allergic inflammation in HMC-1 cells by targeting HDAC4. Mol. Immunol. 2019, 107, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, N.; Zhang, J.; Jiang, Y. IL-13 regulates human nasal epithelial cell differentiation via H3K4me3 modification. J. Inflamm. Res. 2017, 10, 181–188. [Google Scholar] [CrossRef]
- Zheng, Q.; Fan, H.; Meng, Z.; Yuan, L.; Liu, C.; Peng, Y.; Zhao, W.; Wang, L.; Li, J.; Feng, J. Histone demethylase KDM2B promotes triple negative breast cancer proliferation by suppressing p15INK4B, p16INK4A, and p57KIP2 transcription. Acta Biochim. Biophys. Sin. 2018, 50, 897–904. [Google Scholar] [CrossRef]
- Liu, C.C.; Sun, C.; Zheng, X.; Zhao, M.Q.; Kong, F.; Xu, F.L.; Chen, X.J.; Wang, X.X.; Zhang, M.; Xia, M. Regulation of KDM2B and Brg1 on Inflammatory Response of Nasal Mucosa in CRSwNP. Inflammation 2019, 42, 1389–1400. [Google Scholar] [CrossRef]
- Xuan, L.; Luan, G.; Wang, Y.; Lan, F.; Zhang, X.; Hao, Y.; Zheng, M.; Wang, X.; Zhang, L. MicroRNAs regulating mucin type O-glycan biosynthesis and transforming growth factor β signaling pathways in nasal mucosa of patients with chronic rhinosinusitis with nasal polyps in Northern China. Int. Forum Allergy Rhinol. 2019, 9, 106–113. [Google Scholar] [CrossRef]
- Xia, G.; Bao, L.; Gao, W.; Liu, S.; Ji, K.; Li, J. Differentially Expressed miRNA in Inflammatory Mucosa of Chronic Rhinosinusitis. J. Nanosci. Nanotechnol. 2015, 15, 2132–2139. [Google Scholar] [CrossRef]
- Luo, X.Q.; Shao, J.B.; Xie, R.D.; Zeng, L.; Li, X.X.; Qiu, S.Q.; Geng, X.R.; Yang, L.T.; Li, L.J.; Liu, D.B.; et al. Micro RNA-19a interferes with IL-10 expression in peripheral dendritic cells of patients with nasal polyposis. Oncotarget 2017, 8, 48915–48921. [Google Scholar] [CrossRef]
- Colina, R.; Costa-Mattioli, M.; Dowling, R.J.; Jaramillo, M.; Tai, L.H.; Breitbach, C.J.; Martineau, Y.; Larsson, O.; Rong, L.; Svitkin, Y.V.; et al. Translational control of the innate immune response through IRF-7. Nature 2008, 452, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Strecker, G.; Michalski, J.C.; Montreuil, J. Lysosomal catabolic pathway of N-glycosylprotein glycans. Biochimie 1988, 70, 1505–1510. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Li, C.; Zhu, G.; Yuan, W.; Xiao, Z.A. TGF-β1 Induces Epithelial-Mesenchymal Transition of Chronic Sinusitis with Nasal Polyps through MicroRNA-21. Int. Arch. Allergy Immunol. 2019, 179, 304–319. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sun, K.; Tu, Y.; Li, P.; Hao, D.; Yu, P.; Chen, A.; Wan, Y.; Shi, L. miR-200a-3p regulates epithelial-mesenchymal transition and inflammation in chronic rhinosinusitis with nasal polyps by targeting ZEB1 via ERK/p38 pathway. Int. Forum Allergy Rhinol. 2024, 14, 41–56. [Google Scholar] [CrossRef]
- Chhabra, R. miRNA and methylation: A multifaceted liaison. Chembiochem 2015, 16, 195–203. [Google Scholar] [CrossRef]
- Lygeros, S.; Danielides, G.; Grafanaki, K.; Riga, M. Matrix metalloproteinases and chronic rhinosinusitis with nasal polyposis. Unravelling a puzzle through a systematic review. Rhinology 2021, 59, 245–257. [Google Scholar] [CrossRef]
- Lygeros, S.; Danielides, G.; Kyriakopoulos, G.C.; Tsapardoni, F.; Grafanaki, K.; Stathopoulos, C.; Danielides, V. Expression profiles of MMP-9 and EMMPRIN in chronic rhinosinusitis with nasal polyps. Acta Otorhinolaryngol. Ital. 2023, 43, 400–408. [Google Scholar] [CrossRef]
- Lygeros, S.; Danielides, G.; Kyriakopoulos, G.C.; Grafanaki, K.; Tsapardoni, F.; Stathopoulos, C.; Danielides, V. Evaluation of MMP-12 expression in chronic rhinosinusitis with nasal polyposis. Rhinology 2022, 60, 39–46. [Google Scholar] [CrossRef]
- Kou, K.; Okawa, T.; Yamaguchi, Y.; Ono, J.; Inoue, Y.; Kohno, M.; Matsukura, S.; Kambara, T.; Ohta, S.; Izuhara, K.; et al. Periostin levels correlate with disease severity and chronicity in patients with atopic dermatitis. Br. J. Dermatol. 2014, 171, 283–291. [Google Scholar] [CrossRef]
- Tran, N.Q.V.; Kobayashi, Y.; Nakamura, Y.; Ishimaru, K.; Izuhara, K.; Nakao, A. Transcriptomic evidence for T cell-fibroblast-keratinocyte axis via IL-13-periostin-integrin in atopic dermatitis. Allergy 2024, 79, 3521. [Google Scholar] [CrossRef]
- Jia, G.; Erickson, R.W.; Choy, D.F.; Mosesova, S.; Wu, L.C.; Solberg, O.D.; Shikotra, A.; Carter, R.; Audusseau, S.; Hamid, Q.; et al. Periostin is a systemic biomarker of eosinophilic airway inflammation in asthmatic patients. J. Allergy Clin. Immunol. 2012, 130, 647–654.e610. [Google Scholar] [CrossRef] [PubMed]
- Shoda, T.; Futamura, K.; Kobayashi, F.; Saito, H.; Matsumoto, K.; Matsuda, A. Cell type-dependent effects of corticosteroid on periostin production by primary human tissue cells. Allergy 2013, 68, 1467–1470. [Google Scholar] [CrossRef] [PubMed]
- Maintz, L.; Welchowski, T.; Herrmann, N.; Brauer, J.; Traidl-Hoffmann, C.; Havenith, R.; Müller, S.; Rhyner, C.; Dreher, A.; CK-CARE Study Group; et al. IL-13, periostin and dipeptidyl-peptidase-4 reveal endotype-phenotype associations in atopic dermatitis. Allergy 2023, 78, 1554–1569. [Google Scholar] [CrossRef]
- Baumann, R.; Rabaszowski, M.; Stenin, I.; Gaertner-Akerboom, M.; Scheckenbach, K.; Wiltfang, J.; Schipper, J.; Wagenmann, M. The release of IL-31 and IL-13 after nasal allergen challenge and their relation to nasal symptoms. Clin. Transl. Allergy 2012, 2, 13. [Google Scholar] [CrossRef]
- Masuoka, M.; Shiraishi, H.; Ohta, S.; Suzuki, S.; Arima, K.; Aoki, S.; Toda, S.; Inagaki, N.; Kurihara, Y.; Hayashida, S.; et al. Periostin promotes chronic allergic inflammation in response to Th2 cytokines. J. Clin. Investig. 2012, 122, 2590–2600. [Google Scholar] [CrossRef]
- Danielides, G.; Lygeros, S.; Kanakis, M.; Naxakis, S. Periostin as a biomarker in chronic rhinosinusitis: A contemporary systematic review. Int. Forum Allergy Rhinol. 2022, 12, 1535–1550. [Google Scholar] [CrossRef]
- Harb, H.; Chatila, T.A. Mechanisms of Dupilumab. Clin. Exp. Allergy 2020, 50, 5–14. [Google Scholar] [CrossRef]
- Wollenberg, A.; Howell, M.D.; Guttman-Yassky, E.; Silverberg, J.I.; Kell, C.; Ranade, K.; Moate, R.; van der Merwe, R. Treatment of atopic dermatitis with tralokinumab, an anti-IL-13 mAb. J. Allergy Clin. Immunol. 2019, 143, 135–141. [Google Scholar] [CrossRef]
- Nam, D.Y.; Lee, J.M.; Heo, J.C.; Lee, S.H. Mitigation of 2,4-dinitrofluorobenzene-induced atopic dermatitis-related symptoms by Terminalia chebula Retzius. Int. J. Mol. Med. 2011, 28, 1013–1018. [Google Scholar] [CrossRef]
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sørensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, L.; Lv, W.; Yu, H. The NLRP3 inflammasome in allergic diseases: Mechanisms and therapeutic implications. Clin. Exp. Med. 2024, 24, 231. [Google Scholar] [CrossRef] [PubMed]
- Sahdo, B.; Fransén, K.; Asfaw Idosa, B.; Eriksson, P.; Söderquist, B.; Kelly, A.; Särndahl, E. Cytokine profile in a cohort of healthy blood donors carrying polymorphisms in genes encoding the NLRP3 inflammasome. PLoS ONE 2013, 8, e75457. [Google Scholar] [CrossRef]
- Yan, D.; Ye, Y.; Zhang, J.; Zhao, J.; Yu, J.; Luo, Q. Human Neutrophil Elastase Induces MUC5AC Overexpression in Chronic Rhinosinusitis Through miR-146a. Am. J. Rhinol. Allergy 2020, 34, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Yoo, B.; Park, Y.; Park, K.; Kim, H. A 9-year Trend in the Prevalence of Allergic Disease Based on National Health Insurance Data. J. Prev. Med. Public Health 2015, 48, 301–309. [Google Scholar] [CrossRef]
- Dopytalska, K.; Czaplicka, A.; Szymańska, E.; Walecka, I. The Essential Role of microRNAs in Inflammatory and Autoimmune Skin Diseases—A Review. Int. J. Mol. Sci. 2023, 24, 9130. [Google Scholar] [CrossRef]
- Zheng, T.; Yu, J.; Oh, M.H.; Zhu, Z. The Atopic March: Progression from Atopic Dermatitis to Allergic Rhinitis and Asthma. Allergy Asthma Immunol. Res. 2011, 3, 67–73. [Google Scholar] [CrossRef]
- Fu, X.; Hong, C. Osthole attenuates mouse atopic dermatitis by inhibiting thymic stromal lymphopoietin production from keratinocytes. Exp. Dermatol. 2019, 28, 561–567. [Google Scholar] [CrossRef]
- Dai, X.; Sayama, K.; Tohyama, M.; Shirakata, Y.; Hanakawa, Y.; Tokumaru, S.; Yang, L.; Hirakawa, S.; Hashimoto, K. Mite allergen is a danger signal for the skin via activation of inflammasome in keratinocytes. J. Allergy Clin. Immunol. 2011, 127, 806–814, e801–804. [Google Scholar] [CrossRef]
- Haruna, S.; Takeda, K.; El-Hussien, M.A.; Maeda, Y.; Hayama, M.; Shikina, T.; Doi, K.; Inohara, H.; Kikutani, H.; Sakakibara, S. Local production of broadly cross-reactive IgE against multiple fungal cell wall polysaccharides in patients with allergic fungal rhinosinusitis. Allergy 2022, 77, 3147–3151. [Google Scholar] [CrossRef]
- El Gendy, A.; Abo Ali, F.H.; Baioumy, S.A.; Taha, S.I.; El-Bassiouny, M.; Abdel Latif, O.M. NLRP3 inflammasome (rs10754558) gene polymorphism in patients with atopic dermatitis. Egypt. J. Immunol. 2023, 30, 102–109. [Google Scholar] [CrossRef]
- Xiao, Y.; Xu, W.; Su, W. NLRP3 inflammasome: A likely target for the treatment of allergic diseases. Clin. Exp. Allergy 2018, 48, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.H.; Tubridy, K.L.; Agahigian, J.; Furfine, E.; Magill, M.; Kovalchin, J.; Golden, K.; Zarbis-Papastoitsis, G.; Soong, F.; Salapatek, A.M.; et al. A Phase 2 Exploratory Study of a Novel Interleukin-1 Receptor Inhibitor (EBI-005) in the Treatment of Moderate-to-Severe Allergic Conjunctivitis. Eye Contact Lens 2015, 41, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, S.; Wang, W.; Chen, J.; Kong, W.; Wang, Y. Role of P2X7R in eosinophilic and non-eosinophilic chronic rhinosinusitis with nasal polyps. Mol. Med. Rep. 2021, 24, 521. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Boucher, D.; Monteleone, M.; Coll, R.C.; Chen, K.W.; Ross, C.M.; Teo, J.L.; Gomez, G.A.; Holley, C.L.; Bierschenk, D.; Stacey, K.J.; et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J. Exp. Med. 2018, 215, 827–840. [Google Scholar] [CrossRef]
- Shaghayegh, G.; Cooksley, C.; Bouras, G.; Nepal, R.; Houtak, G.; Panchatcharam, B.S.; Fenix, K.A.; Psaltis, A.J.; Wormald, P.J.; Vreugde, S. Staphylococcus aureus biofilm properties and chronic rhinosinusitis severity scores correlate positively with total CD4+ T-cell frequencies and inversely with its Th1, Th17 and regulatory cell frequencies. Immunology 2023, 170, 120–133. [Google Scholar] [CrossRef]
- Huntley, K.S.; Raber, J.; Fine, L.; Bernstein, J.A. Influence of the Microbiome on Chronic Rhinosinusitis With and Without Polyps: An Evolving Discussion. Front. Allergy 2021, 2, 737086. [Google Scholar] [CrossRef]
- Oliveira, D.; Borges, A.; Simões, M. Staphylococcus aureus Toxins and Their Molecular Activity in Infectious Diseases. Toxins 2018, 10, 252. [Google Scholar] [CrossRef]
- Kanemitsu, Y.; Fukumitsu, K.; Kurokawa, R.; Takeda, N.; Ozawa, Y.; Masaki, A.; Ono, J.; Izuhara, K.; Yap, J.M.; Nishiyama, H.; et al. Moulds and Staphylococcus aureus enterotoxins are relevant allergens to affect Type 2 inflammation and clinical outcomes in chronic rhinosinusitis patients. ERJ Open Res. 2020, 6, 00265-2020. [Google Scholar] [CrossRef]
- Toppila-Salmi, S.; Reitsma, S.; Hox, V.; Gane, S.; Eguiluz-Gracia, I.; Shamji, M.; Maza-Solano, J.; Jääskeläinen, B.; Väärä, R.; Escribese, M.M.; et al. Endotyping in Chronic Rhinosinusitis-An EAACI Task Force Report. Allergy 2025, 80, 132–147. [Google Scholar] [CrossRef]
- de Kerckhove, M.; Tanaka, K.; Umehara, T.; Okamoto, M.; Kanematsu, S.; Hayashi, H.; Yano, H.; Nishiura, S.; Tooyama, S.; Matsubayashi, Y.; et al. Targeting miR-223 in neutrophils enhances the clearance of Staphylococcus aureus in infected wounds. EMBO Mol. Med. 2018, 10, e9024. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.M.; Park, J.H.; Yang, H.W.; Moon, J.W.; Lee, H.M.; Park, I.H. miR-29b Regulates TGF-β1-Induced Epithelial-Mesenchymal Transition by Inhibiting Heat Shock Protein 47 Expression in Airway Epithelial Cells. Int. J. Mol. Sci. 2021, 22, 11535. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Cardenas, E.I.; Ekstedt, S.; Arebro, J.; Petro, M.; Karlsson, A.; Hjalmarsson, E.; Arnarson, D.; Ezerskyte, M.; Kumlien Georén, S.; et al. Delayed neutrophil shedding of CD62L in patients with chronic rhinosinusitis with nasal polyps and asthma: Implications for Staphylococcus aureus colonization and corticosteroid treatment. Clin. Transl. Allergy 2024, 14, e12347. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Renert-Yuval, Y.; Brunner, P.M. Atopic dermatitis. Lancet 2025, 405, 583–596. [Google Scholar] [CrossRef]
- Chiang, S.; Lee, S.E. New Concepts in Barrier Dysfunction in CRSwNP and Emerging Roles of Tezepelumab and Dupilumab. Am. J. Rhinol. Allergy 2023, 37, 193–197. [Google Scholar] [CrossRef]
- Potaczek, D.P.; Kabesch, M. Current concepts of IgE regulation and impact of genetic determinants. Clin. Exp. Allergy 2012, 42, 852–871. [Google Scholar] [CrossRef]
- Goetzl, E.J. Th2 cells in rapid immune responses and protective avoidance reactions. FASEB J. 2024, 38, e23485. [Google Scholar] [CrossRef]
- Lemmetyinen, R.E.; Toppila-Salmi, S.K.; But, A.; Renkonen, R.; Pekkanen, J.; Haukka, J.; Karjalainen, J. Comorbidities associated with adult asthma: A population-based matched cohort study in Finland. BMJ Open Respir. Res. 2024, 11, e001959. [Google Scholar] [CrossRef]
- Wojas, O.; Arcimowicz, M.; Furmańczyk, K.; Sybilski, A.; Raciborski, F.; Tomaszewska, A.; Walkiewicz, A.; Samel-Kowalik, P.; Samoliński, B.; Krzych-Fałta, E. The relationship between nasal polyps, bronchial asthma, allergic rhinitis, atopic dermatitis, and non-allergic rhinitis. Postep. Dermatol. Alergol. 2021, 38, 650–656. [Google Scholar] [CrossRef]
- Calabrese, C.; Seccia, V.; Pelaia, C.; Spinelli, F.; Morini, P.; Rizzi, A.; Detoraki, A. S. aureus and IgE-mediated diseases: Pilot or copilot? A narrative review. Expert Rev. Clin. Immunol. 2022, 18, 639–647. [Google Scholar] [CrossRef]
- Wechsler, M.E.; Klion, A.D.; Paggiaro, P.; Nair, P.; Staumont-Salle, D.; Radwan, A.; Johnson, R.R.; Kapoor, U.; Khokhar, F.A.; Daizadeh, N.; et al. Effect of Dupilumab on Blood Eosinophil Counts in Patients with Asthma, Chronic Rhinosinusitis with Nasal Polyps, Atopic Dermatitis, or Eosinophilic Esophagitis. J. Allergy Clin. Immunol. Pract. 2022, 10, 2695–2709. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, M.; Maffei, M.; Patruno, C.; Leone, C.A.; Di Guida, A.; Potestio, L.; Scalvenzi, M.; Fabbrocini, G. Dupilumab effectiveness for the treatment of patients with concomitant atopic dermatitis and chronic rhinosinusitis with nasal polyposis. Dermatol. Ther. 2021, 34, e15120. [Google Scholar] [CrossRef] [PubMed]
- Belliveau, P.P. Omalizumab: A monoclonal anti-IgE antibody. Medscape Gen. Med. 2005, 7, 27. [Google Scholar]
- Ryser, F.S.; Yalamanoglu, A.; Valaperti, A.; Brühlmann, C.; Mauthe, T.; Traidl, S.; Soyka, M.B.; Steiner, U.C. Dupilumab-induced eosinophilia in patients with diffuse type 2 chronic rhinosinusitis. Allergy 2023, 78, 2712–2723. [Google Scholar] [CrossRef]
- Gómez de la Fuente, E.; Alobid, I.; Ojanguren, I.; Rodríguez-Vázquez, V.; Pais, B.; Reyes, V.; Espinosa, M.; Luca de Tena, Á.; Muerza, I.; Vidal-Barraquer, E. Addressing the unmet needs in patients with type 2 inflammatory diseases: When quality of life can make a difference. Front. Allergy 2023, 4, 1296894. [Google Scholar] [CrossRef]
- Caminati, M.; De Corso, E.; Ottaviano, G.; Pipolo, C.; Schiappoli, M.; Seccia, V.; Spinelli, F.R.; Savarino, E.V.; Gisondi, P.; Senna, G. Remission in Type 2 Inflammatory Diseases: Current Evidence, Unmet Needs, and Suggestions for Defining Remission in Chronic Rhinosinusitis with Nasal Polyps. Curr. Allergy Asthma Rep. 2024, 24, 11–23. [Google Scholar] [CrossRef]
- Chung, M.K.; House, J.S.; Akhtari, F.S.; Makris, K.C.; Langston, M.A.; Islam, K.T.; Holmes, P.; Chadeau-Hyam, M.; Smirnov, A.I.; Du, X.; et al. Decoding the exposome: Data science methodologies and implications in exposome-wide association studies (ExWASs). Exposome 2024, 4, osae001. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
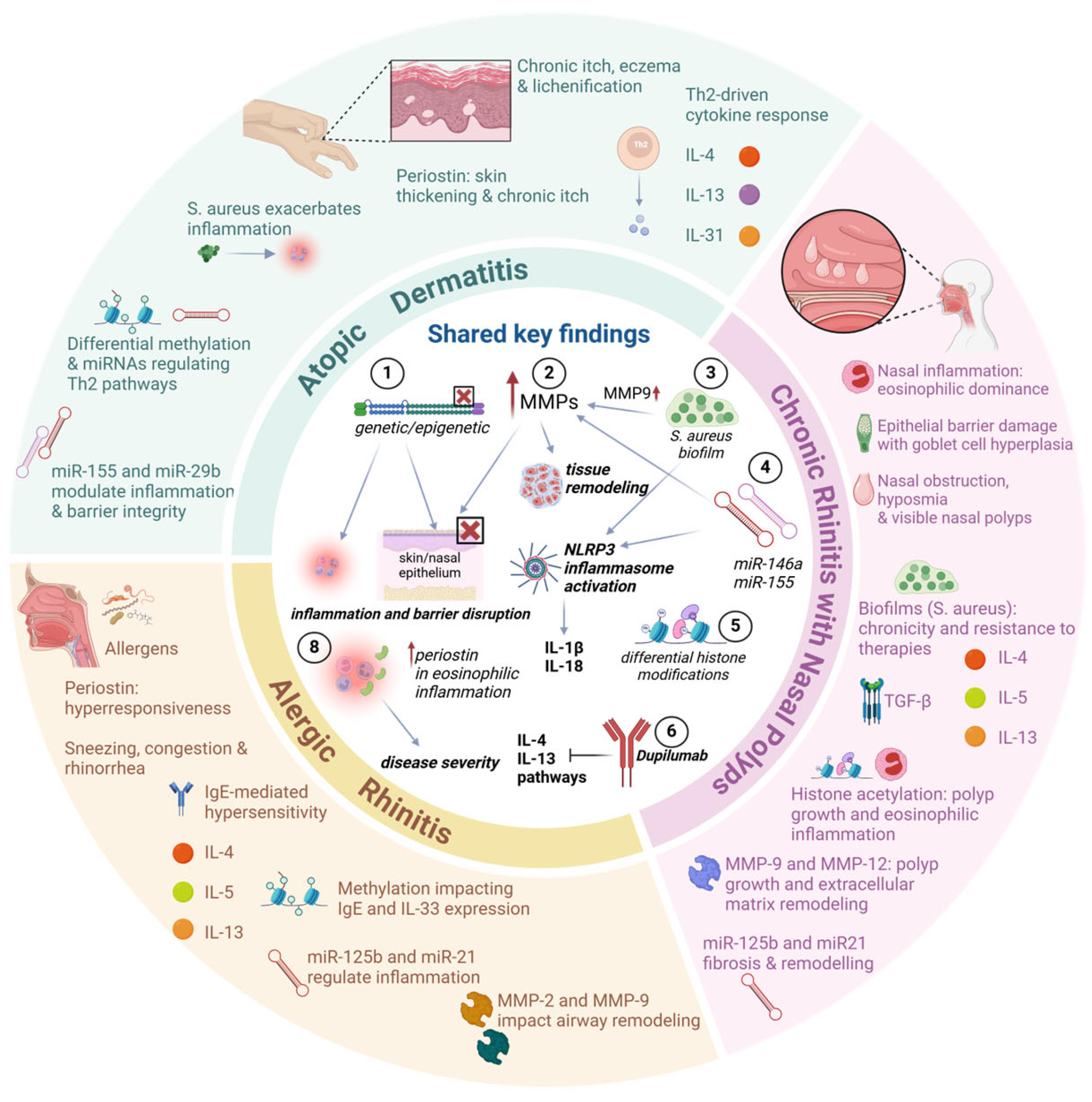
| AD | AR | CRSwNP | Refs | |
|---|---|---|---|---|
| Type of Inflammation | Skin-localized but with a systemic Th2-driven cytokine response | Th2 inflammation in the respiratory tract driven by allergens | Nasal mucosal Th2 inflammation with eosinophilic dominance and polyp formation | [1,7,70] |
| Barrier Dysfunction | Impaired skin barrier due to FLG mutations and Th2 | Barrier dysfunction leading to allergen sensitization | Epithelial damage with goblet cell hyperplasia and biofilm formation | [24,97,147] |
| Role of Periostin | Enhances skin thickening and chronic itching | Promotes airway hyperresponsiveness | Associated with fibrosis and extracellular matrix deposition | [97,148,149] |
| Role of the Microbiome | S. aureus exacerbates inflammation | Dysbiosis linked to IgE-mediated hypersensitivity | Biofilms (S. aureus) promote chronicity and resistance to therapies | [97,148,149] |
| Key Cytokines | IL-4, IL-13, IL-31 | IL-4, IL-5, IL-13 | IL-4, IL-5, IL-13, TGF-β | [7,121,124] |
| Epigenetic Mechanisms | Differential methylation and miRNAs regulating Th2 pathways | Methylation impacting IgE and IL-33 expression | Histone acetylation linked to polyp growth and eosinophilic inflammation | [36,60,69] |
| MicroRNAs | miR-146a miR-155, and miR-29b modulate inflammation and barrier integrity | miR-146a, miR-155 miR-125b, and miR-21 regulate airway inflammation | miR-146a, miR-155 miR-125b, and miR-21 involved in fibrosis and epithelial remodeling | [21,50,86,87,128,129,131,150] |
| MMP Activity | MMP-2 and MMP-9 disrupt the skin’s architecture, worsening barrier dysfunction | MMP-9 and MMP-12 contribute to airway remodeling | MMP-2, MMP-9, and MMP-12 drive polyp growth and extracellular matrix remodeling | [129,130,150,151,152,153] |
| Inflammasome Activation | Inflammasomes (e.g., NLRP3) amplify IL-1β and IL-18 secretion in lesions | Activated by allergens, contributing to airway inflammation | Enhanced by microbial biofilms, perpetuating chronic nasal inflammation | [138,148] |
| Clinical Manifestations | Chronic itch, eczema, and lichenification | Sneezing, congestion, and rhinorrhea | Nasal obstruction, hyposmia, and visible nasal polyps | [10,11,70] |
| Aspect | AD | AR | CRSwNP | Refs |
|---|---|---|---|---|
| Colonization | S. aureus colonizes damaged skin, particularly in barrier-deficient areas. | S. aureus can colonize nasal mucosa, increasing inflammation. | Persistent colonization in nasal polyps, often associated with biofilms. | [97,145] |
| Toxins and Superantigens | Produces α-toxin, β-toxin, and superantigens that trigger T-cell activation and IL-1β release via NLRP3 inflammasome activation. | Superantigens stimulate T-cell responses, amplifying type 2 inflammation. | Superantigens and toxins drive eosinophilic inflammation and Th2 polarization. | [97,146,148] |
| Barrier Dysfunction | Toxins disrupt keratinocyte tight junctions, worsening skin barrier integrity. | Toxins impair nasal epithelial integrity, enhancing allergen penetration. | Biofilms and toxins disrupt mucosal barriers, promoting polyp growth. | [97,147] |
| Inflammasome Activation | Activates the NLRP3 inflammasome in keratinocytes, leading to IL-1β and IL-18 production. | NLRP3 inflammasome activation in nasal epithelial cells amplifies inflammation. | Persistent inflammasome activation exacerbates chronic inflammation and tissue remodeling. | [136,148] |
| Cytokine Amplification | Promotes Th2 cytokine response (IL-4, IL-5, and IL-13) through superantigen activity. | Increases Th2 cytokines, promoting IgE-mediated hypersensitivity. | Enhances Th2 cytokine production, perpetuating eosinophilic inflammation. | [136,148,149] |
| Biofilm Formation | Rare in AD; typically affects acute lesions with heavy bacterial burdens. | Rarely forms; contributes to chronicity in recurrent cases. | Commonly forms in nasal polyps, fostering chronic inflammation and immune evasion. | [148,149] |
| Eosinophilic Recruitment | Enhances eosinophil migration via IL-5 and eotaxins, exacerbating inflammation. | Stimulates eosinophil infiltration in nasal tissues. | Eosinophil-driven inflammation is a hallmark, often worsened by biofilms. | [136,148,149] |
| Epigenetic Influence | Modifies DNA methylation (e.g., hypomethylation of inflammasome genes such as NLRP3). | Alters methylation patterns, enhancing pro-inflammatory gene expression. | Biofilm presence alters histone acetylation and miRNA profiles, amplifying inflammation. | [136,148,149] |
| Aspect | Novel Findings | Clinical Implications | Refs |
|---|---|---|---|
| Genetics in AD, AR, and CRSwNP | Identification of filaggrin mutations in AD, shared loci (e.g., TSLP and IL1B) linked to inflammation and barrier dysfunction | Highlights genetic predispositions contributing to systemic type 2 inflammation and guides genetic risk assessment | [2,42,136] |
| Epigenetics | DNA methylation at TSLP and IL1B, differential histone modifications, miRNA dysregulation (e.g., miR-155 and miR-21) | Targets for developing epigenetic biomarkers and therapies aimed at reversing aberrant gene expression | [36,60,69] |
| Role of the Microbiome | S. aureus biofilms drive NLRP3 inflammasome activation, elevate MMP-9, and disrupt epithelial barriers | Reinforces the need for therapies targeting biofilm-induced inflammation in CRSwNP and AD | [97,148,149] |
| Matrix Metalloproteinases (MMPs) | Elevated MMP-9 and MMP-12 activity linked to tissue remodeling in CRSwNP, barrier dysfunction in AD | Identify MMPs as therapeutic targets for mitigating inflammation and tissue destruction | [118,129,130] |
| MicroRNA Pathways | miR-29b suppression drives MMP upregulation, miR-223 regulates inflammasome activity | Opens avenues for miRNA-based therapeutic interventions to restore regulatory balance | [50,131,143] |
| Periostin in Disease Pathways | Elevated periostin in eosinophilic inflammation linked to disease severity in AD, AR, and CRSwNP | Supports periostin as a biomarker for disease severity and treatment response, especially for biologics | [120,121,126] |
| Biologic Therapies | Dupilumab efficacy in inhibiting IL-4/IL-13 pathways across conditions; emerging TSLP-targeting biologics | Demonstrates success of systemic therapies and the potential for biologic expansion to other pathways. | [127,151,155] |
| Inflammasome Activation | Overactivation of NLRP3 amplifies IL-1β and IL-18 secretion in AD, AR, and CRSwNP | Targets inflammasome pathways for novel anti-inflammatory treatments | [136,138,149] |
| Environmental Influences | Urbanization and pollution exacerbate type 2 inflammation via epigenetic alterations | Suggests environmental interventions to mitigate disease progression and enhance management strategies | [4,13,36] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Danielidi, A.; Lygeros, S.; Anastogianni, A.; Danielidis, G.; Georgiou, S.; Stathopoulos, C.; Grafanaki, K. Genetic and Epigenetic Interconnections Between Atopic Dermatitis, Allergic Rhinitis, and Rhinitis with Nasal Polyps. Allergies 2025, 5, 9. https://doi.org/10.3390/allergies5020009
Danielidi A, Lygeros S, Anastogianni A, Danielidis G, Georgiou S, Stathopoulos C, Grafanaki K. Genetic and Epigenetic Interconnections Between Atopic Dermatitis, Allergic Rhinitis, and Rhinitis with Nasal Polyps. Allergies. 2025; 5(2):9. https://doi.org/10.3390/allergies5020009
Chicago/Turabian StyleDanielidi, Alexandra, Spyridon Lygeros, Alexandra Anastogianni, Gerasimos Danielidis, Sophia Georgiou, Constantinos Stathopoulos, and Katerina Grafanaki. 2025. "Genetic and Epigenetic Interconnections Between Atopic Dermatitis, Allergic Rhinitis, and Rhinitis with Nasal Polyps" Allergies 5, no. 2: 9. https://doi.org/10.3390/allergies5020009
APA StyleDanielidi, A., Lygeros, S., Anastogianni, A., Danielidis, G., Georgiou, S., Stathopoulos, C., & Grafanaki, K. (2025). Genetic and Epigenetic Interconnections Between Atopic Dermatitis, Allergic Rhinitis, and Rhinitis with Nasal Polyps. Allergies, 5(2), 9. https://doi.org/10.3390/allergies5020009