

Design of Perovskite-Type Fluorides Cathodes for Na-ion Batteries: Correlation between Structure and Transport

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
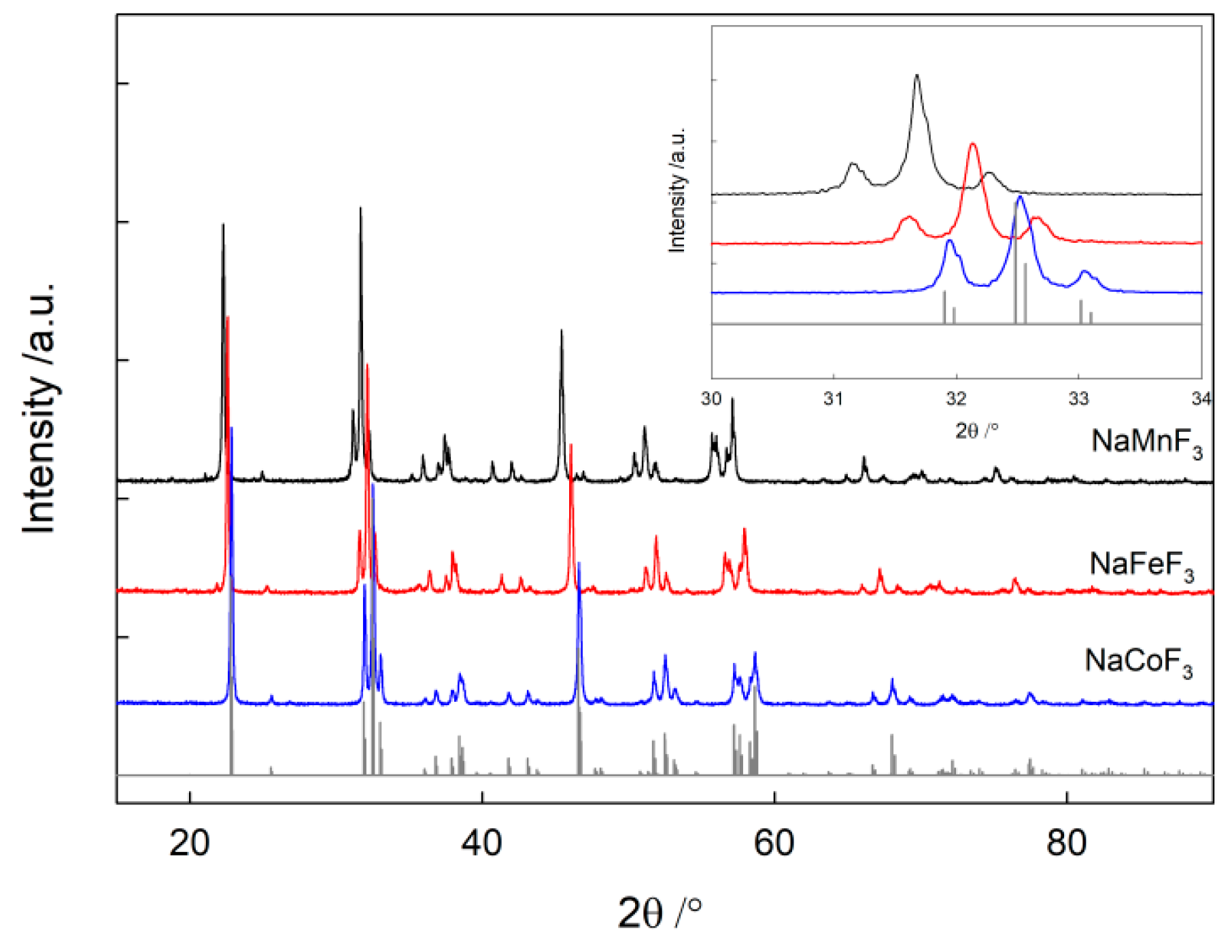
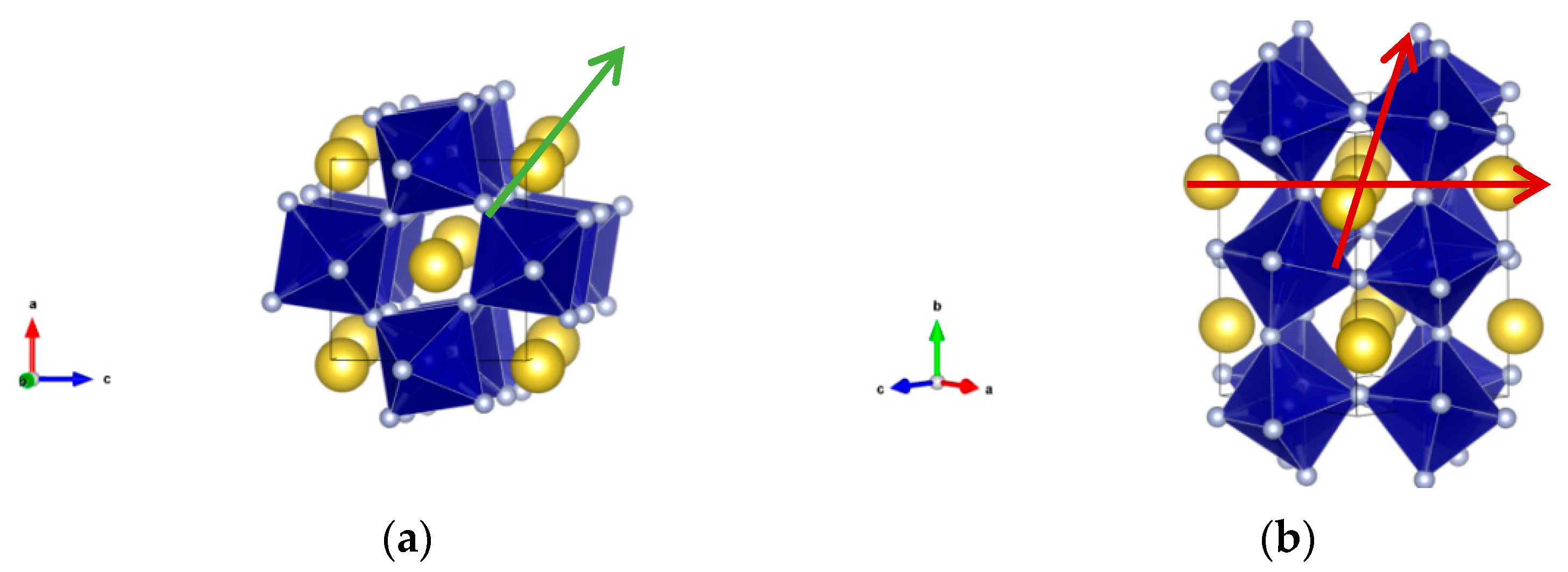
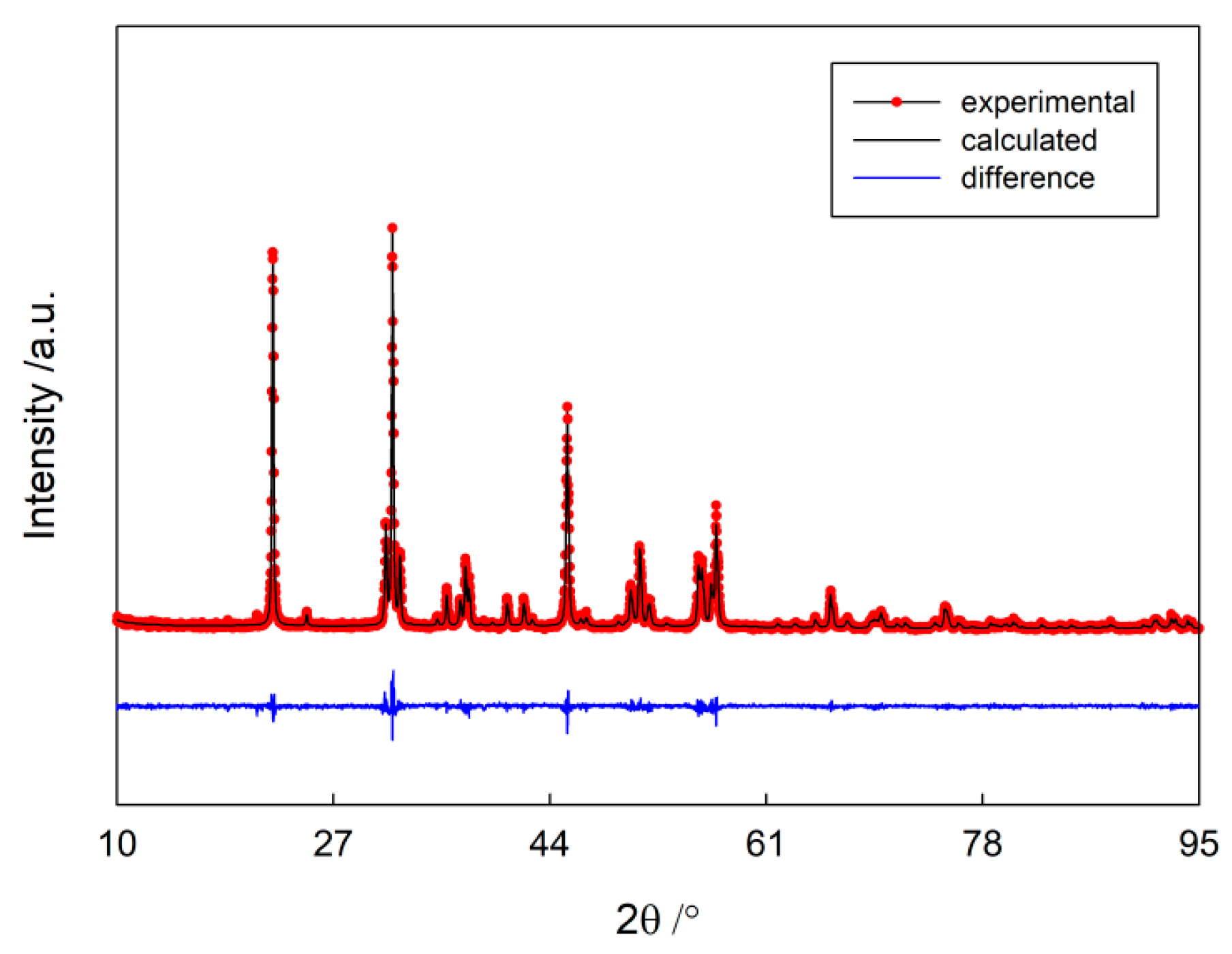
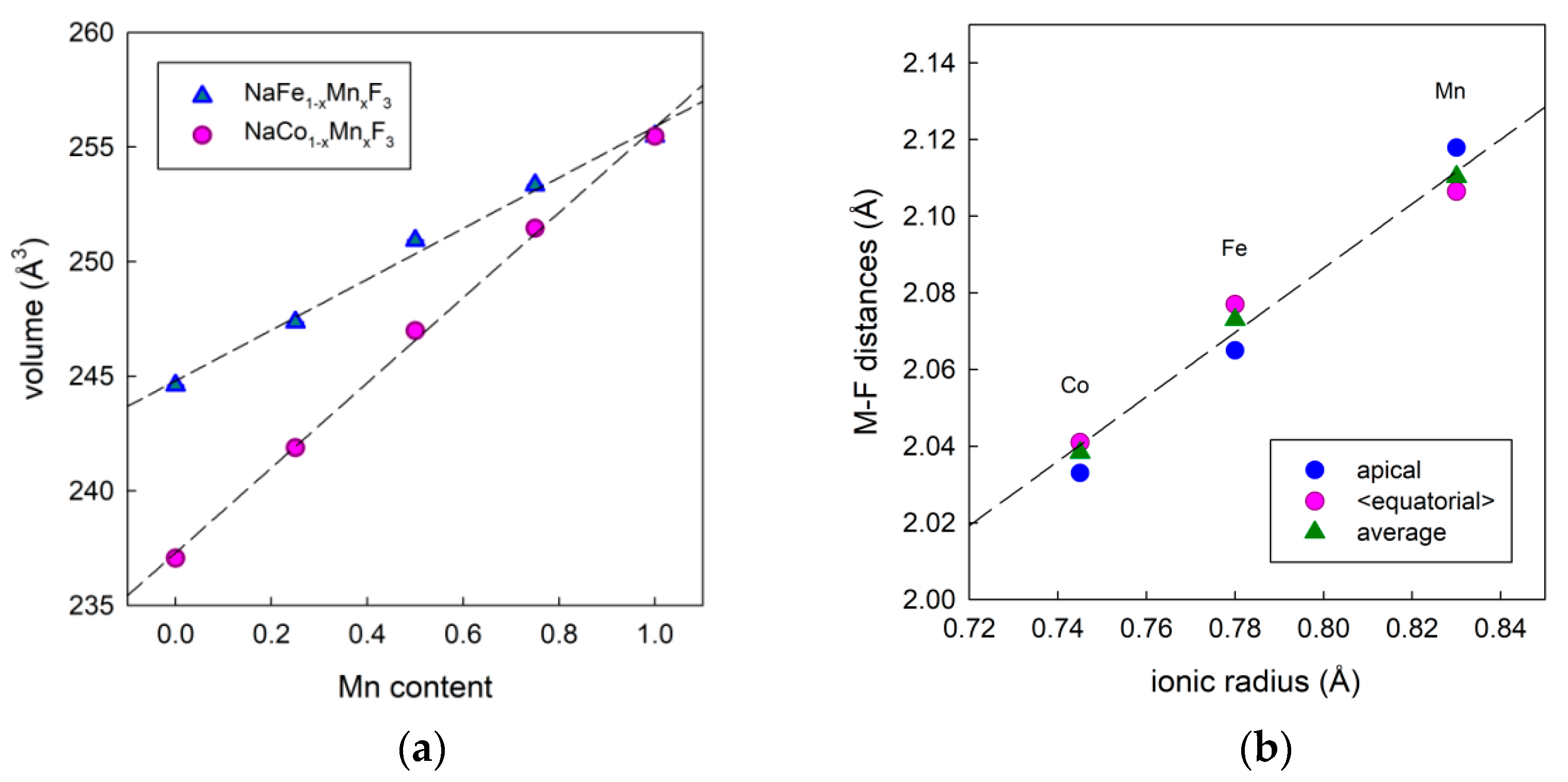
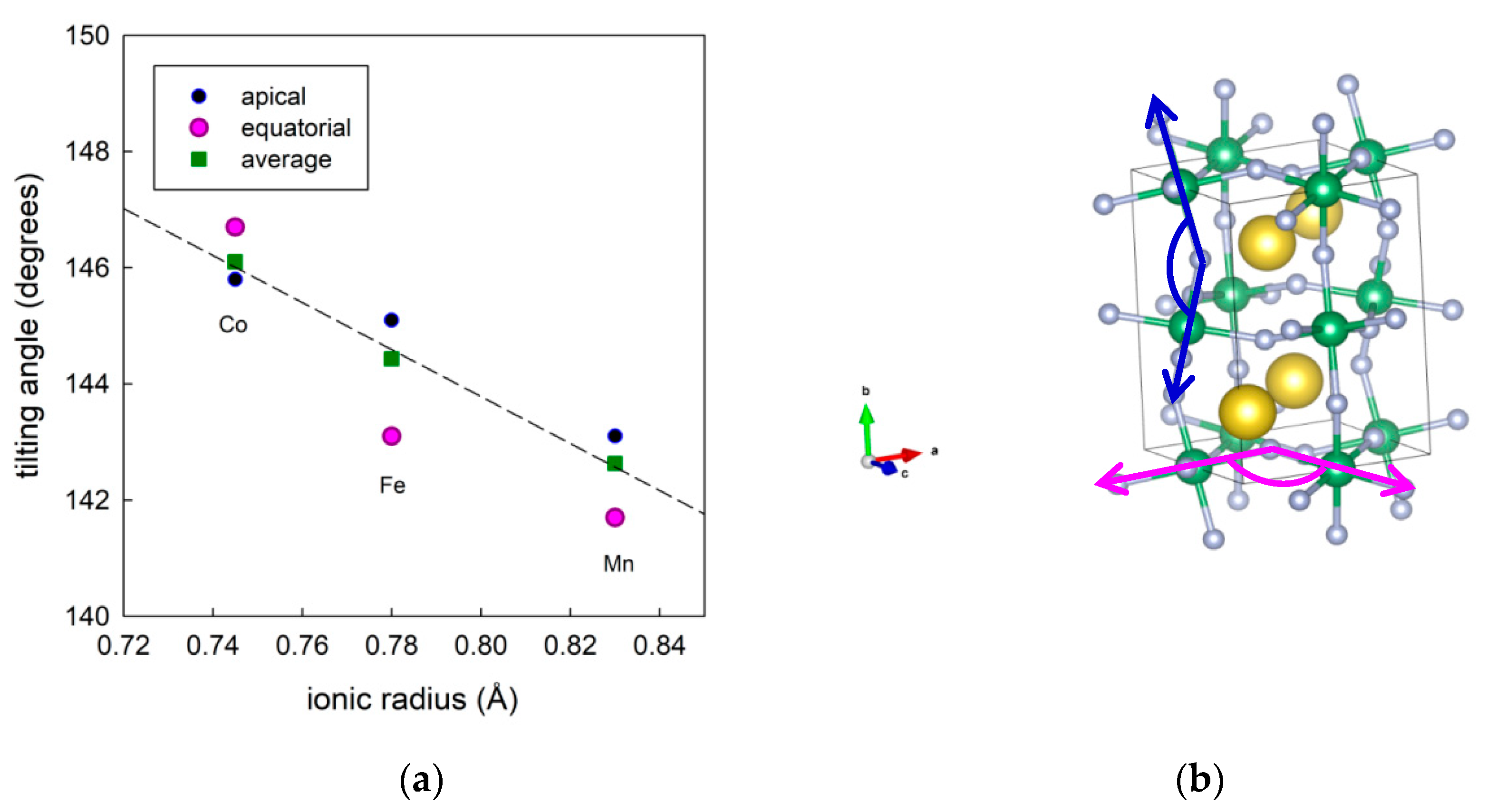
3.1. Materials Synthesis and Characterization
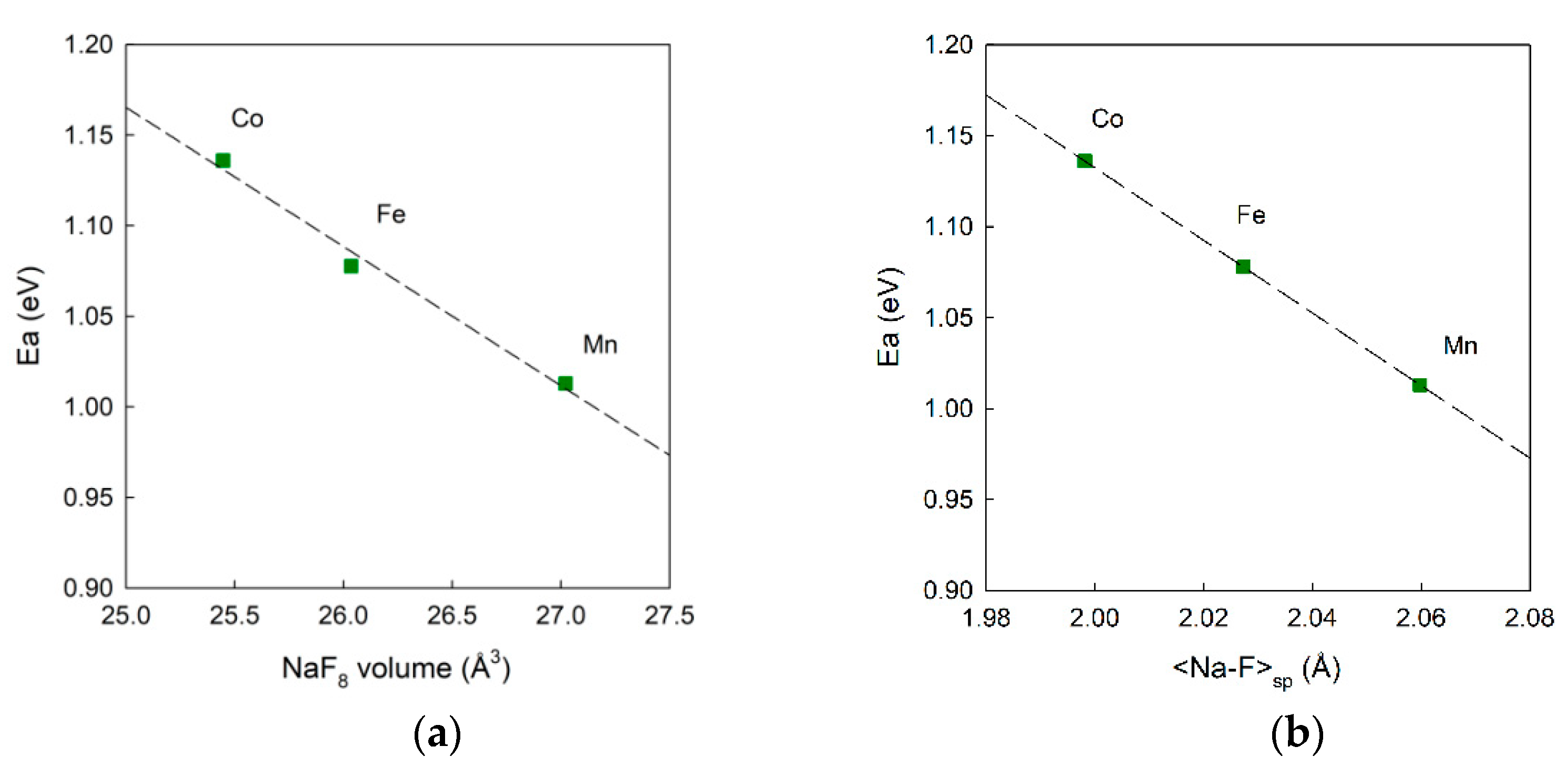
3.2. Materials Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Delmas, C. Sodium and Sodium-Ion Batteries: 50 Years of Research. Adv. Energy Mater. 2018, 8, 1703137. [Google Scholar] [CrossRef]
- Goikolea, E.; Palomares, V.; Wang, S.; de Larramendi, I.R.; Guo, X.; Wang, G.; Rojo, T. Na-Ion Batteries—Approaching Old and New Challenges. Adv. Energy Mater. 2020, 10, 2002055. [Google Scholar] [CrossRef]
- You, Y.; Manthiram, A. Progress in High-Voltage Cathode Materials for Rechargeable Sodium-Ion Batteries. Adv. Energy Mater. 2018, 8, 1701785. [Google Scholar] [CrossRef]
- Li, Y.; Lu, Y.; Zhao, C.; Hu, Y.S.; Titirici, M.M.; Li, H.; Huang, X.; Chen, L. Recent Advances of Electrode Materials for Low-Cost Sodium-Ion Batteries towards Practical Application for Grid Energy Storage. Energy Storage Mater. 2017, 7, 130–151. [Google Scholar] [CrossRef]
- Kitajou, A.; Ishado, Y.; Yamashita, T.; Momida, H.; Oguchi, T.; Okada, S. Cathode Properties of Perovskite-Type NaMF3 (M = Fe, Mn, and Co) Prepared by Mechanical Ball Milling for Sodium-Ion Battery. Electrochim. Acta 2017, 245, 424–429. [Google Scholar] [CrossRef]
- Yamada, Y.; Doi, T.; Tanaka, I.; Okada, S.; Yamaki, J.I. Liquid-Phase Synthesis of Highly Dispersed NaFeF3 Particles and Their Electrochemical Properties for Sodium-Ion Batteries. J. Power Sources 2011, 196, 4837–4841. [Google Scholar] [CrossRef]
- Nava-Avendaño, J.; Arroyo-De Dompablo, M.E.; Frontera, C.; Ayllón, J.A.; Palacín, M.R. Study of Sodium Manganese Fluorides as Positive Electrodes for Na-Ion Batteries. Solid State Ionics 2015, 278, 106–113. [Google Scholar] [CrossRef]
- Kitajou, A.; Komatsu, H.; Chihara, K.; Gocheva, I.D.; Okada, S.; Yamaki, J.I. Novel Synthesis and Electrochemical Properties of Perovskite-Type NaFeF3 for a Sodium-Ion Battery. J. Power Sources 2012, 198, 389–392. [Google Scholar] [CrossRef]
- Kravchyk, K.V.; Zünd, T.; Wörle, M.; Kovalenko, M.V.; Bodnarchuk, M.I. NaFeF3 Nanoplates as Low-Cost Sodium and Lithium Cathode Materials for Stationary Energy Storage. Chem. Mater. 2018, 30, 1825–1829. [Google Scholar] [CrossRef]
- Yuan, L.X.; Wang, Z.H.; Zhang, W.X.; Hu, X.L.; Chen, J.T.; Huang, Y.H.; Goodenough, J.B. Development and Challenges of LiFePO4 Cathode Material for Lithium-Ion Batteries. Energy Environ. Sci. 2011, 4, 269–284. [Google Scholar] [CrossRef]
- Chun, J.; Jo, C.; Lim, E.; Roh, K.C.; Lee, J. Solvothermal Synthesis of Sodium Cobalt Fluoride (NaCoF3) Nanoparticle Clusters. Mater. Lett. 2017, 207, 89–92. [Google Scholar] [CrossRef]
- Rodriguez-Carvajal, J. Recent Advances in Magnetic Structure Determination by Neutron Powder Diffraction. Phys. B 1993, 192, 55–69. [Google Scholar] [CrossRef]
- Lutgert, B.; Babel, D. Kristallstrukturverfeinerungen an Natriumtrifluorometallaten NaMF3 (M =Mg, Co, Ni, Zn): Oktaederkippung Und Toleranzfaktor Orthorhombischer Fluorperowskite. Zeitschrift Anorg. Allg. Chem. 1992, 616, 133–140. [Google Scholar] [CrossRef]
- Gale, J.D.; Rohl, A.L. The General Utility Lattice Program (GULP). Mol. Simul. 2003, 29, 291–341. [Google Scholar] [CrossRef]
- Gale, J.D. GULP: A Computer Program for the Symmetry-Adapted Simulation of Solids. J. Chem. Soc. Faraday Trans. 1997, 93148, 629–637. [Google Scholar] [CrossRef]
- Tripathi, R.; Wood, S.M.; Islam, M.S.; Nazar, L.F. Na-Ion Mobility in Layered Na2FePO4F and Olivine Na[Fe,Mn]PO4. Energy Environ. Sci. 2013, 6, 2257–2264. [Google Scholar] [CrossRef]
- Dick, B.G.; Overhauser, A.W. Theory of the Dielectric Constants of Alkali Halide Crystals. Phys. Rev. 1958, 112, 90–103. [Google Scholar] [CrossRef]
- Mott, N.F.; Littleton, M.J. Conduction in polar crystals. Solid, I. Electrolytic conduciton in salt. Trans. Faraday Soc. 1938, 34, 485–499. [Google Scholar] [CrossRef]
- Bernal, F.L.; Yusenko, K.V.; Sottmann, J.; Drathen, C.; Guignard, J.; Løvvik, O.M.; Crichton, W.A.; Margadonna, S. Perovskite to Postperovskite Transition in NaFeF3. Inorg. Chem. 2014, 53, 12205–12214. [Google Scholar] [CrossRef]
- Shannon, R.D. Revised Effective Ionic Radii and Systematic Studies of Interatomic Distances in Halides and Chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Tealdi, C.; Malavasi, L.; Gozzo, F.; Ritter, C.; Mozzati, M.C.; Chiodelli, G.; Flor, G. Correlation between Transport Properties and Lattice Effects in the NdCoO3-Based Catalysts and Sensor Materials. Chem. Mater. 2007, 19, 4741–4750. [Google Scholar] [CrossRef]
- Khan, W.; Naqvi, A.H.; Gupta, M.; Husain, S.; Kumar, R. Small Polaron Hopping Conduction Mechanism in Fe Doped LaMnO3. J. Chem. Phys. 2011, 135, 054501. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. Commentary: The Materials Project: A Materials Genome Approach to Accelerating Materials Innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef]
- Rong, Z.; Malik, R.; Canepa, P.; Gautam, G.S.; Liu, M.; Jain, A.; Persson, K.; Ceder, G. Materials Design Rules for Multivalent Ion Mobility in Intercalation Structures. Chem. Mater. 2015, 27, 6016–6021. [Google Scholar] [CrossRef]
- Park, K.Y.; Park, I.; Kim, H.; Lim, H.D.; Hong, J.; Kim, J.; Kang, K. Anti-Site Reordering in LiFePO4: Defect Annihilation on Charge Carrier Injection. Chem. Mater. 2014, 26, 5345–5351. [Google Scholar] [CrossRef]
- Islam, M.S.; Driscoll, D.J.; Fisher CA, J.; Slater, P.R. Atomic-Scale Investigation of Defects, Dopants, and Lithium Transport in the LiFePO4 Olivine-Type Battery Material. Chem. Mater. 2005, 17, 5085–5092. [Google Scholar] [CrossRef]
- Gautam, G.S.; Canepa, P.; Urban, A.; Bo, S.; Ceder, G. Influence of Inversion on Mg Mobility and Electrochemistry in Spinels. Chem. Mater. 2017, 29, 7918–7930. [Google Scholar] [CrossRef]
- Morando, C.; Cofrancesco, P.; Tealdi, C. Zn Ion Diffusion in Spinel-Type Cathode Materials for Rechargeable Batteries: The Role of Point Defects. Mater. Today Commun. 2020, 25, 101478. [Google Scholar] [CrossRef]
- Tealdi, C.; Spreafico, C.; Mustarelli, P. Lithium Diffusion in Li1−xFePO4: The Effect of Cationic Disorder. J. Mater. Chem. 2012, 22, 24870. [Google Scholar] [CrossRef]
- Chen, J.; Graetz, J. Study of Antisite Defects in Hydrothermally Prepared LiFePO4 by in Situ X-ray Diffraction. ACS Appl. Mater. Interfaces 2011, 3, 1380–1384. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ea (eV) | |||
|---|---|---|---|
| NaCoF3 | NaFeF3 | NaMnF3 | |
| Path A1 | 1.27 | 1.24 | 1.21 |
| Path A2 | 1.11 | 1.06 | 1.00 |
| Path B | 1.14 | 1.08 | 1.01 |
| NaCoF3 | NaFeF3 | NaMnF3 | |
|---|---|---|---|
| Anti-site isolated (eV) | 1.62 | 1.42 | 1.19 |
| Anti-site cluster (eV) | 0.86 | 0.71 | 0.55 |
| Binding energy (eV) | −0.76 | −0.71 | −0.64 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montalbano, M.; Callegari, D.; Anselmi Tamburini, U.; Tealdi, C. Design of Perovskite-Type Fluorides Cathodes for Na-ion Batteries: Correlation between Structure and Transport. Batteries 2022, 8, 126. https://doi.org/10.3390/batteries8090126
Montalbano M, Callegari D, Anselmi Tamburini U, Tealdi C. Design of Perovskite-Type Fluorides Cathodes for Na-ion Batteries: Correlation between Structure and Transport. Batteries. 2022; 8(9):126. https://doi.org/10.3390/batteries8090126
Chicago/Turabian StyleMontalbano, Michele, Daniele Callegari, Umberto Anselmi Tamburini, and Cristina Tealdi. 2022. "Design of Perovskite-Type Fluorides Cathodes for Na-ion Batteries: Correlation between Structure and Transport" Batteries 8, no. 9: 126. https://doi.org/10.3390/batteries8090126
APA StyleMontalbano, M., Callegari, D., Anselmi Tamburini, U., & Tealdi, C. (2022). Design of Perovskite-Type Fluorides Cathodes for Na-ion Batteries: Correlation between Structure and Transport. Batteries, 8(9), 126. https://doi.org/10.3390/batteries8090126