Abstract
The development of a stable catalyst with excellent catalytic performance for the oxygen evolution reaction (OER) in alkaline environments is a key reaction in various electrochemical technologies. In this work, single-atom catalysts (SACs) systems in which scandium (Sc), a rare earth metal, with different N/C coordination environments (ScNxC3−x@SACs and ScNxC4−x@SACs of Sc) were systematically studied with the help of density functional theory (DFT) calculations. The results of the structural thermodynamic stability analysis indicated that the ScNxC3−x@SACs and ScNxC4−x@SACs systems are more stable with increasing N atom doping concentration around Sc. The ScN3, ScN3C, and ScN4 with better stability were selected as the objects of subsequent research. However, ScN3 and ScN4 form Sc(OH)2N3 and Sc(OH)2N4 structures with double-hydroxyl groups as ligands because of the strong adsorption of OH species, whereas the strong adsorption of OH species by ScN3C causes structural instability. Here, the overpotential (η) of Sc(OH)2N3 was 1.03 V; Sc(OH)2N4 had two reaction paths and the η of path 1 was 0.80 V, which was 0.30 V lower than that of path 2. Therefore, Sc(OH)2N4 can be used as a stable and promising OER catalyst with easy desorption of O2 and good cycle performance. The hydroxyl ligand modification of Sc-NxC3−x@SACs and Sc-NxC4−x@SACs provides a method for studying the catalytic performance of other rare earth elements.
1. Introduction
The current society is confronted with the challenge of increasing energy demand and the associated environmental problems caused by energy consumption. As a result, the global research community has prioritized the search for pollution-free, sustainable, and efficient energy production/energy conversion technologies [1,2,3]. In this regard, the development of rechargeable fuel cells and metal–air battery technologies has been perceived as a promising clean and sustainable energy technology because of its high energy conversion efficiency and power density [4,5]. In these energy conversion technologies, the anodic half-cell reaction, i.e., oxygen evolution reaction (OER), plays a vital role in the efficiency of regenerative fuel cells and metal–air battery technologies [6,7,8]. Due to the instability of transition-metal-based materials in acidic circumstances, electrocatalytic OER is typically carried out in alkaline conditions [9]. Nonetheless, the OER is often deemed the main obstacle in rechargeable fuel cell and metal–air battery technologies because of the slow and sluggish kinetics of this four-electron transfer reaction, which restricts the high efficiency of these energy conversions [10,11].
Extensive research and a wide range of engineered catalysts have been proposed in an effort to increase OER kinetics [12,13,14,15]. Among them, RuO2 and IrO2 have exhibited excellent OER catalytic activities [16,17]. However, both are unstable during the reaction process and are oxidized to form RuO4 and IrO3, respectively, which preferentially dissolve in solution [18]. Furthermore, Ru and Ir are precious metals with scarce resources and high costs, making them unsuitable for large-scale, commercial applications [19]. Thus, it is essential to develop excellent alternative electrocatalysts for OER with better stability and to explore affordable OER electrocatalysts with good catalytic activity.
On the other hand, carbon-based catalysts, such as graphene, have advantages such as a large specific surface area, high acid/alkaline stability, and excellent electronic conductivity [20,21]. In recent years, carbon-based catalysts have become the focus of OER research. However, pure graphene is catalytically inert [20,22]. Heteroatom doping, especially N doping of graphene, is found to be the most effective method to improve the intrinsic activity of the catalyst because of the variation in the spin density and charge of the carbon atom next to the doped N atom, together with high specific surface area, excellent conductivity, and outstanding electrochemical properties [23,24,25,26]. Single-atom catalysts (SACs), in which a central single metal atom surrounded by different doping atoms forms coordination and disperses in the supporting material, have gained special interest for various catalytic reactions because of their excellent stability, maximum utilization of the metallic electrochemically active surface area, strong metal–support interactions, and unique properties [27,28,29]. Metal–organic frameworks (MOFs) have proven to be a promising approach for SAC preparation. Carbonization of MOFs can result in N-doped carbon materials with N/C coordination, which provides an abundance of anchor points for the fixation of a single metal atom, resulting in SAC stability [30,31]. Accordingly, the synthesis of single transition metal–nitrogen moiety-supported carbon catalysts (M–N–C, M = Fe/Co, etc.) as OER catalysts has been extensively investigated, and these are the most widely studied SACs [32,33]. Unfortunately, the instability of Fe–N–C SACs limits their further application owing to the high Fenton catalytic activity of Fe [34]. On the other hand, the OER overpotential in the Co-N-Gra system is 0.69 V. In addition, due to cobalt’s toxicity, poisoning can happen during the manufacturing and recycling processes, and cobalt leakage also results in battery damage [35].
Rare earth (RE) elements, such as Sc, Nd, Gd, and Ce, have attracted increasing attention in the field of electrocatalytic applications because of their unique multi-electron properties and electronic structure, high electrical conductivity, good stability, abundant reserves, and excellent electrocatalytic activity [36,37,38,39,40,41,42]. In particular, Sc atoms have the characteristics of a small atomic radius, suitability for single/double atom vacancy doping, and multi-electron properties [43,44,45]. Moreover, it has been well established that SACs with a Sc atom as the active site of the catalytic center have a good catalytic effect on CO oxidation and the reduction of CO2 and N2 [46,47,48]. Sc@SACs are synthesized on a carbon support with high yields in the literature. Catalytic activities are observed in Sc1/NC SACs despite the fact that Sc-based nanomaterials are typically inert to room temperature electrochemical processes. This is because N and C coordination alters the local electronic structure of Sc single atoms. The catalytic properties of rare earth single atoms not only showcase the miraculous impact of SACs, but also advocate for the use of rare earth catalysts in electrochemical reactions that take place at room temperature [46,47,48]. Therefore, Sc@SACs systems with varied N/C coordinations as OER catalysts for increased OER kinetics are worth investigating. However, there has been little research on the use of Sc for OER applications. As a result, we investigated the effect of Sc and its coordination environment on the OER in alkaline media.
In this work, Sc@SACs systems with different N/C coordination ratios (4 ScNxC3−x@SACs and 7 ScNxC4−x@SACs) were studied in detail by performing density functional theory (DFT) calculations. Based on the calculation of the thermodynamic formation energy, the influence of N/C coordination on ScNxC3−x@SACs and ScNxC4−x@SACs was studied. Thermodynamic stability screening indicated that the formation energies of the ScN3, ScN3C, and ScN4 structures were negative, making them more stable than other structures. Further, simulation analysis in an alkaline environment found that ScN3 and ScN4 can exist as Sc(OH)2N3 and Sc(OH)2N4, respectively, because of the strong adsorption of OH species. However, the Sc (OH)2N3C structure formed by ScN3C was unstable. The catalytic reactions of Sc(OH)2N3 and Sc(OH)2N4 structures with double-hydroxyl ligand modification for the OER under alkaline conditions were also studied. Finally, the influence of N atom doping concentration, also known as different N/C coordination and hydroxyl ligand effects, on the catalytic performance of the structure was described.
2. Computational Details
2.1. Methods
Dmol3 [49] is the algorithm used for any calculations involving density functional theory with no restrictions on spin. Electronic exchange and correlation effects were described using the generalized gradient approximation [50] with the Perdew–Burke–Ernzerhof functional. After taking into account the ion–electron interaction in the DFT semi-core pseudopotentials [51] and the double numerical plus polarization [52] basis sets, we proceeded to the core treatment. Grimme’s [53] method was used to calculate the dispersion’s share. Energy convergence tolerances were
Ha, maximum force tolerances were 0.002 Ha Å−1, and displacement tolerances were 0.005 Å. To hasten the convergence, an orbital occupancy smear of 0.005 Ha was used. The calculations used a cutoff radius for global orbits in actual space that was of fine quality. Electronic structure calculations were performed on a 15 × 15 × 1 k-point grid, whereas geometry optimization and energy calculations were performed on a 5 × 5 × 1 k-point grid in the Brillouin zone. For this exercise, we assumed that water has a dielectric constant of 78.54 and utilized the conductor-like screening model [54] to mimic this environment.
2.2. Models
To begin, a 5 × 5 × 1 periodic graphene supercell was built and separated by a 30 Å vacuum layer to eliminate periodic interactions. By substituting a Sc atom for either one or two carbon atoms, ScNxC3−x@SACs and ScNxC4−x@SACs were created. In place of the C atoms around the central Sc atom, the extremely electronegative N atoms acted as anchors. There were four structures with three coordinations (Sc-NxC3−x@SACs) and seven structures with four coordinations (Sc-NxC4−x@SACs). Three distinct architectures of ScN2C2 are depicted in Figure 1. ScN2C2-h is a hexa-atomic ring composed of two nitrogen atoms, two carbon atoms, and a Sc metal atom, whereas ScN2C2-p is a penta-atomic ring composed of two nitrogen atoms, two carbon atoms, and a Sc metal atom. The two N atoms in ScN2C2-o are situated on either side of the Sc atom.
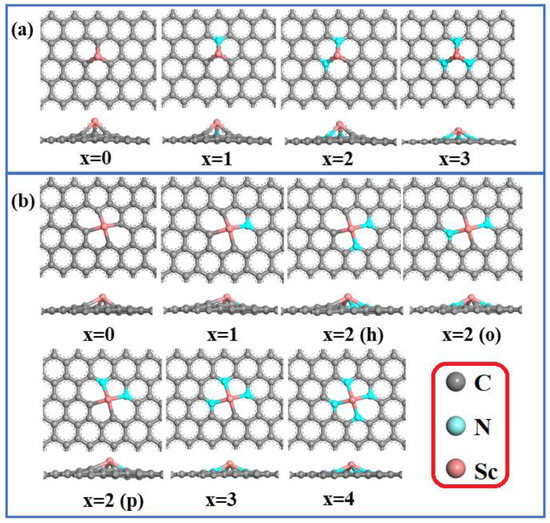
Figure 1.
Optimized geometries of the (a) Sc-NxC3−x@SACs and (b) Sc-NxC4−x@SACs structures.
2.3. Computational Contents
- (1)
- Formation energy ()
The of different ScNxC3−x@SACs and ScNxC4−x@SACs structures can be calculated as follows:
where and are the total energies of the optimized structures of ScNxC3−x@SACs and ScNxC4−x@SACs, respectively. represents the total energy of a single layer of a 5 × 5 × 1 graphene supercell. , , and are the chemical potentials of C, Sc, and N atoms, respectively. is equal to the energy of each carbon atom in the 5 × 5 × 1 graphene supercell, is the half energy of the isolation N2 molecule, and is the energy of one Sc atom, which can be obtained from the total energy of the Sc unit cell divided by the number of atoms. x + 1, x + 2 and x are the numbers of C atoms discharged from the original graphene and doped N atoms, respectively.
- (2)
- Adsorption energy ()
The of intermediates can be calculated as follows:
where , , and are the total energies of the ScNxC3−x@SACs/ScNxC4−x@SACs with n adsorbed species, the ScNxC3−x@SACs and ScNxC4−x@SACs, and the isolated n species, respectively.
- (3)
- Gibbs free energy variation ()
The of each reaction step can be calculated as follows [55]:
where , , and represent the variation in the reaction energy, zero-point energy (ZPE), and entropy, respectively. T is 298.15 K, which represents the room temperature. , where is the number of electron transfers during these elementary reactions, U is the electrode potential, and is the free energy contribution related to the applied electrode potential U. , where is the Boltzmann constant, and the value of pH is 14 in an alkaline environment.
- (4)
- Reaction steps of the OER in an alkaline environmentOH + *→OH* + eOH* + OH→O* + H2O (l) + eO* + OH→OOH* + eOOH* + OH→O2 (g) + * + H2O + e
* represents the adsorption sites on the catalyst surface. *OOH, *O, and *OH are the oxygen-containing intermediate products of the OER process. g and l represent the gas and liquid phases, respectively.
The overpotential of the OER is defined as follows (ƞOER):
ƞOER = [∆G1, ∆G2, ∆G3, ∆G4]max/e − 0.402 V
3. Results and Discussion
3.1. Stability of Sc and N Co-Doped Graphene
First, the constructed structures (ScNxC3−x@SACs and ScNxC4−x@SACs) were optimized to obtain the formation energies () of different Sc and N co-doped graphene structures, which can be used to analyze the influence of the N atom doping concentration and different coordination environments on structural stability. The formation energies of the different structures are shown in Figure 2, and the detailed values are listed in Table S1. The results showed that the structures of ScN3, ScN3C, and ScN4 were more stable than the other structures because of the negative value of the formation energy. In general, lower
values indicate higher structural stability. Furthermore, with the increase in the N atom doping concentration, the values of the ScNxC3−x@SACs and ScNxC4−x@SACs systems with three and four coordinations, respectively, decreased, and the structures were more stable, which is consistent with the phenomena of other systems studied by predecessors [56,57]. Figure 2 shows a close to negative correlation between the concentration of N atoms and formation energy, regardless of the structure of ScNxC3−x@SACs and ScNxC4−x@SACs. Moreover, in the four-coordination ScNxC4−x@SACs system, although the structures of the three kinds of ScN2C2 catalysts were different, their formation energies were similar, which is consistent with previous results [57]. However, Sc was not in the same horizontal plane as the underlying graphene in either optimized structures of ScNxC3−x@SACs or ScNxC4−x@SACs. The reason for this is that the atomic radius of Sc is larger, and the Sc atom bulges on the surface, making some structures with less N atom doping concentration more unstable. However, the bulge of the Sc atom in the stable structure is more favorable for the adsorption of reactants. Because the incorporation of N atoms can alter the structure’s electronegativity, the charge of C and N atoms coupled to Sc is higher, which results in the structures exhibiting increased levels of stability. Moreover, we demonstrated that the structures of ScN3 with three coordinations and ScN4 with four coordinations were more stable. Above all, different N/C coordination affected the stability of ScNxC3−x@SACs and ScNxC4−x@SACs, with more N atoms resulting in a more stable structure.
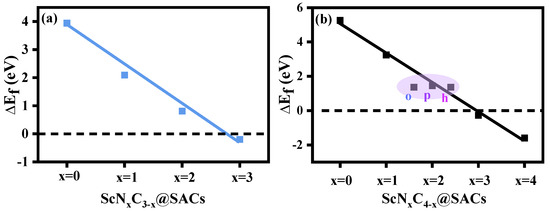
Figure 2.
Formation energy () of the various structures of (a) ScNxC3−x@SACs and (b) ScNxC4−x@SACs.
The partial densities of states (PDOS) of the ScNxC3−x@SACs and ScNxC4−x@SACs systems are shown in Figure 3. The analysis of Figure 3 shows that all the models passed through the Fermi level, indicating that all the structures had excellent conductivity. Furthermore, an effective overlap of the s, p, and d orbitals was observed, indicating that Sc formed bonds with the neighboring N and C atoms. In addition, the PDOS of the central part connected to Sc was studied. The overlap of the s, p, and d orbitals in the PDOS of the central part was more obvious, indicating that the Sc atom formed a bond with the base. The peak value of the d orbital rose with the increase of N atom doping concentration, which is similar to the law of formation energy in the ScNxC3−x@SACs system. However, in the ScNxC4−x@SACs system, although the peak value of the d orbital of ScN3C was equivalent to three types of ScN2C2 structures, it still showed an overall upward trend. Hence, different N/C coordination can significantly affect the electron distribution of the structure. The higher the concentration of N atoms is, the higher the peak value of the d orbital.
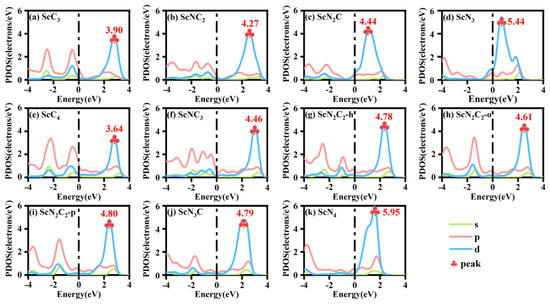
Figure 3.
Partial densities of states (PDOS) of various structures of (a–d) ScNxC3−x@SACs and (e–k) ScNxC4−x@SACs.
Based on Mulliken charge analysis, charge transfer occurred between the Sc, N, and C atoms. The electron transfer number is shown in Table S2, which indicates that the ScNxC3−x@SACs and ScNxC4−x@SACs structures formed chemical bonds, and the stability of the structure was explained from the side. In addition, different coordination structures followed the same law: with an increase in the N atom doping concentration, more electrons are lost by the Sc atom and gained by the N atoms, and the structure becomes more stable, which is consistent with the law of formation energy. Mulliken charge analysis for three-coordination ScN3 and four-coordination ScN4 with more stable and more negative values of the formation energy is further discussed, as shown in Figure 4. The charge distribution was symmetrical, corresponding to a symmetrical structure, which is consistent with the conclusion reached by other scholars on the charge distribution of symmetrical graphene-based materials [58,59]. The darker the color was, the more the charges on the structures gain or lose.
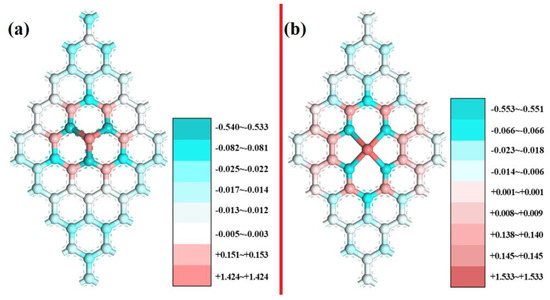
Figure 4.
Mulliken charge analysis of (a) ScN3 and (b) ScN4.
3.2. Adsorption Properties of the Intermediates
The adsorption capacity of the intermediates on the catalysts reflects their catalytic performance. Therefore, a series of adsorption configurations was obtained by constructing different intermediates and optimizing them in an alkaline environment. The adsorption of OH species was the first priority, and it indicated the availability of the reaction. In an alkaline environment, owing to the existence of a large amount of OH species and H2O in the solution, the adsorption of OH species occurs as the first step in the OER. The detailed adsorption structures for different numbers of OH species and H2O molecules are shown in Table S3. Therefore, calculation of the adsorption energy of ScNxC3−x@SACs and ScNxC4−x@SACs for OH species and H2O and the comparison of adsorption capacity are prerequisites for subsequent work. As shown in Table S4, the value of the adsorption energy of different structures for OH species was greater than that for water, which also indicates that during the follow-up of the reaction process, H2O can be desorbed in the structure to facilitate the reaction cycle. Moreover, with an increase in the concentration of doped N atoms, the adsorption capacity of the ScNxC3−x@SACs structures for OH species also increased. Although the rule of the ScNxC4−x@SACs system was not obvious, it still showed a similar general rule. Among them, N/C coordination in all N environments had the strongest adsorption capacity for OH species in ScNxC3−x@SACs and ScNxC4−x@SACs. The results are shown in Figure 5.
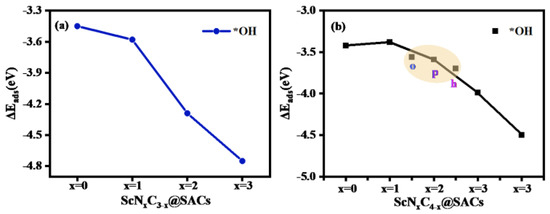
Figure 5.
Adsorption energy (, eV) of various (a) ScNxC3−x@SACs and (b) ScNxC4−x@SACs structures with adsorbed OH species.
However, based on the structures of the model, we found that the structures had a strong adsorption capacity for OH species. As the amount of OH species adsorption increased, some structures in addition to ScN3 and ScN4 in the ScNxC3−x@SACs and ScNxC4−x@SACs systems became increasingly unstable, as illustrated in Table S3. This structural instability resulted in structural deformation, which is not conducive to the study of the reaction and increases the experimental difficulty. However, after absorbing sufficient OH species, the structures of ScN3 and ScN4 could exist stably as Sc(OH)2N3 and Sc(OH)2N4. When the structure adsorbed enough OH species, further adsorption of OH species no longer occurred on the Sc atom, and they reacted with the ligand OH to generate intermediate products, as shown in Table S3. The detailed results are as follows: through structural optimization and DFT calculations, we found that three-coordination ScN3 could adsorb up to three OH species, whereas four-coordination ScN4 could adsorb up to four. When ScN4 adsorbed a fifth OH species, it no longer adsorbed it on the Sc atom as a ligand but formed an OOH structure with a hydroxyl ligand and generated H2O. While the ScN3 structure with three coordination groups adsorbed a fourth OH species, it no longer adsorbed it on the Sc atom. As the number of adsorbed OH species increased, the adsorption capacity of both for OH species decreased, as shown in Figure 6. Hence, modification of the hydroxyl ligand weakened the adsorption strength of the structure to OH species.
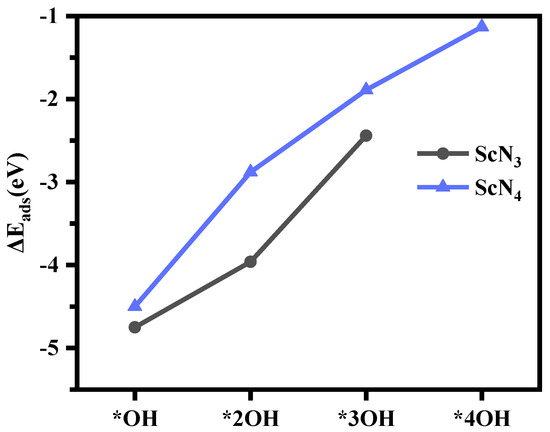
Figure 6.
The adsorption energy (, eV) of ScN3 and ScN4 with different numbers of OH species.
Therefore, ScN3 and ScN4 structures are two types of bases that can stably exist with two hydroxyl ligands modified in an alkaline environment and can be regarded as Sc(OH)2N3 and Sc(OH)2N4, respectively. A structural diagram of both is shown in Figure 7. Owing to the modification of the two hydroxyl ligands, the OH species adsorption capacity of the structure was reduced, which is beneficial for promoting the next reaction. Catalysts modified with hydroxyl ligand clusters displayed excellent OER performance [60,61]. Therefore, follow-up studies should be conducted.
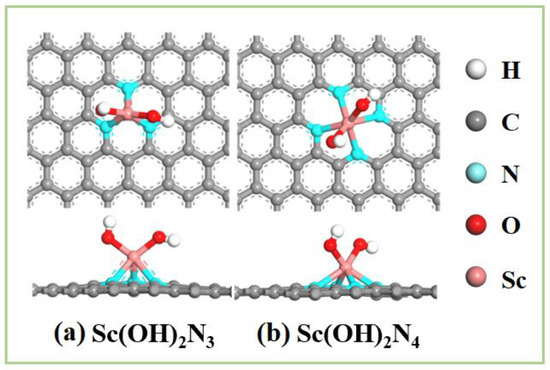
Figure 7.
Diagram of the structures of (a) Sc(OH)2N3 and (b) Sc(OH)2N4.
The cyclicity of the OER is highly dependent on the desorption of the product O2, and as the reverse process of adsorption, the degree of desorption is determined by the adsorption energy. The more positive the value of the adsorption energy is, the easier the product desorption. Next, the adsorption energies of O2 on two bases with different numbers of OH ligands were calculated, and the adsorption configurations are listed in Table S5. Among them, the desorption of O2 occurred in two forms: one was side-on (both oxygen atoms formed bonds with Sc atoms), and the other was end-on (only one oxygen atom formed a bond with the Sc atom). As shown in Figure 8, the ScN3 and ScN4 structures with double-hydroxyl ligands effectively desorbed O2 so that the catalytic reaction was repeated. Moreover, both were more favorable for O2 desorption with an increase in the number of hydroxyl ligands, and the value of the adsorption energy of O2 adsorbed by Sc(OH)2N4 was positive, as shown in Table S6. In addition, an adsorption model of the intermediates was constructed, as shown in Table S7. It can be seen from the optimized structures (Table S7) that the adsorption configurations of Sc(OH)2N3 and Sc(OH)2N4 for O2 all existed in end-on model. With the modification of two hydroxyl ligands, the adsorption capacity of the base for the next OH species was greater than that for water. The adsorption energies of Sc(OH)2N4 and Sc(OH)2N3 structures for intermediate products (OOH, O, and OH) are shown in Table S8. The values expressed that the intermediate products (OOH, O, and OH) could be effectively adsorbed on the base. The stability of Sc atoms with multiple adsorbates is shown in Table 1. This shows that the catalytic process of the formation of intermediate products (O, OH, and OOH) was stable, and the formation energy was negative.
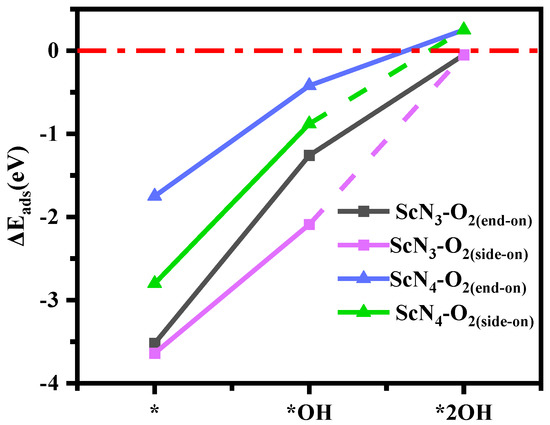
Figure 8.
Adsorption energy (ΔE, eV) of ScN3 and ScN4 with different numbers of OH ligands for adsorbed O2.

Table 1.
Formation energy data (, eV) of reactive species on Sc(OH)2N3 and Sc(OH)2N4.
3.3. The Catalytic Activity of Sc(OH)2N3 and Sc(OH)2N4
To further investigate the catalytic performance of the Sc(OH)2N3 and Sc(OH)2N4 catalysts for the OER process, the Gibbs free energy change () and overpotential (η) of each step of the reaction were calculated. First, *+OH→*OH and *OH+OH→*2OH were calculated, and the results are listed in Table S9. These values clearly indicated that ScN3 and ScN4 spontaneously generated Sc(OH)2N3 and Sc(OH)2N4, respectively, with two hydroxyl groups as ligands in an alkaline environment. It was further proven that ScN3 and ScN4 have a strong adsorption capacity for OH species. Figure 9 lists the and ⴄ of Sc(OH)2N3 and Sc(OH)2N4 catalysts for each step of reaction in the OER, and the detailed data are shown in Table S10. The potential determining step (PDS) of the reaction with Sc(OH)2N3 as the catalyst was *OH + OH→*O, and the overpotential was 1.03 V. This could be related to the strong adsorption of OH species on the catalyst. Because of the strong adsorption of OH species, formation of *O became more difficult, resulting in a higher overpotential. Two reaction paths were found when Sc(OH)2N4 was used as the catalyst. In the second step, there were two reaction pathways: *OH + OH→*O and *OH + OH→*2OH. The first reaction path was the same as that of the Sc(OH)2N3 catalyst, and the second reaction path changed to *2OH + OH→*OOH in the third step. The potential determining step of both reaction paths was the second step: path 1: *OH + OH→*O, path 2: *OH + OH→*2OH. The overpotentials were 0.80 V and 1.10 V, respectively. Additionally, because the ΔG2 of path 1 was 1.20 eV, which was 0.3 eV smaller than that of path 2, the reaction tended to proceed to the first reaction path when Sc(OH)2N4 was used as the catalyst. It can be concluded that Sc(OH)2N4 with the reaction path *→*OH→*O→*O + *OH→*OOH + *OH→* is a suitable catalyst for the OER.
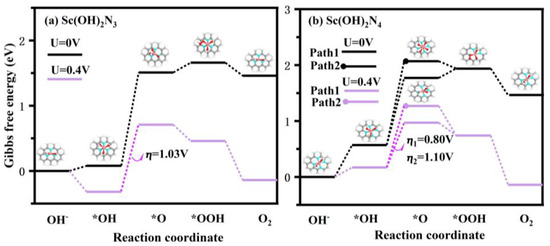
Figure 9.
Possible reaction pathways of (a) Sc(OH)2N3 and (b) Sc(OH)2N4 structures. Pink atoms: Sc, gray atoms: C, blue atoms: N, red atoms: O, white atoms: H.
In addition, the possibility of producing H2O2 via side reactions was analyzed. For Sc(OH)2N3, the *OOH + OH→*O + H2O2 reaction ( = 3.58 eV) was endothermic and required more energy than the *OOH→*( = −0.00 eV) reaction, which was exothermic. For Sc(OH)2N4, the product of H2O2 was *OOH + OH→*O + H2O2 ( = 3.56 eV) and *2OH + OH→*OH + H2O2 ( = 2.23 eV), nor is it greater than the energy needed for *OOH→* ( = −0.34 eV) and *2OH + OH→*OOH ( = 0.17 eV). The specific values are listed in Table S11. Hence, the likelihood of side reactions forming H2O2 was very low. Therefore, Sc(OH)2N4 is a promising catalyst for the OER in alkaline environment.
3.4. The Origin of Catalytic Activity of Sc(OH)2N3 and Sc(OH)2N4
The PDOS of Sc(OH)2N3 and Sc(OH)2N4 and that of other atoms connected to Sc are shown in Figure 10. The two structures passed the Fermi level, proving the electrical conductivity of Sc(OH)2N3 and Sc(OH)2N4. In addition, by studying the PDOS of s, p, and d orbitals, it is found that there is a certain degree of overlap between p and d energies, indicating that the two hydroxyl ligands formed bonds with the Sc atoms. By comparing the PDOS in the central region of Sc(OH)2N3 and Sc(OH)2N4, the peak shape of the p orbital of Sc(OH)2N3 was found to be sharper than that of Sc(OH)2N4. This is because the coordination number of Sc(OH)2N4 and the number of doped N atoms are greater. The number of electrons obtained by the N atom was higher in the Sc(OH)2N4 structure, which is consistent with the structure obtained by Mulliken charge analysis, as shown in Figure S1.
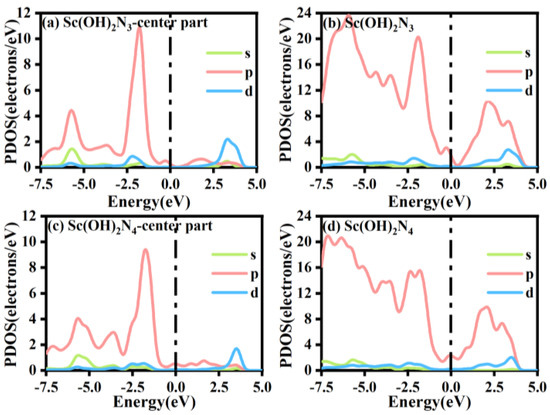
Figure 10.
The PDOS of (a) Sc(OH)2N3-center part, (b) Sc(OH)2N3, (c) Sc(OH)2N4-center part, and (d) Sc(OH)2N4.
Moreover, by comparing the PDOS of Sc(OH)2N3 and Sc(OH)2N4 with ScN3 and ScN4, we found that the number of electrons in the p orbital of the structure decreased with the modification of the two hydroxyl ligands, which can be obtained by comparing the area of the PDOS of the two structures. The peak deformation of the p orbital was sharper with the double-hydroxyl ligand modification. Second, the analysis of the PDOS comparison of Sc(OH)2N4 with ScN4 and Sc(OH)2N3 with ScN3 shows that the peaks of the 3d orbitals of Sc(OH)2N3 and Sc(OH)2N4 moved to the right, and the peak value dropped by almost three times. The structure without double-hydroxyl ligands had sharper 3d orbital peaks. This indicates that the presence of hydroxyl ligands can effectively change the properties of the structure and enhance its catalytic activity. In addition, a charge chromatic difference diagram was created by analyzing the Mulliken charges of Sc(OH)2N3 and Sc(OH)2N4, as shown in Figure S1. Compared with the results of Mulliken charge analysis of ScN4 and ScN3, the symmetry of the electron gain and loss distributions of Sc(OH)2N3 and Sc(OH)2N4 decreased under the modification of double-hydroxyl ligands. Compared with the Mulliken charge analysis structures of Sc(OH)2N3 and Sc(OH)2N4, the gain and loss charges of the main atoms were similar. Each N atom in the structure lost approximately the same amount of charge, but the N atom in Sc(OH)2N4 gained charge by a factor of three compared to the N atom in Sc(OH)2N3. All these conclusions can be drawn from Table S12.
4. Conclusions
In this study, we explored the potential of Sc as an OER catalyst. Through DFT calculations, the structures of the ScNxC3−x@SACs and ScNxC4−x@SACs systems were optimized. We concluded that the structures of both the ScNxC3−x@SACs and ScNxC4−x@SACs systems were more stable with increasing N concentration. Hence, the concentration of N atoms can significantly affect the structural stability. In consideration of the N/C coordination in all N environments of ScNxC3−x@SACs and ScNxC4−x@SACs systems, ScN3 and ScN4 were the most stable structures. Moreover, the ScNxC3−x@SACs and ScNxC4−x@SACs systems exhibited a strong adsorption capacity for OH species in alkaline environments, and Sc(OH)2N3 and Sc(OH)2N4 structures with double-hydroxyl ligands could exist stably in alkaline environments. Sc(OH)2N3 and Sc(OH)2N4 could effectively desorb O2 and H2O to ensure a repeated catalytic sequence for the OER. The potential determining step (PDS) of Sc(OH)2N3 was *OH + OH→*O, and its overpotential (η) was 1.03 V. Sc(OH)2N4 had two reaction paths, and the potential determining steps of the two paths were as follows: path 1: *OH + OH→*O; path 2: *OH + OH→*2OH (η1 = 0.80 V; η2 = 1.10 V). In conclusion, Sc(OH)2N4 can be used as an effective catalyst for the OER because of its reasonable overpotential and structural stability. Meanwhile, PDOS and charge transfer analysis showed that double-hydroxyl ligands, as special modified ligands, could improve the catalytic activity of the reaction. Furthermore, different coordination structures affect catalytic activity. The method of preparing SACs based on metal–organic frameworks makes possible the experimental study of Sc@SACs. It is expected that our research will promote the development of rare earth catalysis.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/batteries9030175/s1.
Author Contributions
Conceptualization, Y.L., M.L., Y.H. and C.L.; methodology, Y.L., M.L. and Y.H.; software, Y.L., M.L. and Y.H.; validation, S.G.P. and C.L.; formal analysis, R.K.; investigation, Y.L.; resources, C.L.; data curation, Y.L.; writing—original draft preparation, Y.L.; writing—review and editing, C.L., R.K., T.-G.L. and S.G.P.; supervision, C.L. and S.G.P.; project administration, C.L. and S.G.P.; funding acquisition, C.L. and S.G.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Natural Science Foundation of Jiangxi Province (Grant nos. 20212BAB203015 and 20212BCJL23053), the Research Foundation of the Education Department of Jiangxi Province (Grant no. GJJ200808), and the Program of Qingjiang Excellent Young Talents (JXUSTQJYX2020002), Jiangxi University of Science and Technology. The authors also thank the National Research Foundation of Korea (NRF), funded by the Korean government, Ministry of Science, and ICT (MSIT) (No. 2021R1F1A1046648), Republic of Korea.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare that they have no known competing financial interests or personal relationships that could have influenced the work reported in this study.
References
- Güney, T. Renewable energy consumption and sustainable development in high-income countries. Int. J. Sustain. Dev. World Ecol. 2021, 28, 376–385. [Google Scholar] [CrossRef]
- Güney, T. Renewable energy, non-renewable energy and sustainable development. Int. J. Sustain. Dev. World Ecol. 2019, 26, 389–397. [Google Scholar] [CrossRef]
- Hao, J.; Yuan, L.; Zhu, Y.; Jaroniec, M.; Qiao, S. Triple-Function Electrolyte Regulation toward Advanced Aqueous Zn-Ion Batteries. Adv. Mater. 2022, 34, 2206963. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.Z.; Rehman, A.-U.; Siddique, S. Prospects and challenges of graphene based fuel cells. J. Energy Chem. 2019, 39, 217–234. [Google Scholar] [CrossRef]
- Zhang, J.; Xia, Z.; Dai, L. Carbon-based electrocatalysts for advanced energy conversion and storage. Sci. Adv. 2015, 1, e1500564. [Google Scholar] [CrossRef]
- Wang, K.; Wang, X.; Li, Z.; Yang, B.; Ling, M.; Gao, X.; Lu, J.; Shi, Q.; Lei, L.; Wu, G.; et al. Designing 3d dual transition metal electrocatalysts for oxygen evolution reaction in alkaline electrolyte: Beyond oxides. Nano Energy 2020, 77, 105162. [Google Scholar] [CrossRef]
- Dang, N.K.; Tiwari, J.N.; Sultan, S.; Meena, A.; Kim, K.S. Multi-site catalyst derived from Cr atoms-substituted CoFe nanoparticles for high-performance oxygen evolution activity. Chem. Eng. J. 2021, 404, 126513. [Google Scholar] [CrossRef]
- Zheng, X.; Yang, J.; Xu, Z.; Wang, Q.; Wu, J.; Zhang, E.; Dou, S.; Sun, W.; Wang, D.; Li, Y. Ru–Co Pair Sites Catalyst Boosts the Energetics for the Oxygen Evolution Reaction. Angew. Chem. Int. Ed. 2022, 61, e202205946. [Google Scholar] [CrossRef]
- An, L.; Wei, C.; Lu, M.; Liu, H.; Chen, Y.; Scherer, G.G.; Fisher, A.C.; Xi, P.; Xu, Z.J.; Yan, C.H. Recent Development of Oxygen Evolution Electrocatalysts in Acidic Environment. Adv. Mater. 2021, 33, e2006328. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Wu, Y.; Wang, Y.; Zhang, H.-N.; Zhu, L.-H.; Wang, X.-C. Electronic properties of double-atom catalysts for electrocatalytic oxygen evolution reaction in alkaline solution: A DFT study. Nanoscale 2022, 14, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Long, X.; Xiao, J.; Zhang, Z.; Liu, G.; Tong, H.; Liu, Z.; Li, N.; Qian, D.; Li, J.; et al. Rationally constructing CoO and CoSe2 hybrid with CNTs-graphene for impressively enhanced oxygen evolution and DFT calculations. Chem. Eng. J. 2021, 422, 129982. [Google Scholar] [CrossRef]
- Charles, V.; Anumah, A.O.; Adegoke, K.A.; Adesina, M.O.; Ebuka, I.P.; Gaya, N.A.; Ogwuche, S.; Yakubu, M.O. Progress and challenges pertaining to the earthly-abundant electrocatalytic materials for oxygen evolution reaction. Sustain. Mater. Technol. 2021, 28, e00252. [Google Scholar] [CrossRef]
- Ji, Y.; Dong, H.; Liu, C.; Li, Y. The progress of metal-free catalysts for the oxygen reduction reaction based on theoretical simulations. J. Mater. Chem. A 2018, 6, 13489–13508. [Google Scholar] [CrossRef]
- Suen, N.-T.; Hung, S.-F.; Quan, Q.; Zhang, N.; Xu, Y.-J.; Chen, H.M. Electrocatalysis for the oxygen evolution reaction: Recent development and future perspectives. Chem. Soc. Rev. 2017, 46, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, X.F.; Zang, S.; Lou, X.W. Non-Noble-Metal-Based Electrocatalysts toward the Oxygen Evolution Reaction. Adv. Funct. Mater. 2020, 30, 1910274. [Google Scholar] [CrossRef]
- Sun, W.; Zhou, Z.; Zaman, W.Q.; Cao, L.-M.; Yang, J. Rational Manipulation of IrO2 Lattice Strain on α-MnO2 Nanorods as a Highly Efficient Water-Splitting Catalyst. ACS Appl. Mater. Interfaces 2017, 9, 41855–41862. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Suntivich, J.; May, K.J.; Perry, E.E.; Shao-Horn, Y. Synthesis and Activities of Rutile IrO2 and RuO2 Nanoparticles for Oxygen Evolution in Acid and Alkaline Solutions. J. Phys. Chem. Lett. 2012, 3, 399–404. [Google Scholar] [CrossRef]
- Chen, Y.; Rui, K.; Zhu, J.; Dou, S.X.; Sun, W. Recent Progress on Nickel-Based Oxide/(Oxy)Hydroxide Electrocatalysts for the Oxygen Evolution Reaction. Chem. Eur. J. 2019, 25, 703–713. [Google Scholar] [CrossRef]
- Ďurovič, M.; Hnát, J.; Bouzek, K. Electrocatalysts for the hydrogen evolution reaction in alkaline and neutral media. A comparative review. J. Power Sources 2021, 493, 229708. [Google Scholar] [CrossRef]
- Wang, C.; Tong, H.; Lu, J.; Liu, B.; Zheng, F.; Tao, W.; Zhang, W.; Chen, Q. Boosting oxygen evolution reaction on graphene through engineering electronic structure. Carbon 2020, 170, 414–420. [Google Scholar] [CrossRef]
- Sadeghi, S.; Amani, M. Co-doped triel–pnicogen graphene as metal-free catalyst for CO oxidation: Role of multi-center covalency. J. Mol. Model. 2019, 25, 77. [Google Scholar] [CrossRef]
- Zhou, X.; Kang, L. A DFT study of graphene-FeNx (x = 4, 3, 2, 1) catalysts for acetylene hydrochlorination. Colloids Surfaces A Physicochem. Eng. Asp. 2021, 618, 126495. [Google Scholar] [CrossRef]
- Ede, S.R.; Luo, Z. Tuning the intrinsic catalytic activities of oxygen-evolution catalysts by doping: A comprehensive review. J. Mater. Chem. A 2021, 9, 20131–20163. [Google Scholar] [CrossRef]
- Li, F.; Shu, H.; Liu, X.; Shi, Z.; Liang, P.; Chen, X. Electrocatalytic Activity and Design Principles of Heteroatom-Doped Graphene Catalysts for Oxygen-Reduction Reaction. J. Phys. Chem. C 2017, 121, 14434–14442. [Google Scholar] [CrossRef]
- Bai, J.; Zhu, Q.; Lv, Z.; Dong, H.; Yu, J.; Dong, L. Nitrogen-doped graphene as catalysts and catalyst supports for oxygen reduction in both acidic and alkaline solutions. Int. J. Hydrogen Energy 2013, 38, 1413–1418. [Google Scholar] [CrossRef]
- Wang, T.; Sang, X.; Zheng, W.; Yang, B.; Yao, S.; Lei, C.; Li, Z.; He, Q.; Lu, J.; Lei, L.; et al. Gas Diffusion Strategy for Inserting Atomic Iron Sites into Graphitized Carbon Supports for Unusually High-Efficient CO 2 Electroreduction and High-Performance Zn–CO2 Batteries. Adv. Mater. 2020, 32, 2002430. [Google Scholar] [CrossRef]
- Lee, W.H.; Ko, Y.-J.; Kim, J.-Y.; Min, B.K.; Hwang, Y.J.; Oh, H.-S. Single-atom catalysts for the oxygen evolution reaction: Recent developments and future perspectives. Chem. Commun. 2020, 56, 12687–12697. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.; Lee, Y.J.; Jan, A.; Choi, S.M.; Park, M.Y.; Choi, S.; Hwang, J.Y.; Hong, S.; Park, S.G.; Chang, H.J.; et al. Highly active and thermally stable single-atom catalysts for high-temperature electrochemical devices. Energy Environ. Sci. 2020, 13, 4903–4920. [Google Scholar] [CrossRef]
- Kim, J.; Roh, C.-W.; Sahoo, S.K.; Yang, S.; Bae, J.; Han, J.W.; Lee, H. Highly Durable Platinum Single-Atom Alloy Catalyst for Electrochemical Reactions. Adv. Energy Mater. 2018, 8, 1701476. [Google Scholar] [CrossRef]
- Zou, L.; Wei, Y.-S.; Hou, C.-C.; Li, C.; Xu, Q. Single-Atom Catalysts Derived from Metal–Organic Frameworks for Electrochemical Applications. Small 2021, 17, 2004809. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zhang, J.; Chen, J.; Chen, Y.; Zhang, C.; Luo, Y.; Wang, G.; Wang, R. Bi-functional electrocatalysis through synergetic coupling strategy of atomically dispersed Fe and Co active sites anchored on 3D nitrogen-doped carbon sheets for Zn-air battery. J. Catal. 2021, 397, 223–232. [Google Scholar] [CrossRef]
- Kattel, S.; Wang, G. Reaction Pathway for Oxygen Reduction on FeN4 Embedded Graphene. J. Phys. Chem. Lett. 2014, 5, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Kattel, S.; Wang, G. A density functional theory study of oxygen reduction reaction on Me–N4 (Me = Fe, Co, or Ni) clusters between graphitic pores. J. Mater. Chem. A 2013, 1, 10790–10797. [Google Scholar] [CrossRef]
- Ding, S.; Lyu, Z.; Sarnello, E.; Xu, M.; Fang, L.; Tian, H.; Karcher, S.E.; Li, T.; Pan, X.; McCloy, J.; et al. A MnOx enhanced atomically dispersed iron–nitrogen–carbon catalyst for the oxygen reduction reaction. J. Mater. Chem. A 2022, 10, 5981–5989. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Z.; Lu, Z.; Wang, W. Bifunctional CoNx embedded graphene electrocatalysts for OER and ORR: A theoretical evaluation. Carbon 2018, 130, 112–119. [Google Scholar] [CrossRef]
- Wang, R.; Lu, G.; Qiao, W.; Sun, Z.; Zhuang, H.; Yu, J. Catalytic effect of praseodymium oxide additive on the microstructure and electrical property of graphite anode. Carbon 2015, 95, 940–948. [Google Scholar] [CrossRef]
- Kim, H.J.; Shin, D.; Jeong, H.; Jang, M.G.; Lee, H.; Han, J.W. Design of an Ultrastable and Highly Active Ceria Catalyst for CO Oxidation by Rare-Earth- and Transition-Metal Co-Doping. ACS Catal. 2020, 10, 14877–14886. [Google Scholar] [CrossRef]
- Li, J.; Chu, B.; Xie, Z.; Deng, Y.; Zhou, Y.; Dong, L.; Li, B.; Chen, Z. Mechanism and DFT Study of Degradation of Organic Pollutants on Rare Earth Ions Doped TiO2 Photocatalysts Prepared by Sol-Hydrothermal Synthesis. Catal. Lett. 2022, 152, 489–502. [Google Scholar] [CrossRef]
- Chen, W.; Jiang, C.; Zhang, J.; Xu, J.; Xu, L.; Xu, X.; Li, J.; Cui, C. Rare-Earth-Catalyzed Selective 1,4-Hydrosilylation of Branched 1,3-Enynes Giving Tetrasubstituted Silylallenes. J. Am. Chem. Soc. 2021, 143, 12913–12918. [Google Scholar] [CrossRef]
- Abbott, D.F.; Pittkowski, R.K.; Macounova, K.; Nebel, R.; Marelli, E.; Fabbri, E.; Castelli, I.E.; Krtil, P.; Schmidt, T.J. Design and Synthesis of Ir/Ru Pyrochlore Catalysts for the Oxygen Evolution Reaction Based on Their Bulk Thermodynamic Properties. ACS Appl. Mater. Interfaces 2019, 11, 37748–37760. [Google Scholar] [CrossRef]
- Zhu, M.; Zhao, C.; Liu, X.; Wang, X.; Zhou, F.; Wang, J.; Hu, Y.; Zhao, Y.; Yao, T.; Yang, L.-M.; et al. Single Atomic Cerium Sites with a High Coordination Number for Efficient Oxygen Reduction in Proton-Exchange Membrane Fuel Cells. ACS Catal. 2021, 11, 3923–3929. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, J.; Jin, C.; Wang, N.; Wang, F. Hexagonal La2O3 Nanocrystals Chemically Coupled with Nitrogen-Doped Porous Carbon as Efficient Catalysts for the Oxygen Reduction Reaction. Chem. A Eur. J. 2020, 26, 12606–12614. [Google Scholar] [CrossRef] [PubMed]
- Murr, L.E. Summarizing Atom and Ion Structure: The Periodic Table of the Elements. In Handbook of Materials Structures, Properties, Processing and Performance; Springer International Publishing: Cham, Switzerland, 2015; pp. 83–95. [Google Scholar] [CrossRef]
- Gismondi, P.; Kuzmin, A.; Unsworth, C.; Rangan, S.; Khalid, S.; Saha, D. Understanding the Adsorption of Rare-Earth Elements in Oligo-Grafted Mesoporous Carbon. Langmuir 2022, 38, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tang, Y.; Lee, J.-M.; Fu, G. Recent advances in rare-earth-based materials for electrocatalysis. Chem Catal. 2022, 2, 967–1008. [Google Scholar] [CrossRef]
- Liu, J.; Kong, X.; Zheng, L.; Guo, X.; Liu, X.; Shui, J. Rare Earth Single-Atom Catalysts for Nitrogen and Carbon Dioxide Reduction. ACS Nano 2020, 14, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.-Z. Graphdiyne-supported single-atom Sc and Ti catalysts for high-efficient CO oxidation. Carbon 2016, 108, 343–350. [Google Scholar] [CrossRef]
- Wang, Q.-Y.; Tong, Y.-C.; Yan, P.-J.; Xu, X.-J.; Li, Z. Attachment of CO to a (6, 6) CNT with a Sc adsorbate atom. Struct. Chem. 2019, 30, 399–408. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Delley, B. Hardness conserving semilocal pseudopotentials. Phys. Rev. B 2002, 66, 155125. [Google Scholar] [CrossRef]
- Delley, B. An all-electron numerical method for solving the local density functional for polyatomic molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Delley, B. The conductor-like screening model for polymers and surfaces. Mol. Simul. 2006, 32, 117–123. [Google Scholar] [CrossRef]
- Yang, K.; Zaffran, J.; Yang, B. Fast prediction of oxygen reduction reaction activity on carbon nanotubes with a localized geometric descriptor. Phys. Chem. Chem. Phys. 2020, 22, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhu, G.; Yang, D.; Jia, D.; Jin, F.; Wang, W. Systematic exploration of N, C configurational effects on the ORR performance of Fe–N doped graphene catalysts based on DFT calculations. RSC Adv. 2019, 9, 22656–22667. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.; Luo, M.; Chen, M.; Qi, X.; Liu, J.; Liu, C.; Peera, S.G.; Liang, T. Exploring the oxygen electrode bi-functional activity of Ni–N–C-doped graphene systems with N, C co-ordination and OH ligand effects. J. Mater. Chem. A 2020, 8, 20453–20462. [Google Scholar] [CrossRef]
- Revanappa, S.K.; Soni, I.; Siddalinganahalli, M.; Jayaprakash, G.K.; Flores-Moreno, R.; Nanjegowda, C.B. A Fukui Analysis of an Arginine-Modified Carbon Surface for the Electrochemical Sensing of Dopamine. Materials 2022, 15, 6337. [Google Scholar] [CrossRef]
- Jayaprakash, G.K.; Swamy, B.E.K.; Flores-Moreno, R.; Pineda-Urbina, K. Theoretical and Cyclic Voltammetric Analysis of Asparagine and Glutamine Electrocatalytic Activities for Dopamine Sensing Applications. Catalysts 2023, 13, 100. [Google Scholar] [CrossRef]
- Qin, R.; Zhou, L.; Liu, P.; Gong, Y.; Liu, K.; Xu, C.; Zhao, Y.; Gu, L.; Fu, G.; Zheng, N. Alkali ions secure hydrides for catalytic hydrogenation. Nat. Catal. 2020, 3, 703–709. [Google Scholar] [CrossRef]
- Liang, Z.; Luo, M.; Chen, M.; Liu, C.; Peera, S.G.; Qi, X.; Liu, J.; Kumar, U.P.; Liang, T.L.T. Evaluating the catalytic activity of transition metal dimers for the oxygen reduction reaction. J. Colloid Interface Sci. 2020, 568, 54–62. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).