1. Introduction
The demand for plastics remains undiminished. After a brief period of stagnation in 2020, global plastics production increased again and reached a new peak of 390.7 Mt in 2022. The largest consumer of plastics in Europe by far is the packaging sector with 39.1%, followed by the building and construction sector with 21.3% and the automotive industry with 8.6% [
1]. In a 2008 directive, the European Parliament issued guidelines for member states to increase the rates of reuse and recycling of waste materials, with the target of reaching a 55% recycling rate by 2025 and a 60% rate by 2030 [
2]. Further, with a 2018 decision, the European Parliament urged the European Commission to revise and strengthen the general requirements of the Packaging and Packaging Waste Directives (PPWD) by the end of 2020 to implement the circular economy strategy in the plastics industry. Although legislative pressure on the plastics industry has recently increased, the proportion of recycled plastics in the EU stagnates at roughly 8% [
1]. If the above targets are to be achieved, the recycling of plastics will become increasingly important, and plastic as a material must be shifted into a sustainable circular economy [
3]. Life-cycle analyses (LCAs) have shown that beverages multi-packaged in post-consumer recycled plastic cause 73% less greenhouse gas emissions and consume 90% less energy than their cardboard counterparts [
4]. These results accord with the goals of the European Green Deal, which aims to reduce greenhouse gases to 45% of the 1990 levels by 2030 and to achieve climate neutrality by 2050 [
5,
6].
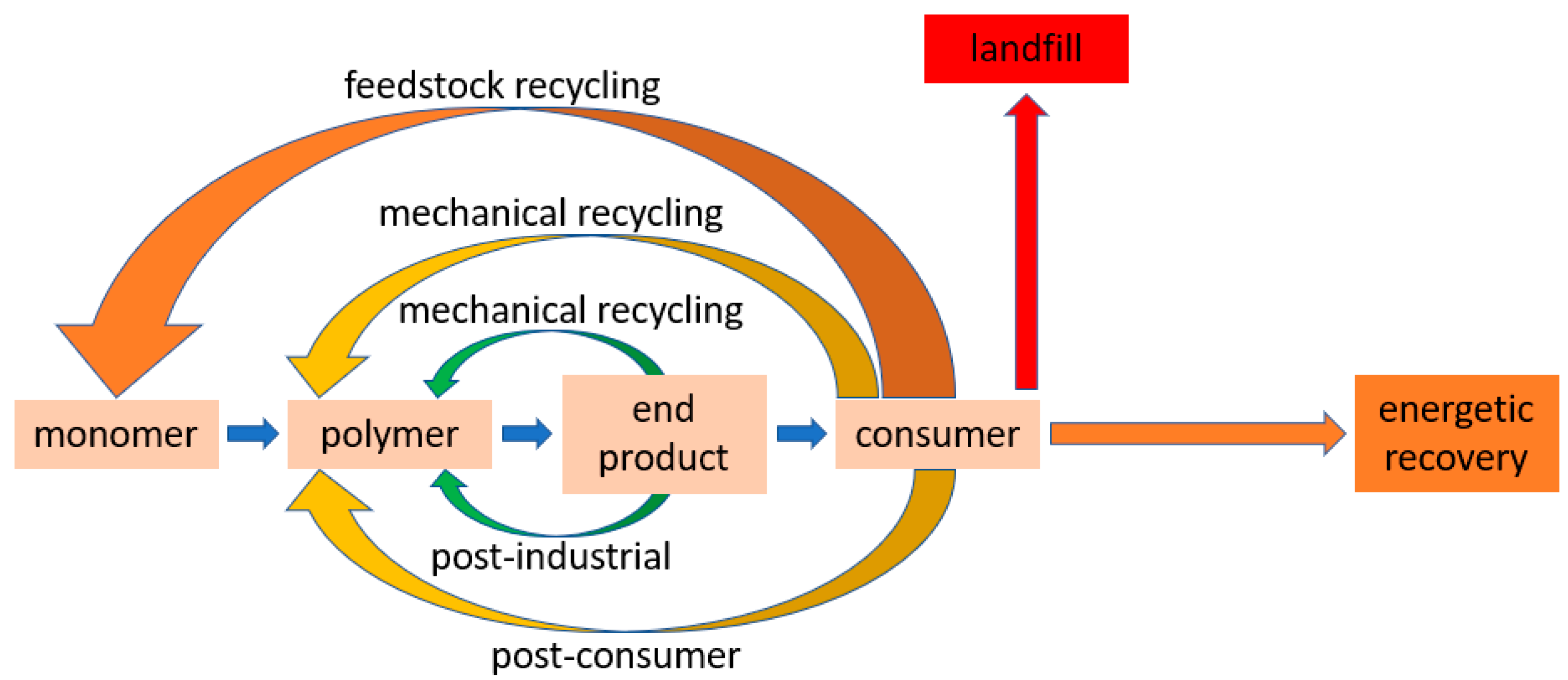
Fundamentally, distinctions are made between post-industrial and post-consumer plastic waste and between the recovery and recycling terms (
Figure 1). Post-consumer waste is generated by the end consumer, while post-industrial waste is generated during production. Recovery is a generic term and describes all ways of recovering plastic waste. Recycling is a sub-term and describes all options of material reuse and resource recycling.
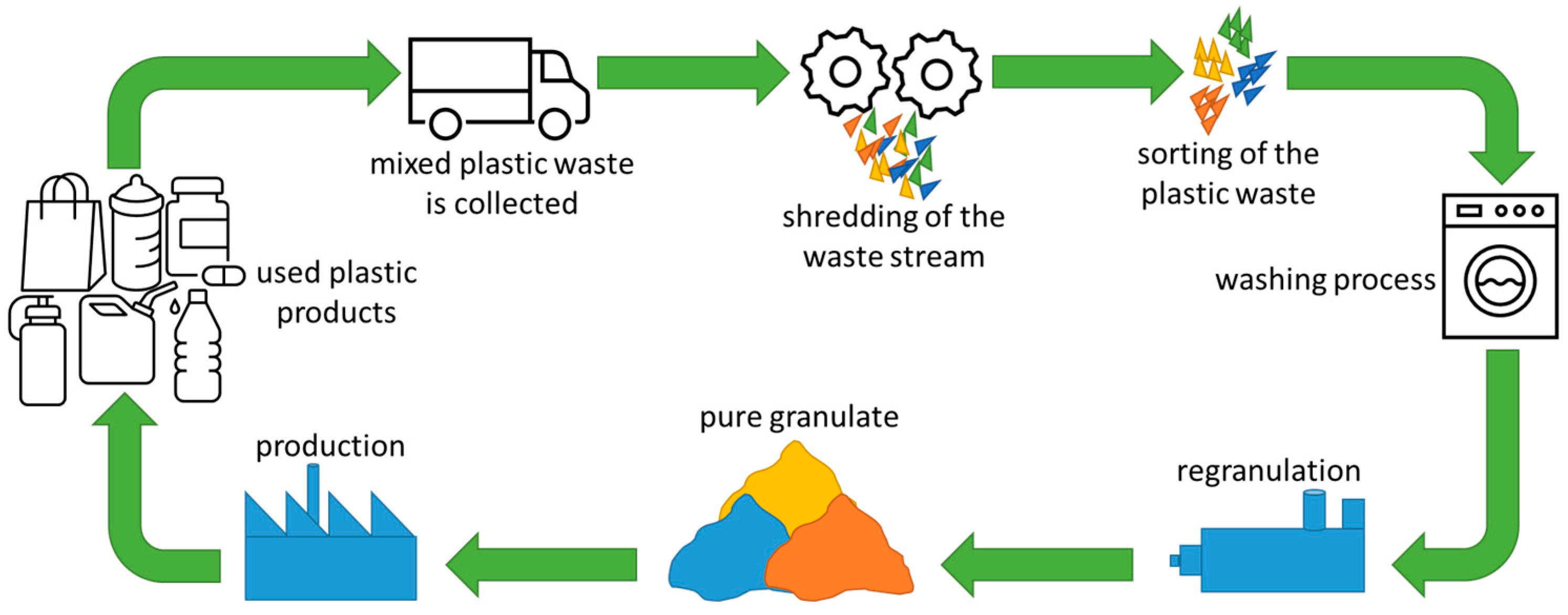
Figure 2 shows an overview of a mechanical recycling process. Post-consumer plastic waste is commonly collected by disposal companies. The plastic waste is often shredded before it is sent to a sorting process, where it is initially roughly sorted according to density, shape, size, and color [
7,
8,
9]. Here, ferrous metals, for example, can be separated from the waste stream by using a magnet. Paper and label scraps are typically separated from plastics in an air classifier. A vibrating sieve can be employed to sort by size, and optical sensors detect color differences. The waste is then sorted automatically by polymer type, most commonly by means of near-infrared (NIR) spectroscopy. This optical surface technique, however, has problems with detecting black or very dark surfaces and potentially with multilayer films [
10,
11]. The sorted plastic waste is then sent for washing, where organic and adhesive residues and cellulose fibers are removed using media such as hot or cold water, with or without the addition of chemicals.
In the next step, the pretreated plastic waste is converted into granules in an extrusion or compounding process, where material quality can be improved, for example, by filtration or degassing and by the targeted addition of virgin materials or additives. In mechanical recycling, extrusion is the most important process for the regranulation of plastic waste. The direct processing of preconditioned plastic waste is often problematic due to the low bulk density of regrind and film material and the material variability of the input stream, especially in post-consumer waste. The end product made from recyclate is returned to the consumer, thus closing the material cycle. Consistently high recyclate quality is a basic requirement for achieving high recycling rates in future.
Products made of plastic come in different colors, and much plastic waste is also contaminated with volatile substances. This may not only affect the quality of the finished product; the product may also smell unpleasant and thus fail to meet the consumer’s expectations. In addition, plastic products entering the recycling chain have usually been exposed to combinations of various environmental influences during their service life, such as heat, light, moisture and oxygen, and mechanical stress, which causes material degradation by photooxidation processes [
12]. Material degradation, due to thermomechanical processes both in use and in processing, results in the formation of low-molecular-weight volatile compounds, which are commonly referred to as volatile organic compounds (VOCs). These VOCs can affect not only the product properties (e.g., bubble formation and poorer mechanical performance), but also manufacturing processes (e.g., strong foaming of the melt and corrosion of the processing machine). A critical step in many polymer processing operations is therefore degassing—the removal of volatile components, such as reaction by-products, residual monomers, solvents, moisture and other impurities, from the polymeric phase [
13]. For this purpose, various methods and devices are available in industries, which can be classified into two main categories: (i) rotating devolatilizers (e.g., vented extruders) and (ii) non-rotating devolatilizers (e.g., falling-strand devolatilizers) [
14]. Although polymers can be degassed in the solid state (e.g., the removal of odors during the drying process), volatile components are often removed from the melt phase because VOCs evaporate more easily in this aggregate state (e.g., devolatilization in a degassing extruder). In this context, extrusion (and compounding) is of particular importance, where vacuum is used to extract volatile components via free surfaces in a partially filled screw channel.
There are two mechanisms that describe the mass transfer of volatile substances in polymeric phases: (i) diffusion-dominated film degassing and (ii) bubble-dominated foam degassing [
15]. For bubble forming, the material must be supersaturated with volatile components and subjected to shear stress. In general, foam degassing is more efficient than film degassing because the free surfaces for volatilization of unwanted substances are many times larger [
16]. In regranulation processes, mass transfer is commonly assumed to be diffusion-driven [
14,
15,
17,
18]. The diffusion coefficient describes the mass transport of molecules due to their random motion [
19,
20] and is a measure of the velocity at which diffusion processes occur. Regardless of whether the mass transport is diffusion-based (film degassing) or bubble-dominated (foam degassing), a proportion of mass is always transported via diffusion. From a general viewpoint, there is no universal measurement technique for determining, experimentally, the diffusion coefficients of VOCs in polymeric matrices. The diffusion coefficient depends on various variables, including (i) the molecular size of the diffusing substance, (ii) temperature, (iii) pressure, (iv) the viscosities of the medium in which the substance diffuses and (v) the concentration of the diffusing substance. The effects of these parameters depend on the aggregate state of the medium in which the molecules move, that is, in gas, liquid or solid [
21]. The measurement of mass transfer properties is complex because it is difficult to measure the concentrations of volatile substances. Existing experimental techniques can be divided into direct and indirect methods [
22]:
Direct methods determine the diffusion coefficient by measuring the concentration of the diffusing substance as a function of penetration depth. These methods are based directly on Fick’s diffusion laws.
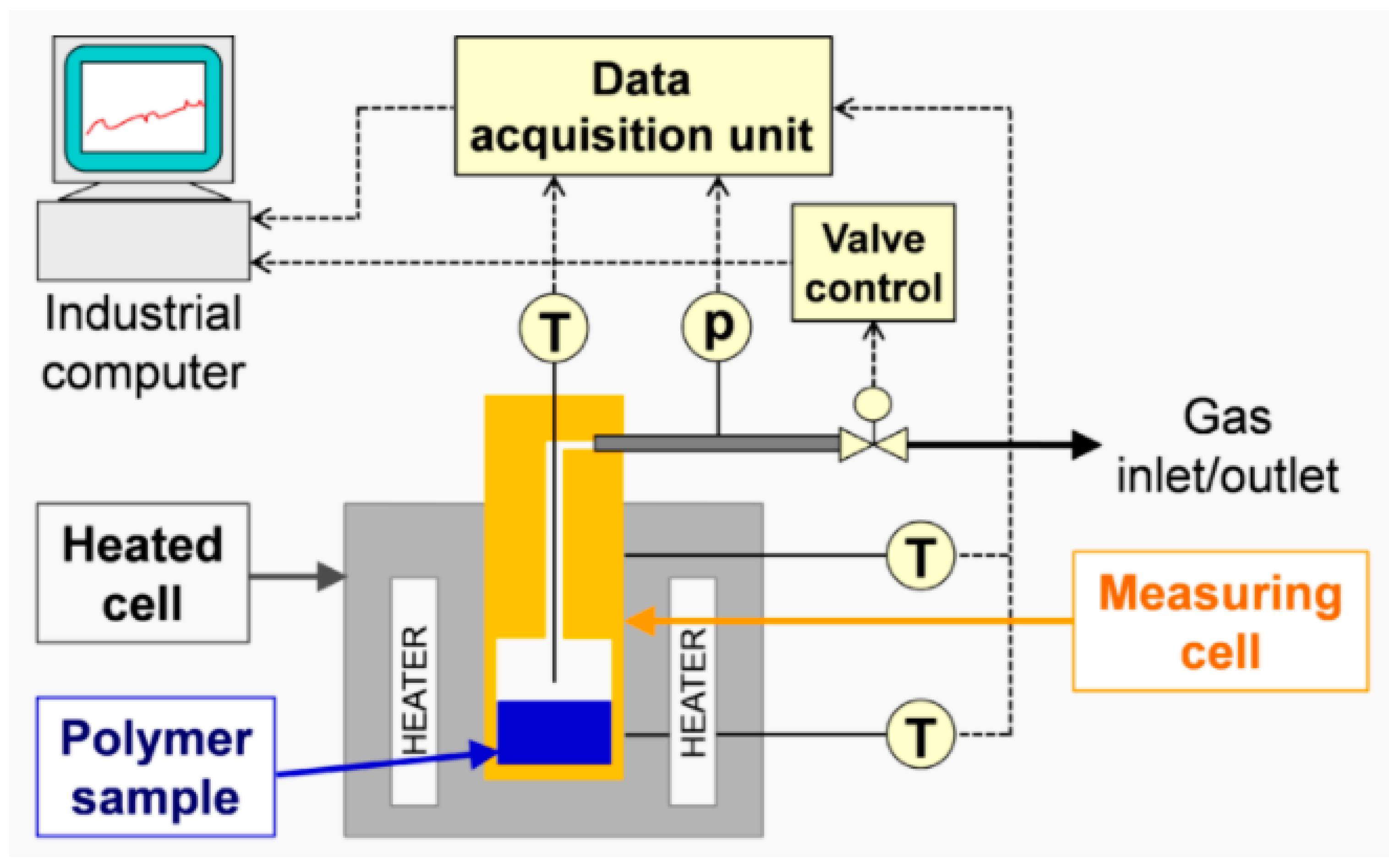
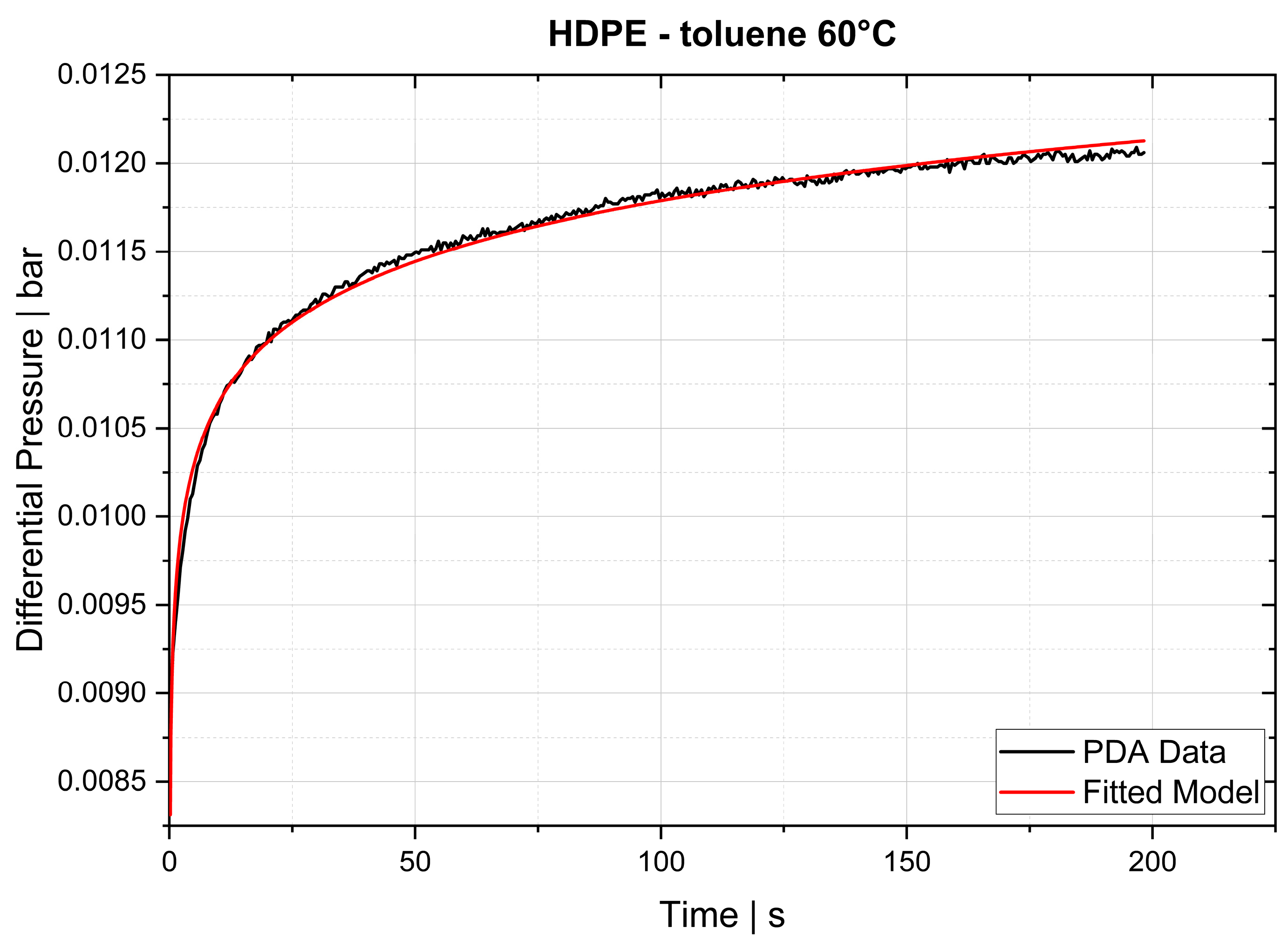
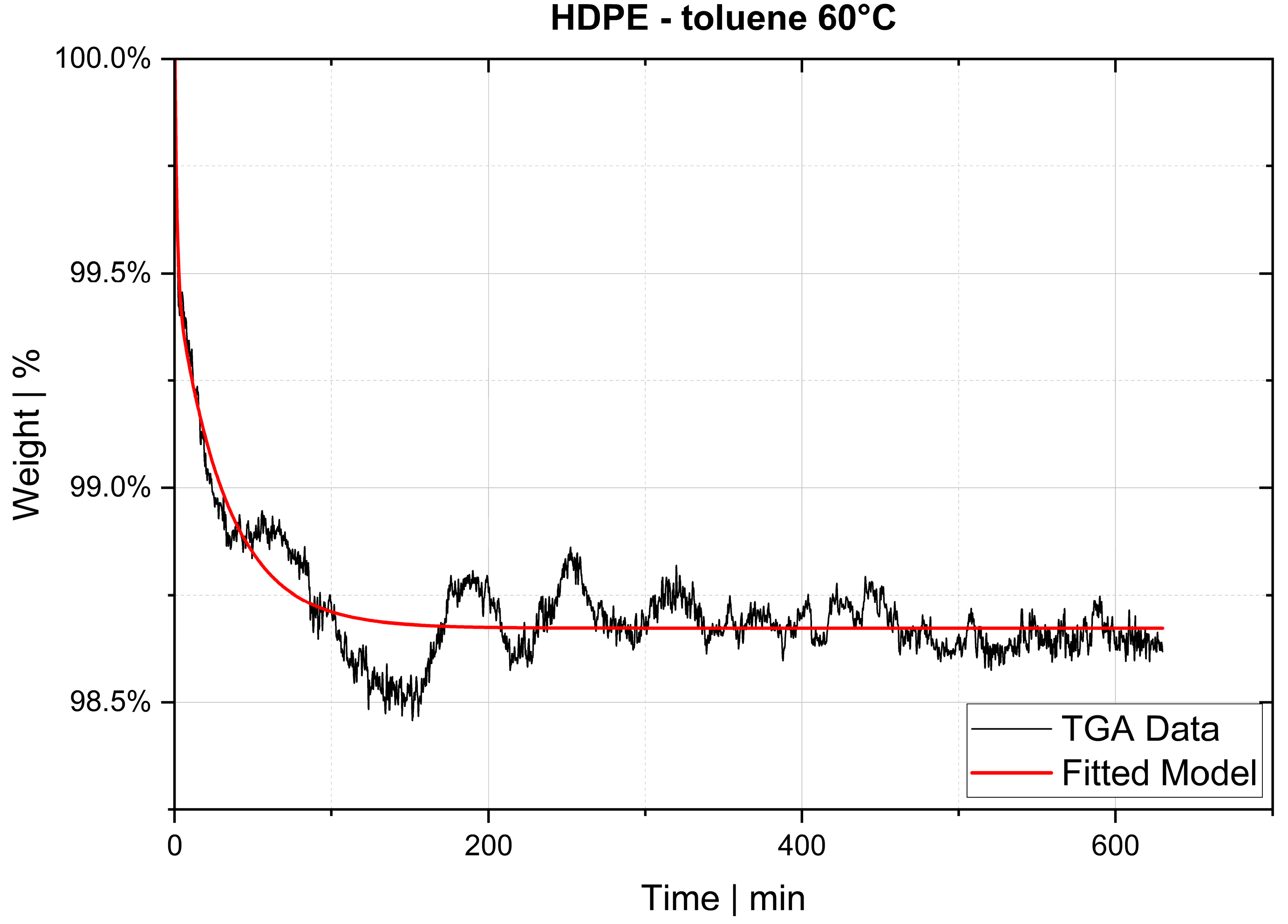
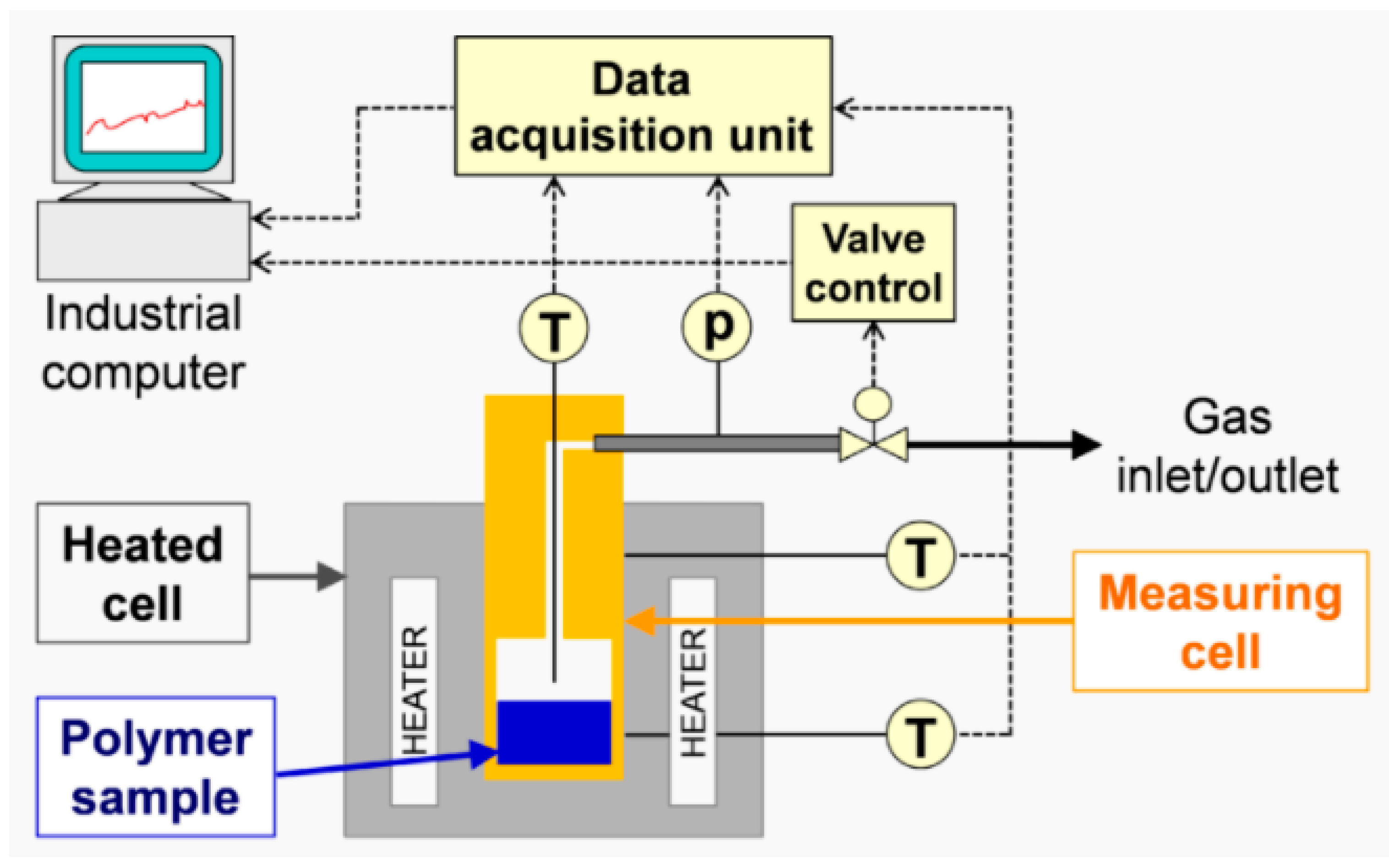
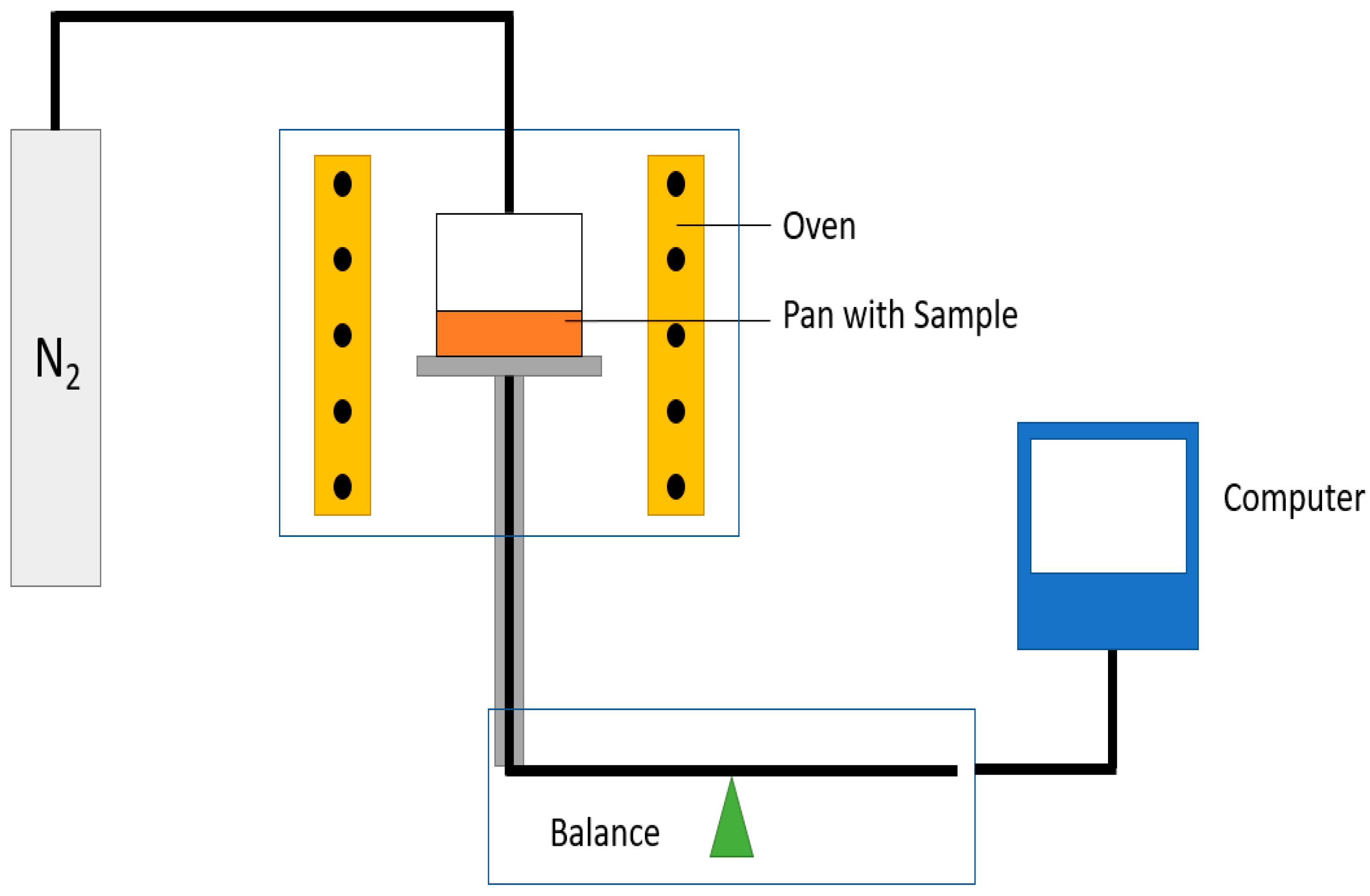
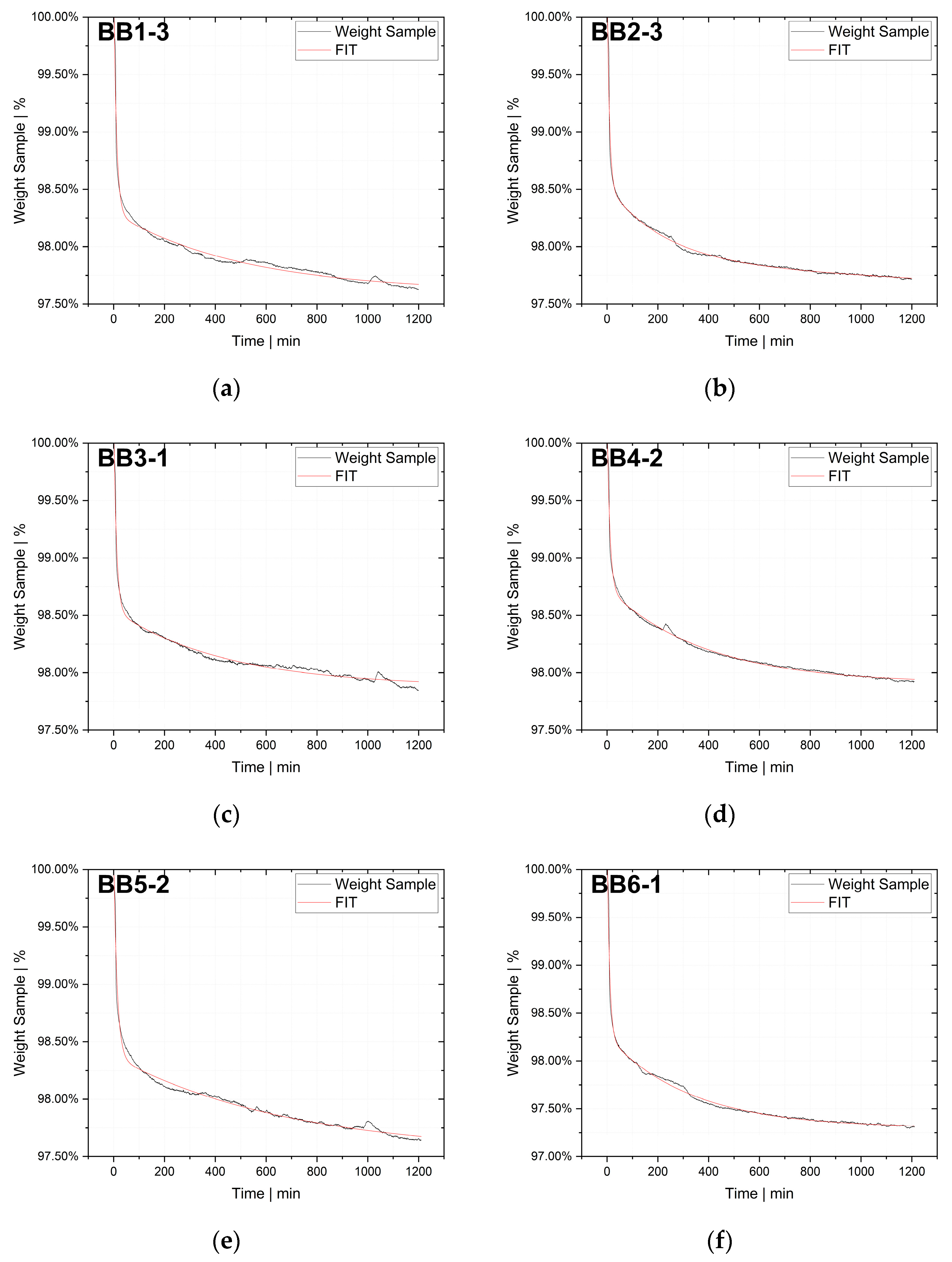
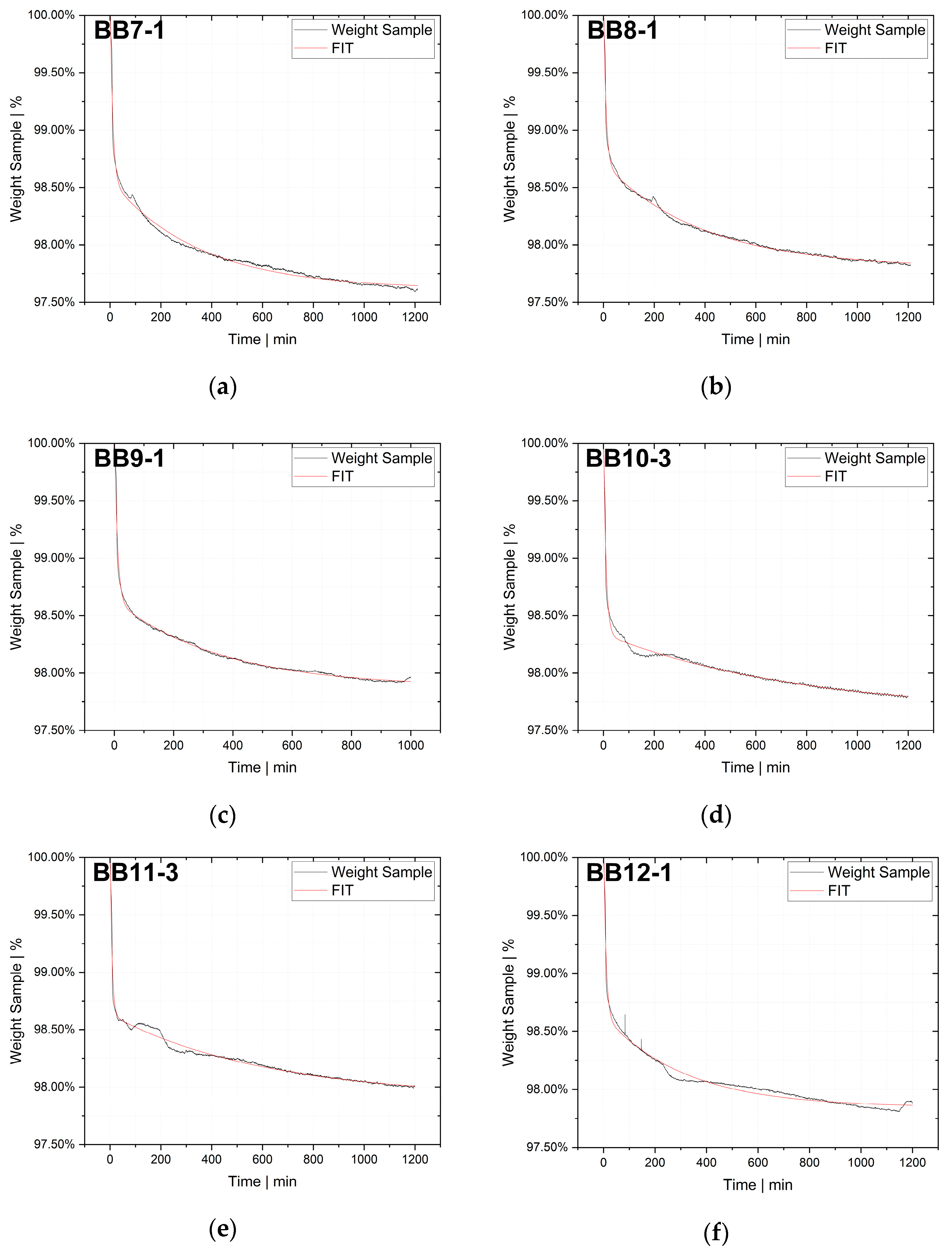
Indirect methods measure the changes in a parameter that depends on the rate of diffusion, for instance, the rate of change in solution volume or the movement of the interface (e.g., gas–liquid interface), the rate of pressure drop in a closed cell or the rate of mass loss in a closed cell. The two techniques used in this study, thermogravimetric analysis (TGA) and pressure decay apparatus (PDA), are also indirect methods.
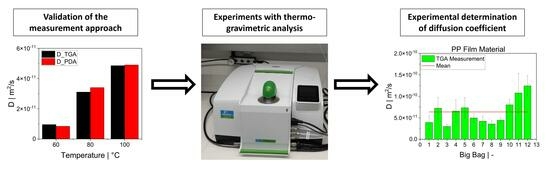
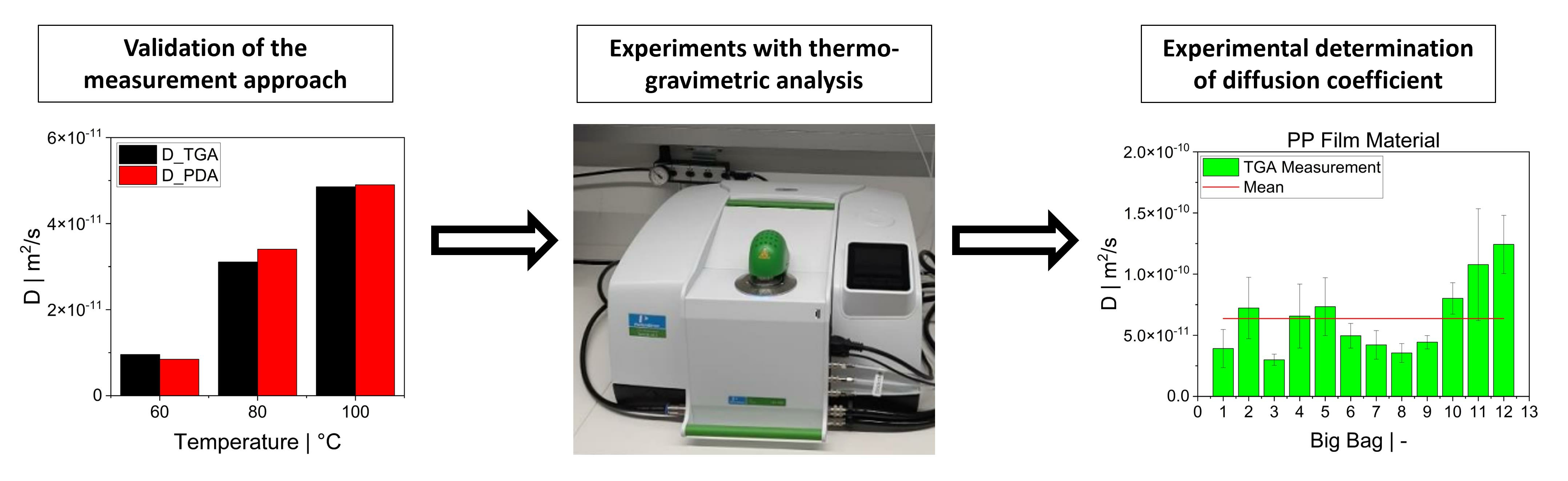
The focus of this study was on the assessment of a practically workable experimental method for evaluating diffusion coefficients under real process conditions. As the material feedstock in mechanical recycling usually exhibits some level of variation and contains unknown volatile substances, it is not possible to predict the diffusion coefficients for all binary mixtures involved. Therefore, our aim was to determine an “average” diffusion coefficient over all volatile substances at process temperatures. We present a feasible method using TGA to determine diffusion coefficients in the melt phase. Previous experimental studies on the measurement of diffusion coefficients sought to reproduce the literature values and/or were in most cases limited to a specific polymer/solvent system [
23,
24,
25]. However, reproducing these experiments is difficult because the exact measurement conditions and the experimental procedure are in most cases poorly documented. Even the concentration of the substance in the sample depends on the test environment, as the equilibrium state is a function of, for instance, temperature and pressure. Rather than validating the TGA method by using the literature values, we took a different approach by applying a second experimental method. First, the TGA method was validated with experimental results obtained from a pressure decay apparatus (PDA). For this purpose, samples of high-density polyethylene (HDPE) and polypropylene (PP) were prepared and saturated with toluene. The same samples were used in both methods. The results of the PDA served as a reference value for the TGA. After successful validation, TGA was used to determine diffusion coefficients of post-industrial PP recyclates in the melt phase.
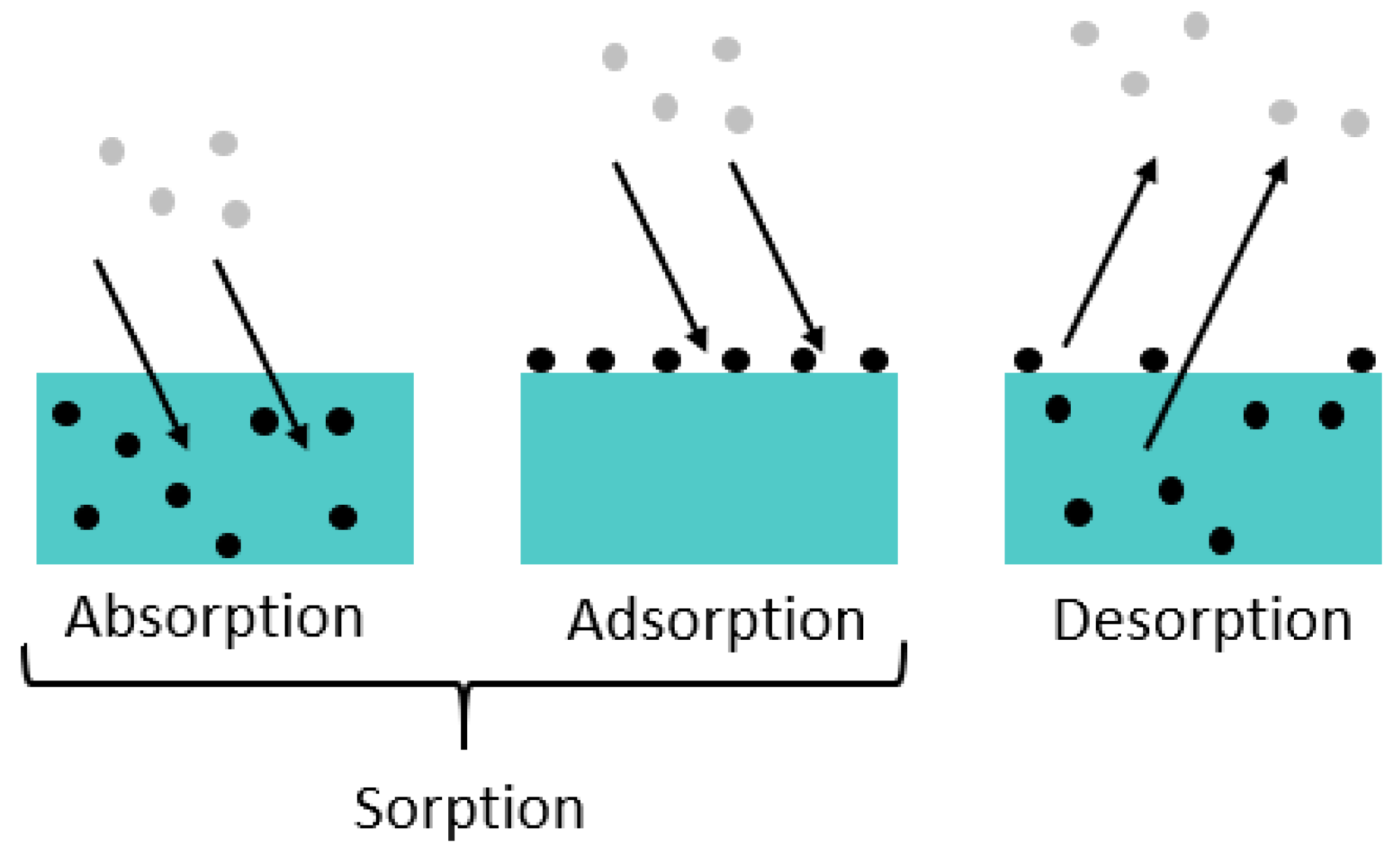
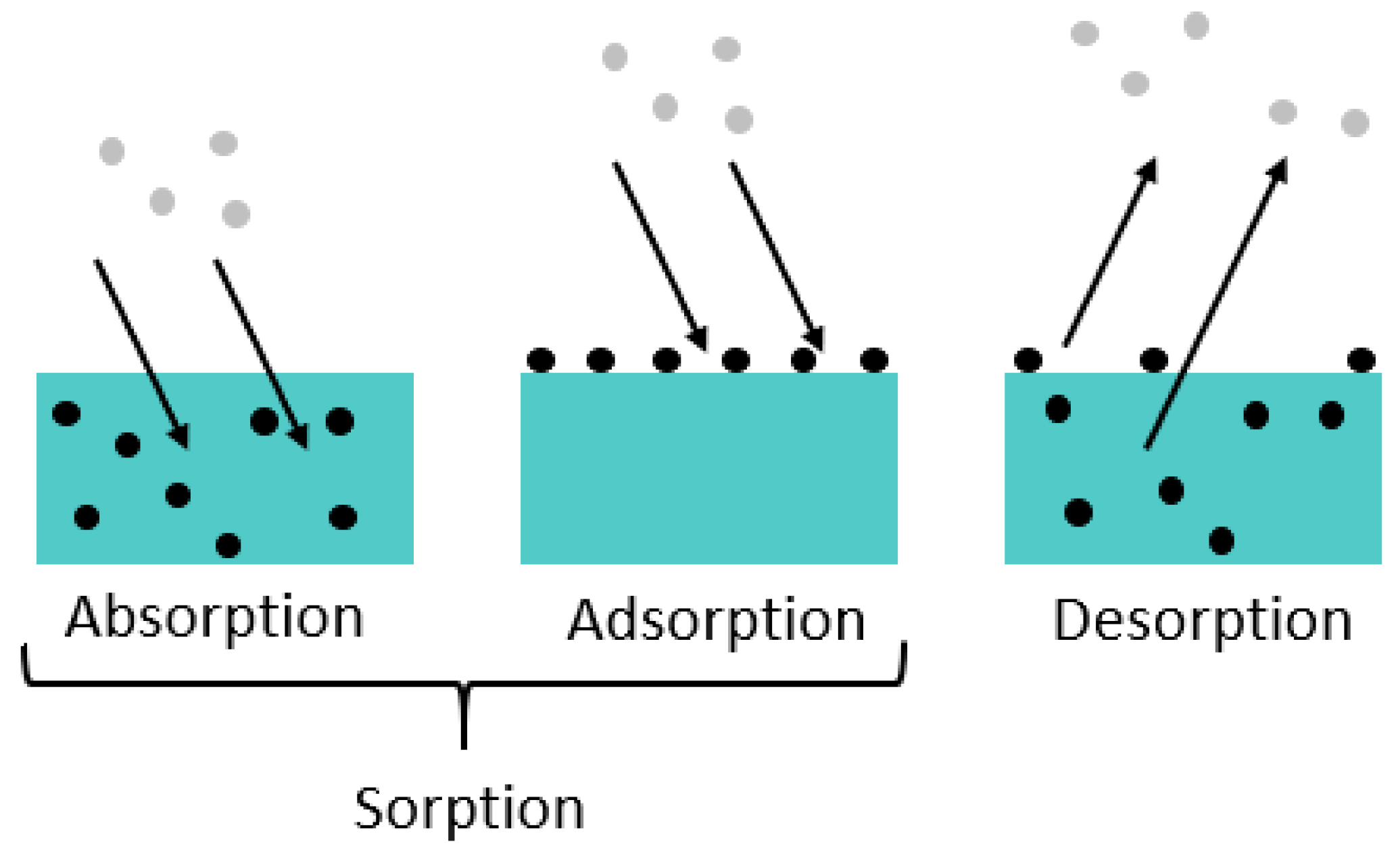
Diffusion is a physical process in which molecules of a substance become evenly distributed in a system. The Brownian motion causes the molecules to move from areas of higher concentration to areas of lower concentration. Three phenomena can be distinguished (see
Figure 3 [
26,
27,
28]): Sorption is a generic term and describes the accumulation of a particular substance in (i.e., absorption) or on (i.e., adsorption) another medium. Desorption is the reverse process, where the molecules of the dissolved substance leave, for example, the surface of a solid or the liquid in which they were dissolved.
2. Mathematical Background
Diffusion-based mass transport was described mathematically by Adolph Fick in 1855. He recognized analogies between diffusion and heat transfer by heat conduction. By adapting the equations of heat conduction, Fick established two relationships—still known today as Fick’s diffusion laws—that describe diffusion in isotropic materials [
29]. To determine the diffusion coefficient, both methods we employed (TGA and PDA) use Fick’s second law, which describes the relationship between local and temporal changes in the concentration of a substance and states (see Equation (1)) that the dispersion of a gas or liquid is proportional to the spatial concentration gradient of that substance. The proportionality factor is called the diffusion coefficient (
D) and is a measure of the velocity at which the substance is transported.
Solving Equation (1) strongly depends on the initial and boundary conditions [
29]. If the initial concentration is
or constant (i.e.,
or
) and the sample is symmetrical, then the boundaries can be defined as
or
. For a membrane, the following assumptions are made:
In the area , there is a homogeneous initial concentration , which means that .
At the interfaces , the equilibrium concentration establishes spontaneously and prevails.
The diffusion coefficient is constant, and is independent of the concentration in the sample ().
With these assumptions, Fick’s second diffusion law from Equation (1) results in the following trigonometric series:
If
is the mass of substance diffused from the sample at time
and
is the total mass of substance diffused from the sample at the thermodynamic equilibrium (steady state), the solution can be rewritten as:
Since it is assumed that the diffusion coefficient is independent of concentration (
), the mean value of the measurement is subsequently determined [
29]. An analysis of TGA measurements requires making the following assumptions:
5. Summary and Conclusions
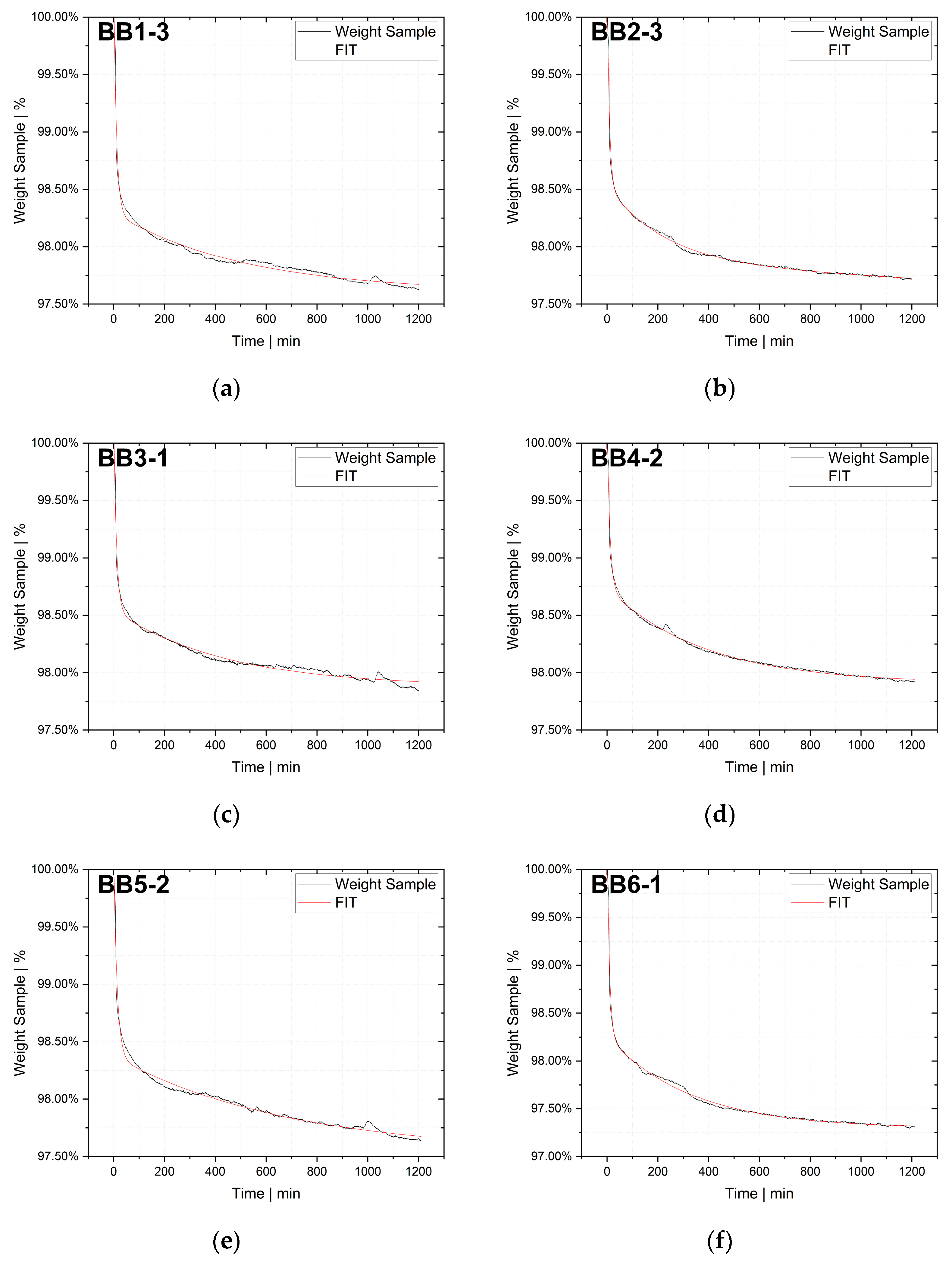
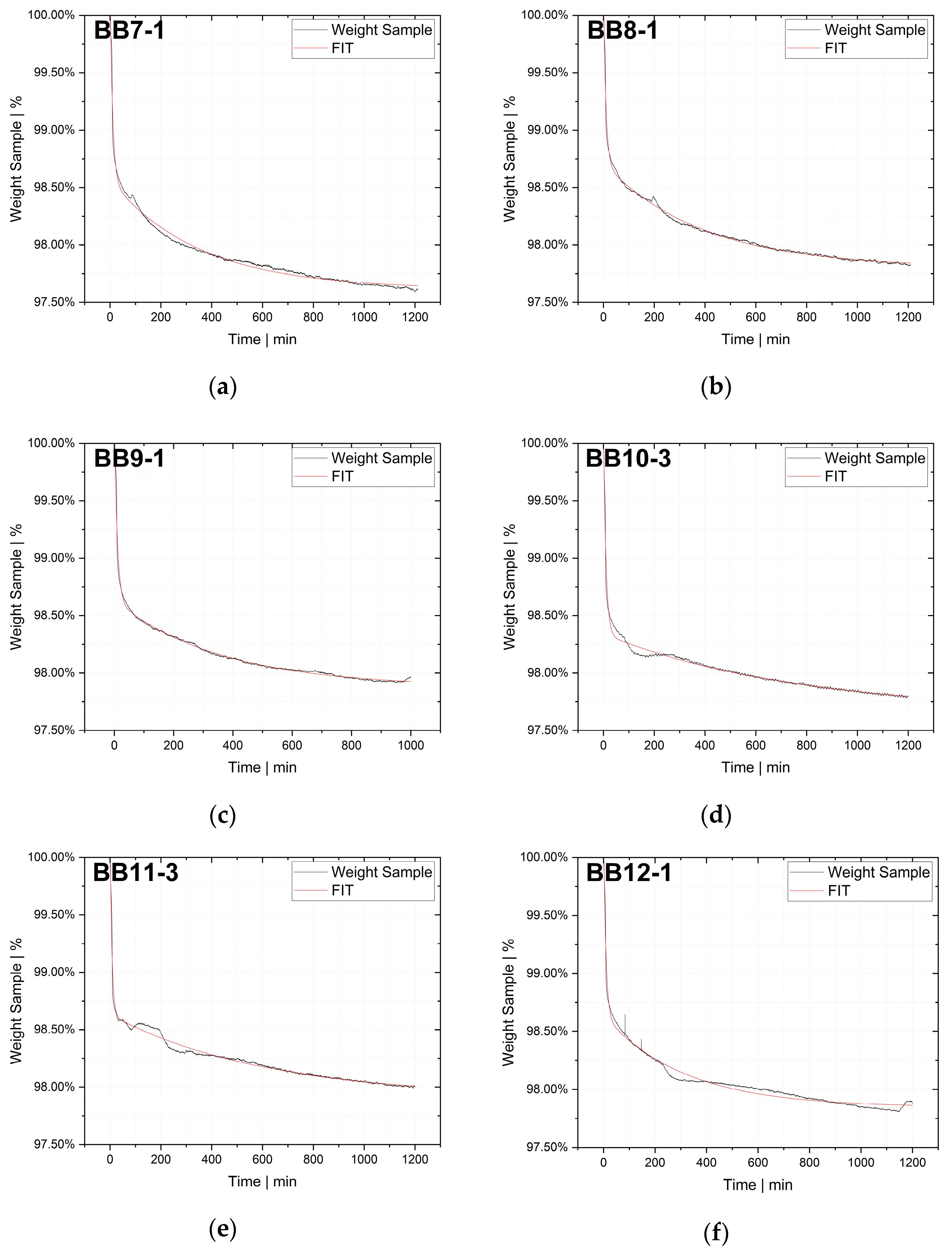
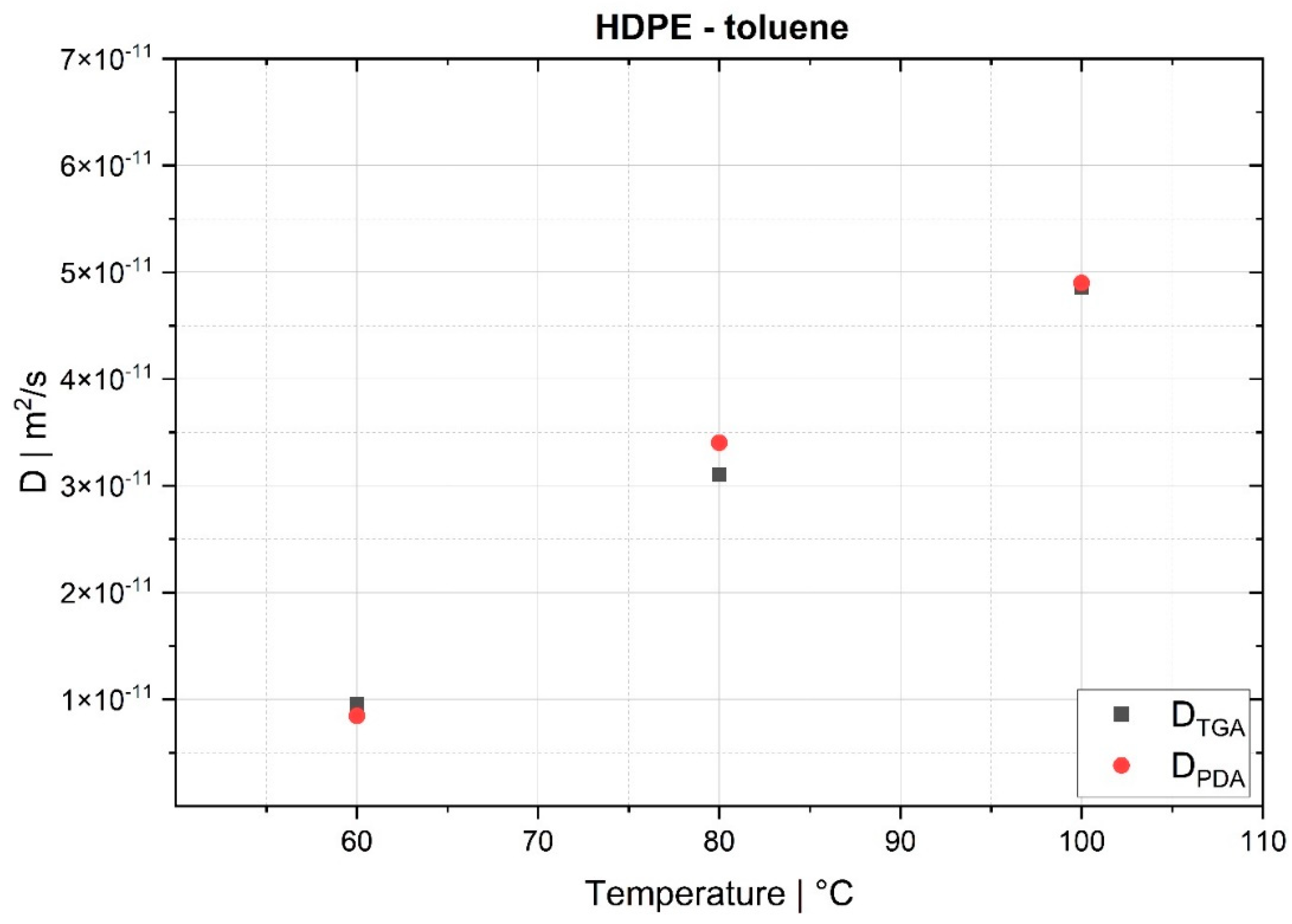
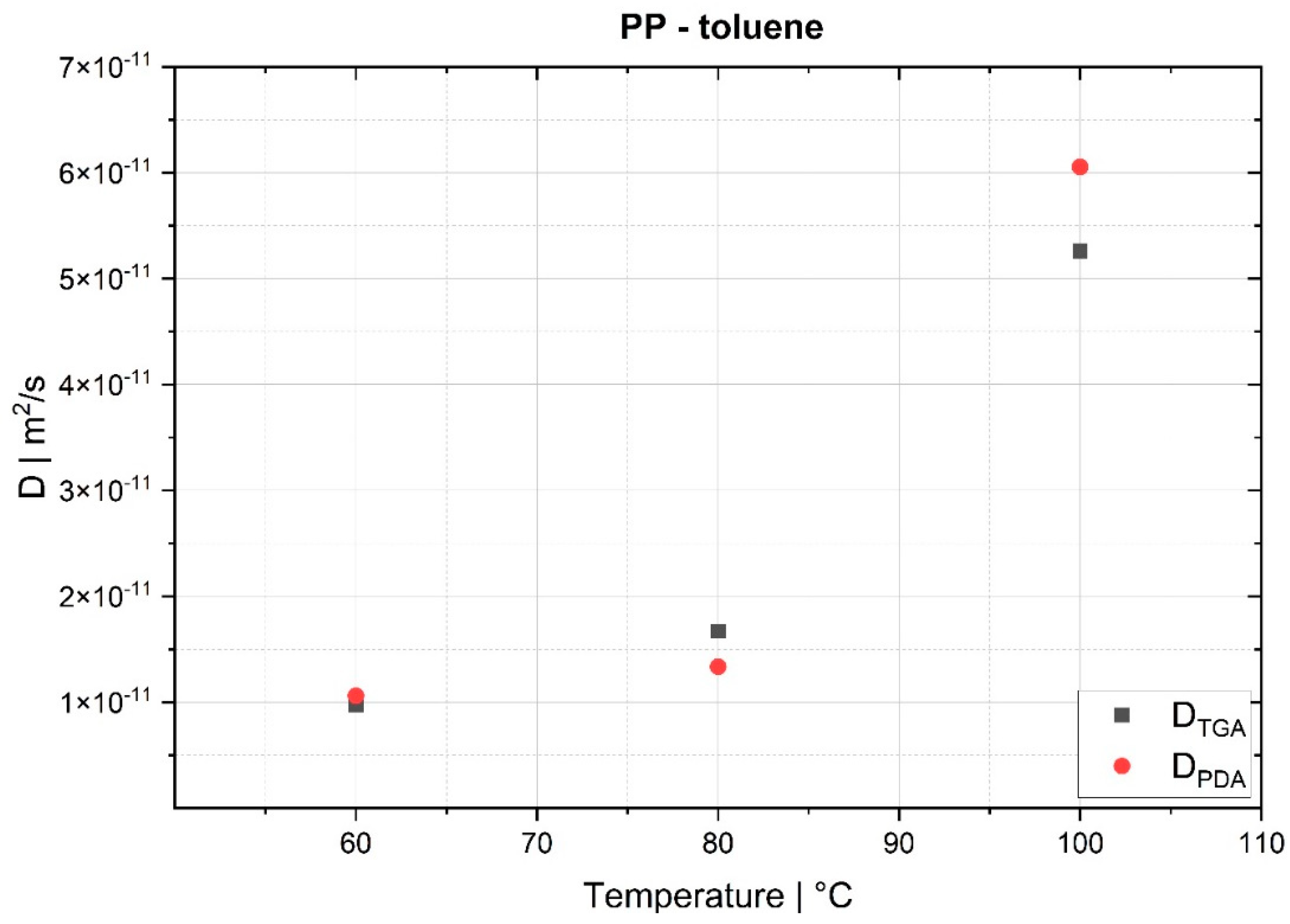
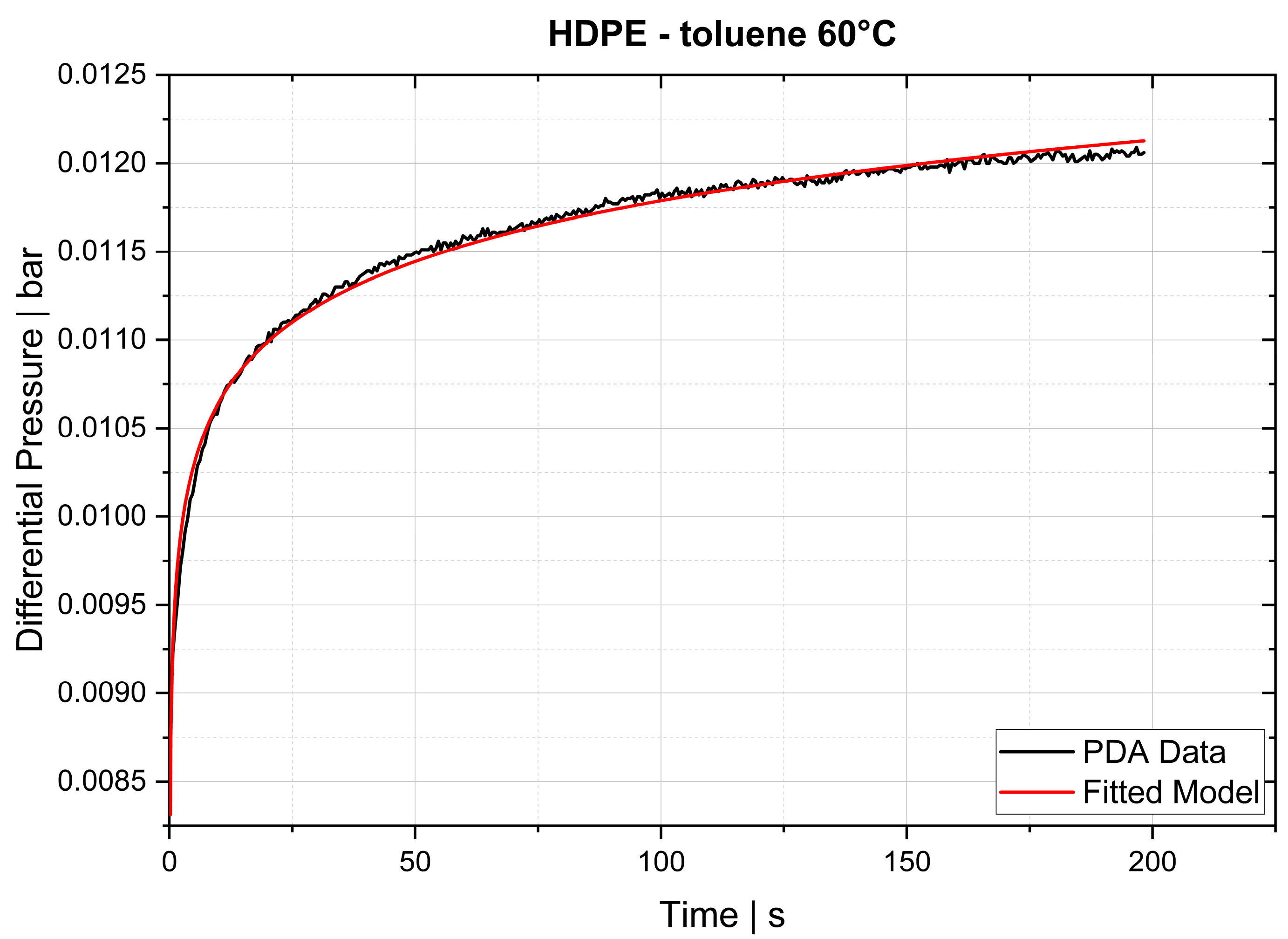
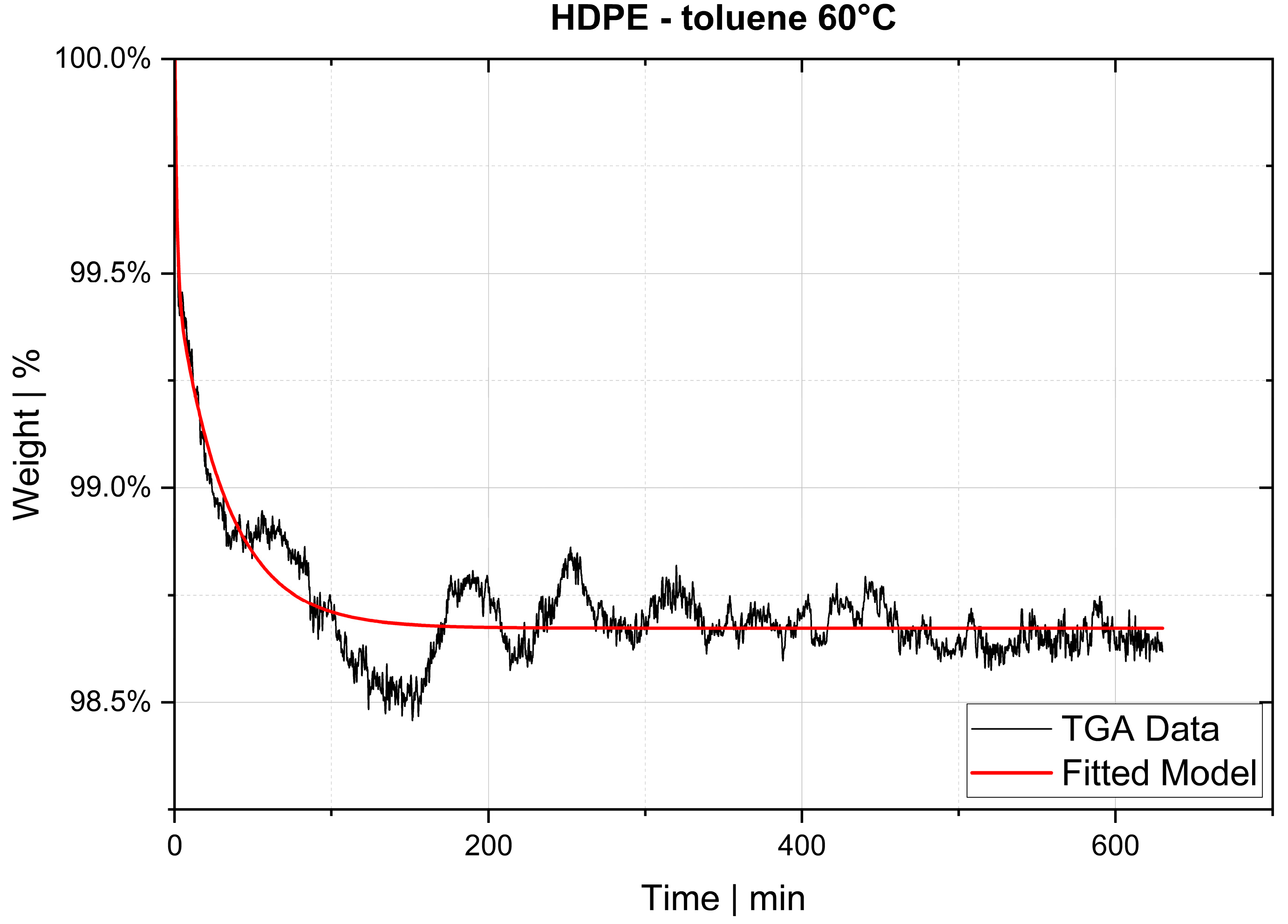
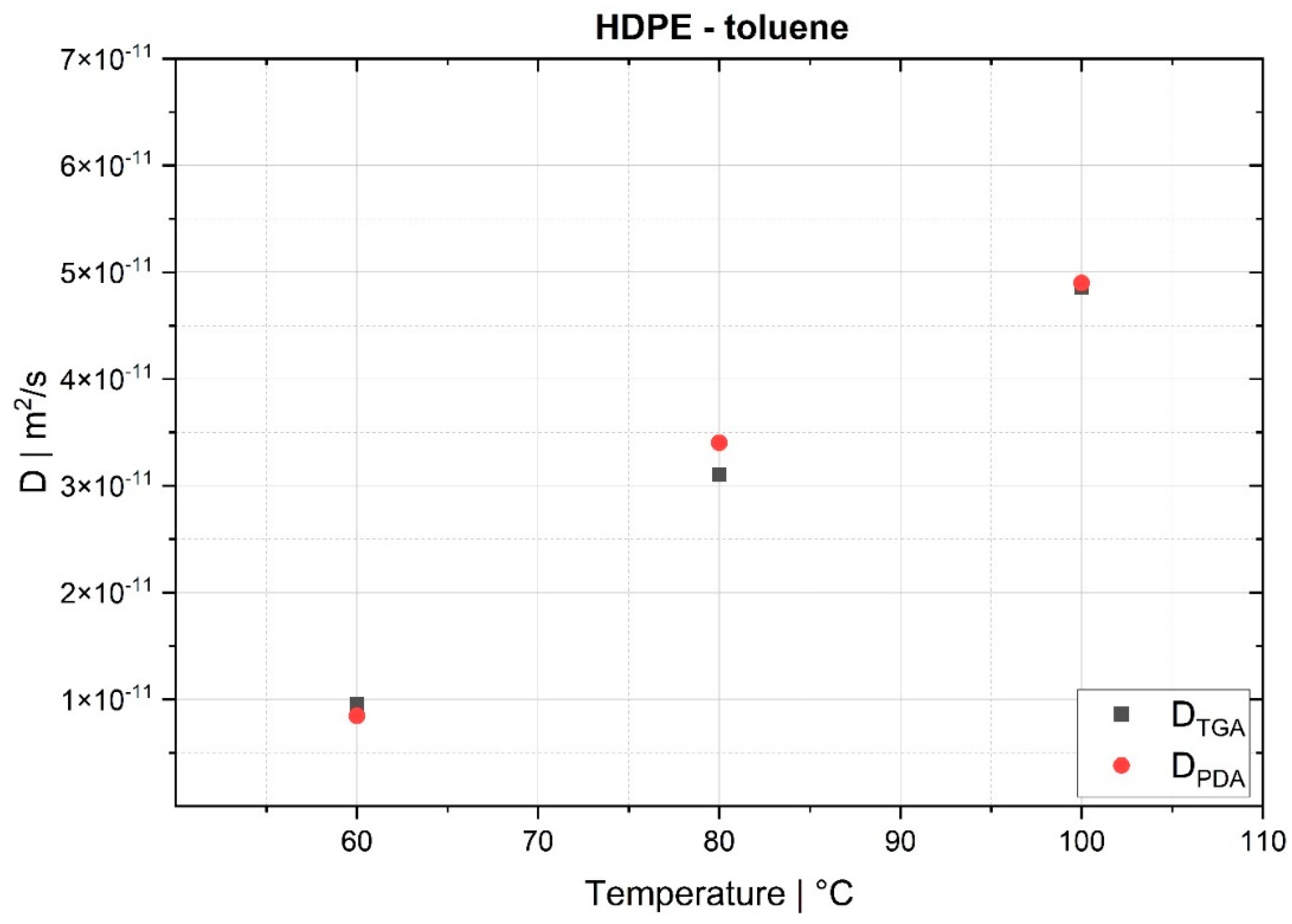
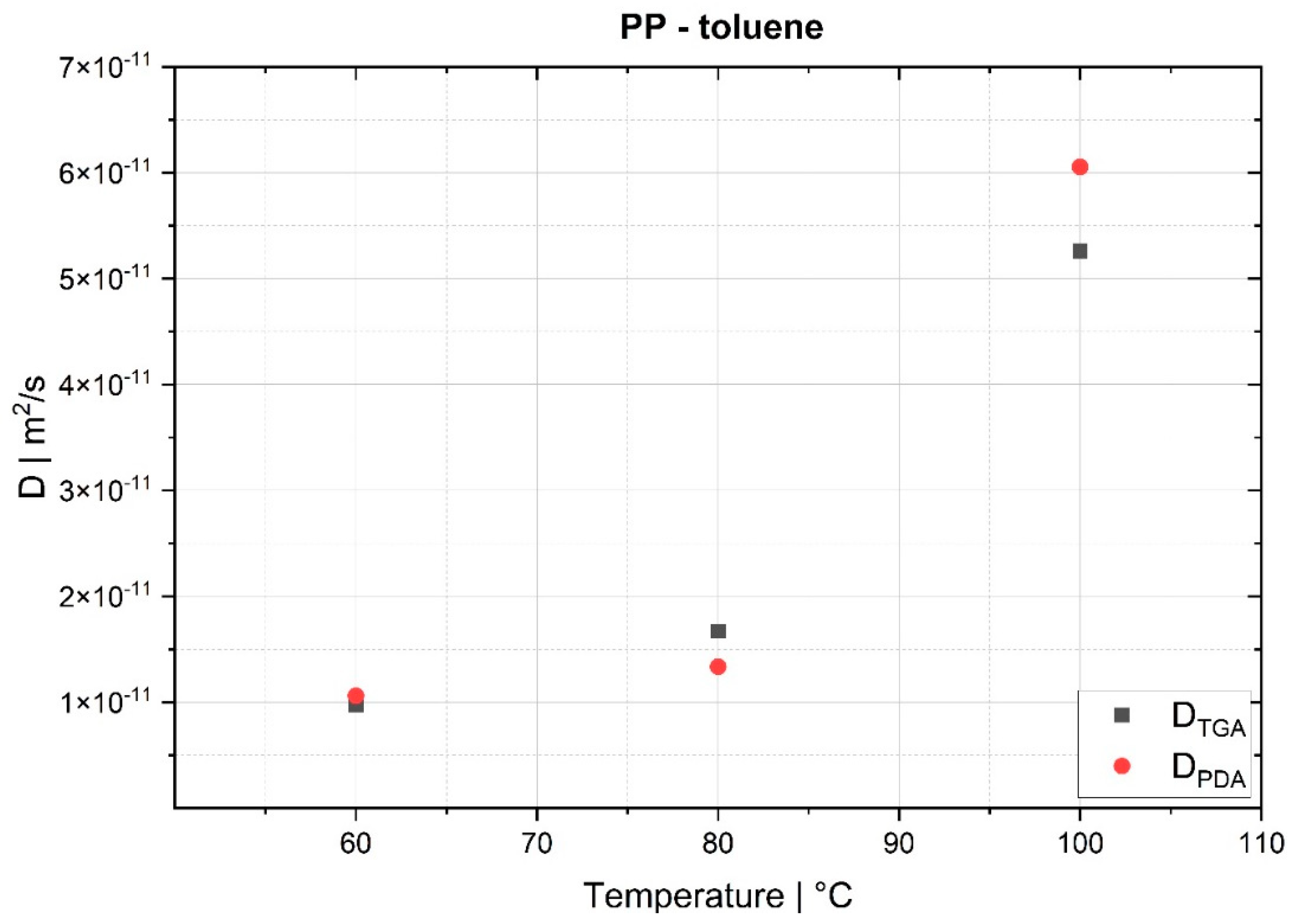
The desorption experiments in this study show that thermogravimetric analysis (TGA) offers a practical way to determine the diffusion coefficient experimentally. To validate the TGA method, both pressure decay apparatus tests and thermogravimetric analyses were performed at 60 °C, 80 °C and 100 °C. The two techniques yielded comparable results, with an average difference of 7.4% for the HDPE-toluene system and 14.7% for the PP-toluene system. Thus, TGA has potential for use in determining the diffusion coefficient in desorption experiments. The selection of a suitable method depends on many factors (e.g., application, material, and availability); in principle, both methods are suited for determining , but each method has its own advantages.
On the one hand, the great advantages of the pressure decay method are (i) that it delivers results very quickly, as the measurements take only a few minutes, and (ii) that it allows for both sorption and desorption experiments to be carried out. TGA, on the other hand, offers the advantage of being an almost universal measuring technique and thus widely used in laboratories and research institutions, rendering the acquisition of new equipment unnecessary in most cases. TGA devices are easy to operate, and the amount of work per measurement is low. Further, as demonstrated, the method shows potential for determining in the melt under process conditions. However, TGA allows for only desorption tests to be performed, and measurements take several hours, which is a considerable disadvantage compared to the pressure decay method.
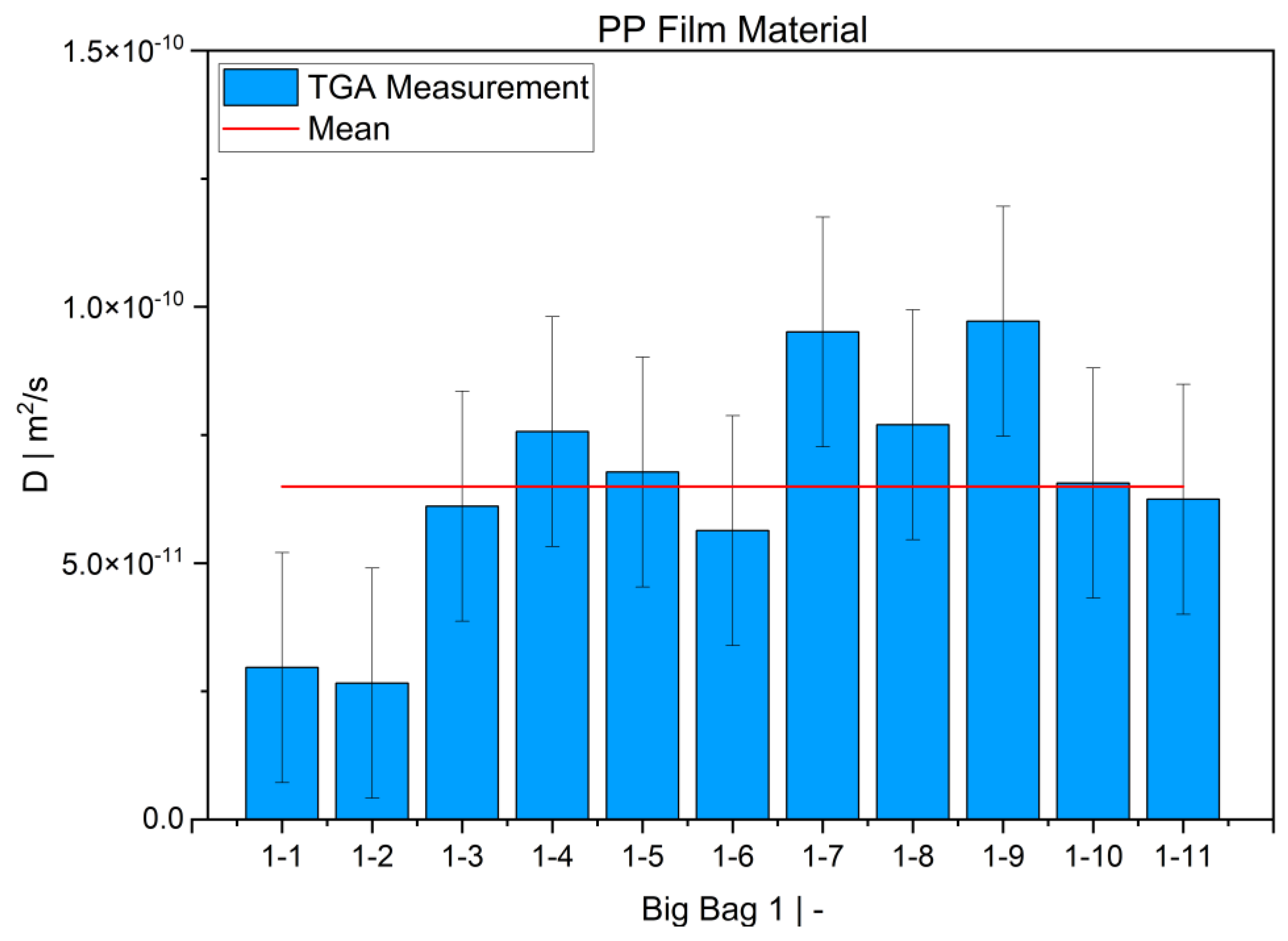
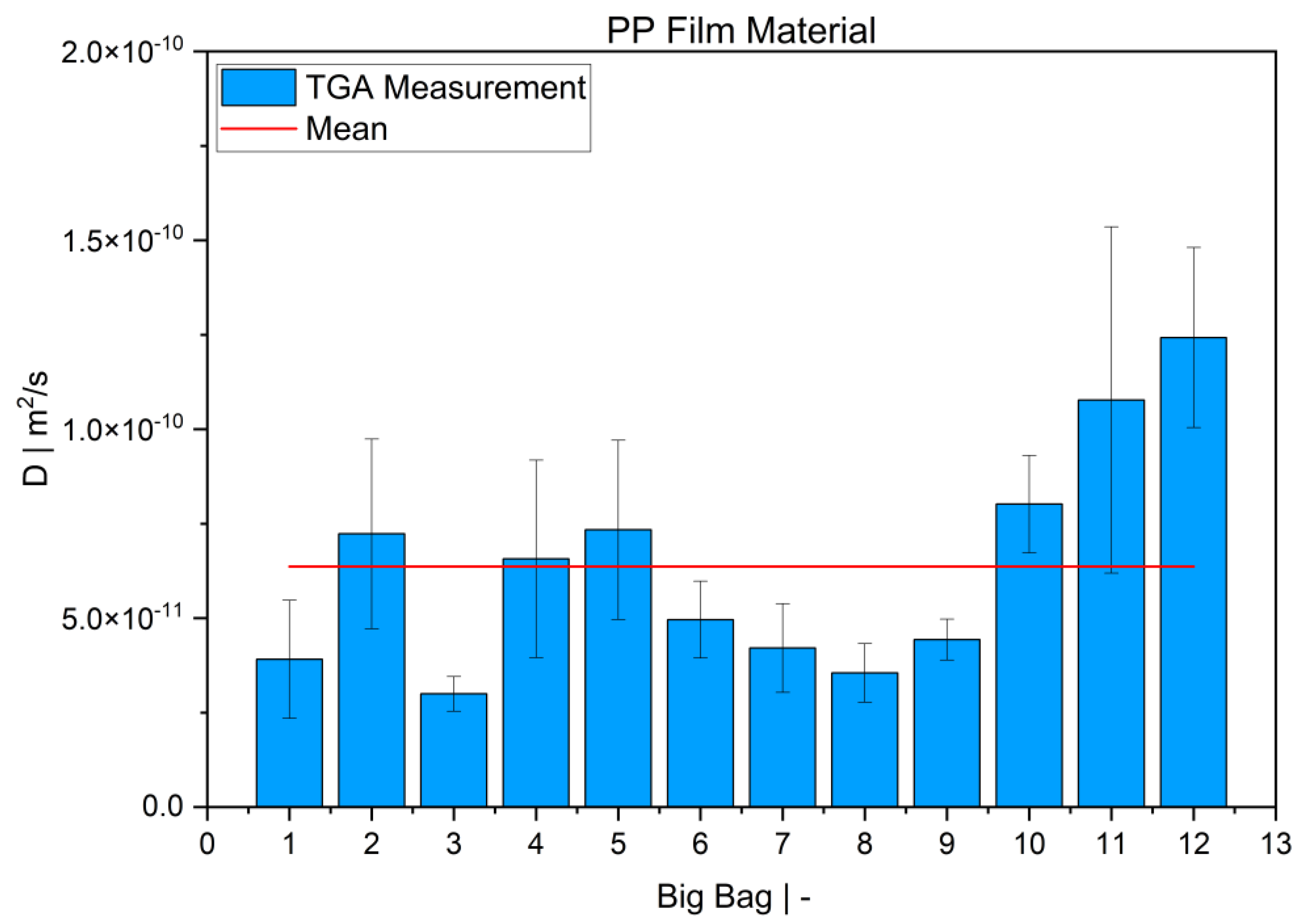
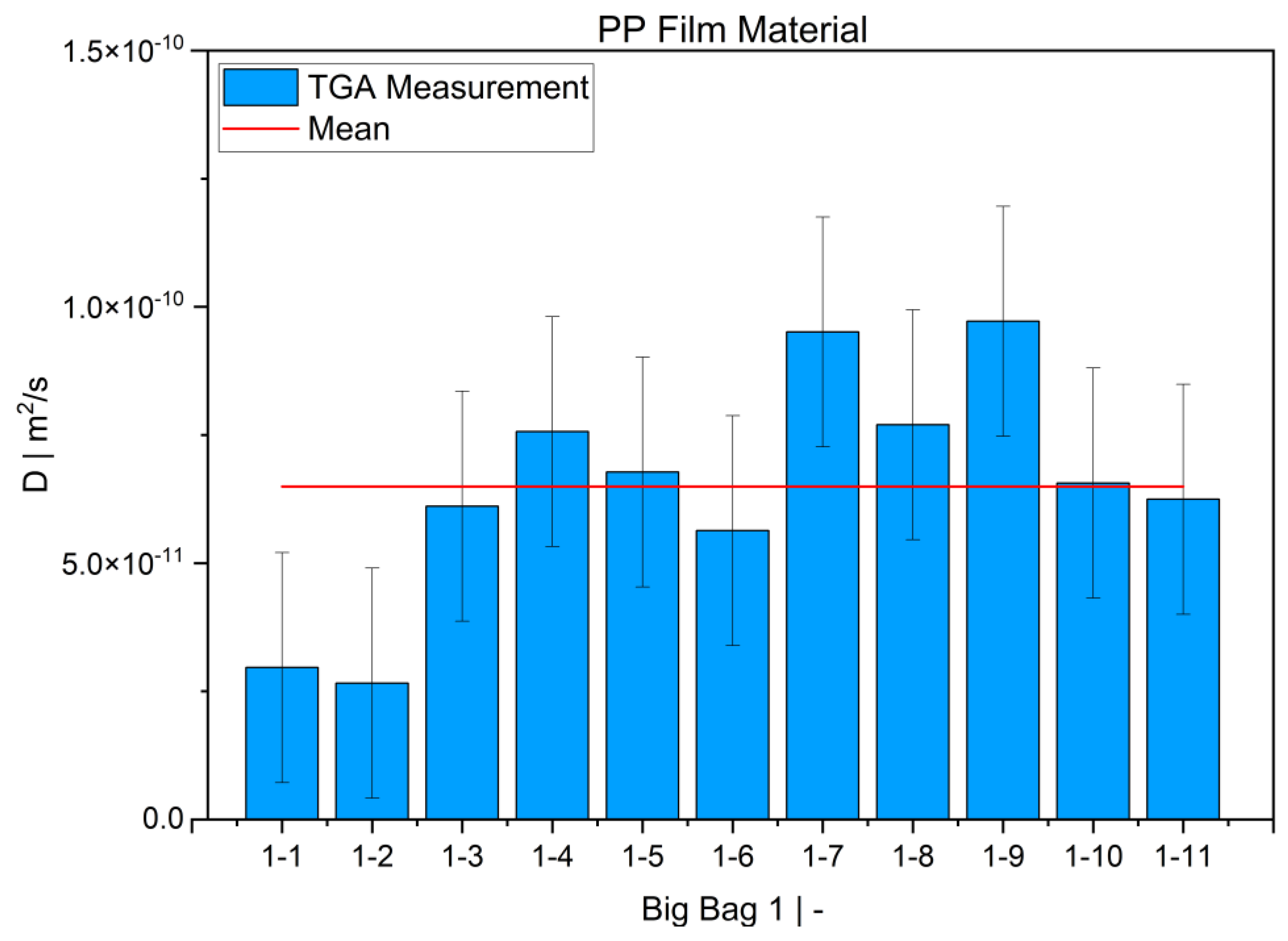
TGA was used in this study to determine the diffusion coefficient in the melt of plastic waste material at 220 °C. Care was taken to ensure that the measurements were statistically representative. All 12 big bags contributed material to the 36 TGA measurements carried out, from which the mean value was then determined. Overall, the standard deviations were low, thus indicating the reproducibility of the measurements. For big bag 1, 8 additional measurements (11 measurements in total) were taken to confirm the normal distribution of the TGA measurements in one big bag.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}