
Quantitative Lithiation Depth Profiling in Silicon Containing Anodes Investigated by Ion Beam Analysis

 ,
,  , ,
, ,  and
and
Abstract
:
1. Introduction
2. IBA Method in View of Lithium Analysis
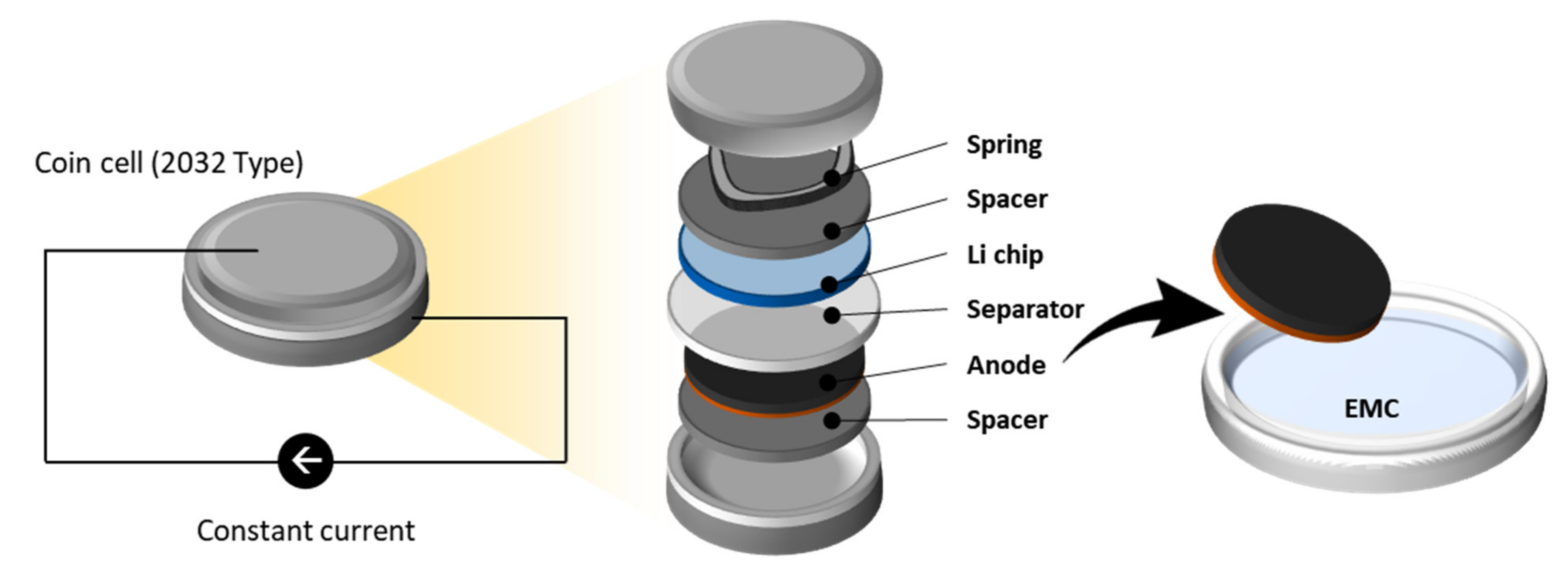
3. Sample Preparation
3.1. Material and Electrode Preparation
3.2. Lithiation Process
4. Experimental Setup
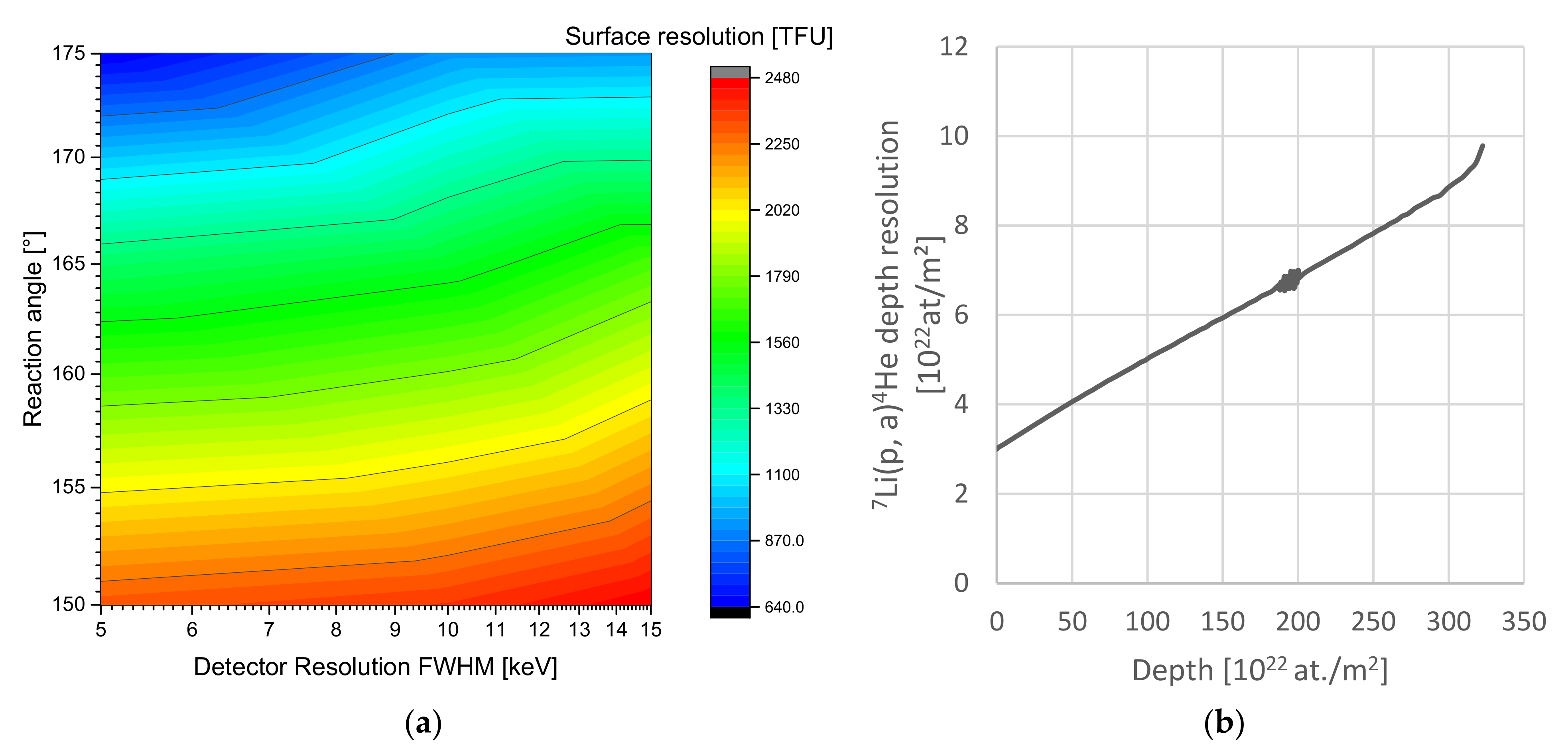
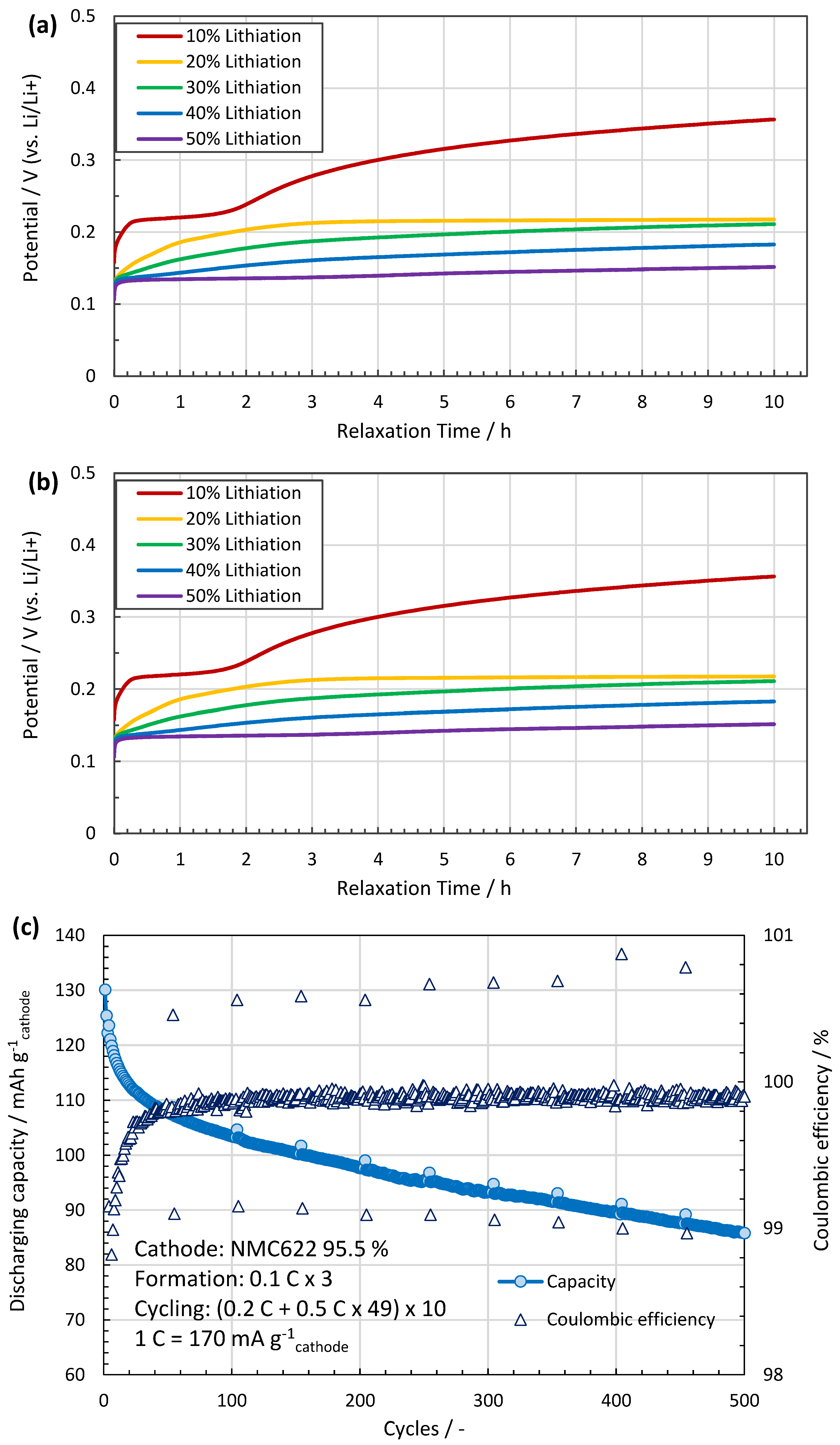
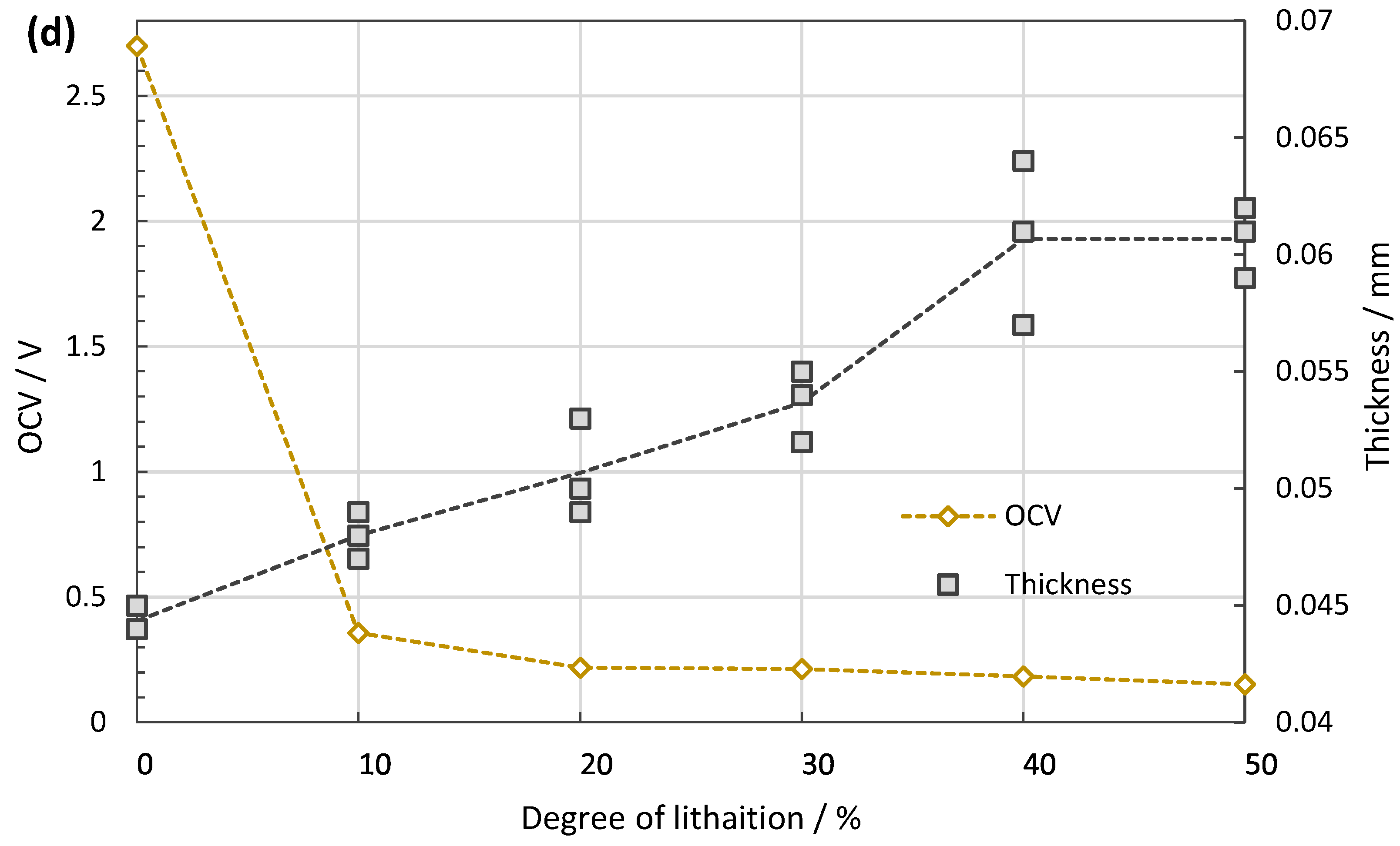
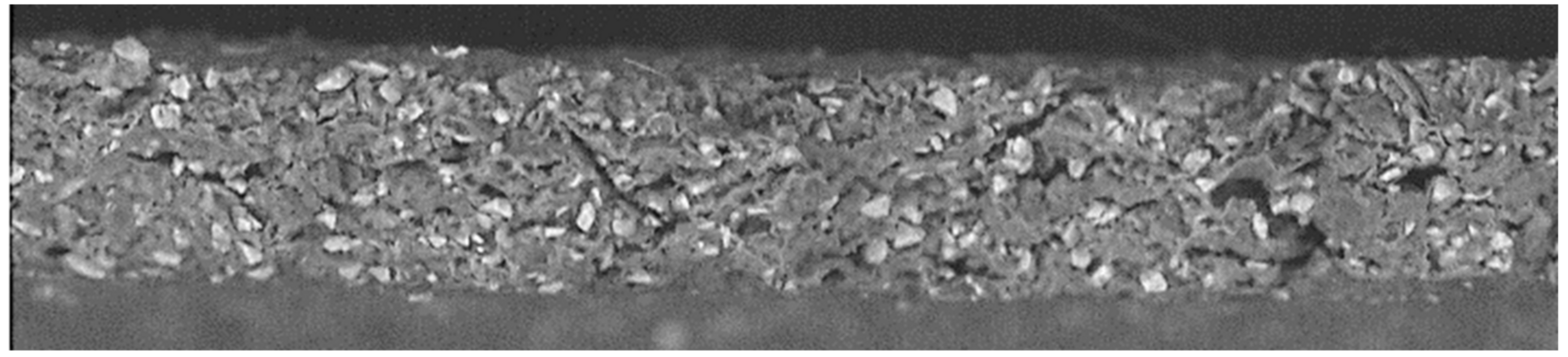
5. Results
6. Conclusions
6.1. IBA Method
6.2. Si-Doped Anodes for Li Batteries
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Obrovac, M.N.; Christensen, L. Structural Changes in Silicon Anodes during Lithium Insertion/Extraction. Electrochem. Solid-state Lett. 2004, 7, A93–A96. [Google Scholar] [CrossRef]
- Ko, M.; Oh, P.; Chae, S.; Cho, W.; Cho, J. Considering Critical Factors of Li-rich Cathode and Si Anode Materials for Practical Li-ion Cell Applications. Small 2015, 11, 4058–4073. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.W.; Aurbach, D. Promise and reality of post-lithium-ion batteries with high energy densities. Nat. Rev. Mater. 2016, 1, 16013. [Google Scholar] [CrossRef]
- Liu, N.; Li, W.; Pasta, M.; Cui, Y. Nanomaterials for electrochemical energy storage. Front. Phys. 2014, 9, 323–350. [Google Scholar] [CrossRef]
- Müller, S.; Pietsch, P.; Brandt, B.-E.; Baade, P.; De Andrade, V.; De Carlo, F.; Wood, V. Quantification and modeling of mechanical degradation in lithium-ion batteries based on nanoscale imaging. Nat. Commun. 2018, 9, 1599. [Google Scholar] [CrossRef]
- Jin, Y.; Zhu, B.; Lu, Z.; Liu, N.; Zhu, J. Challenges and Recent Progress in the Development of Si Anodes for Lithium-Ion Battery. Adv. Energy Mater. 2017, 7, 1700715. [Google Scholar] [CrossRef] [Green Version]
- Chae, S.; Ko, M.; Kim, K.; Ahn, K.; Cho, J. Confronting Issues of the Practical Implementation of Si Anode in High-Energy Lithium-Ion Batteries. Joule 2017, 1, 47–60. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.-K.; Kim, J.; Jung, Y.S.; Kang, K. Scalable Fabrication of Silicon Nanotubes and their Application to Energy Storage. Adv. Mater. 2012, 24, 5452–5456. [Google Scholar] [CrossRef]
- Cui, L.-F.; Yang, Y.; Hsu, C.-M.; Cui, Y. Carbon−Silicon Core−Shell Nanowires as High Capacity Electrode for Lithium Ion Batteries. Nano Lett. 2009, 9, 3370–3374. [Google Scholar] [CrossRef]
- Baasner, A.; Reuter, F.; Seidel, M.; Krause, A.; Pflug, E.; Härtel, P.; Dörfler, S.; Abendroth, T.; Althues, H.; Kaskel, S. The Role of Balancing Nanostructured Silicon Anodes and NMC Cathodes in Lithium-Ion Full-Cells with High Volumetric Energy Density. J. Electrochem. Soc. 2020, 167, 020516. [Google Scholar] [CrossRef]
- Maddipatla, R.; Loka, C.; Choi, W.J.; Lee, K.-S. Nanocomposite of Si/C Anode Material Prepared by Hybrid Process of High-Energy Mechanical Milling and Carbonization for Li-Ion Secondary Batteries. Appl. Sci. 2018, 8, 2140. [Google Scholar] [CrossRef] [Green Version]
- Hwang, T.H.; Lee, Y.M.; Kong, B.-S.; Seo, J.-S.; Choi, J.W. Electrospun Core–Shell Fibers for Robust Silicon Nanoparticle-Based Lithium Ion Battery Anodes. Nano Lett. 2012, 12, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Xiao, X.; Cheng, Y.-T.; Verbrugge, M.W. Atomic Layered Coating Enabling Ultrafast Surface Kinetics at Silicon Electrodes in Lithium Ion Batteries. J. Phys. Chem. Lett. 2013, 4, 3387–3391. [Google Scholar] [CrossRef]
- Kovalenko, I.; Zdyrko, B.; Magasinski, A.; Hertzberg, B.; Milicev, Z.; Burtovyy, R.; Luzinov, I.; Yushin, G. A Major Constituent of Brown Algae for Use in High-Capacity Li-Ion Batteries. Science 2011, 334, 75–79. [Google Scholar] [CrossRef]
- Kwon, T.-W.; Choi, J.W.; Coskun, A. The emerging era of supramolecular polymeric binders in silicon anodes. Chem. Soc. Rev. 2018, 47, 2145–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Liu, J.; He, S.; Huang, C.; Gan, L.; Gong, Z.; Long, M. A novel high-performance 3D polymer binder for silicon anode in lithium-ion batteries. J. Phys. Chem. Solids 2019, 135, 109113. [Google Scholar] [CrossRef]
- Fridman, K.; Sharabi, R.; Elazari, R.; Gershinsky, G.; Markevich, E.; Salitra, G.; Aurbach, D.; Garsuch, A.; Lampert, J. A new advanced lithium ion battery: Combination of high performance amorphous columnar silicon thin film anode, 5V LiNi0.5Mn1.5O4 spinel cathode and fluoroethylene carbonate-based electrolyte solution. Electrochem. Commun. 2013, 33, 31–34. [Google Scholar] [CrossRef]
- Aupperle, F.; von Aspern, N.; Berghus, D.; Weber, F.; Eshetu, G.G.; Winter, M.; Figgemeier, E. The Role of Electrolyte Additives on the Interfacial Chemistry and Thermal Reactivity of Si-Anode-Based Li-Ion Battery. ACS Appl. Energy Mater. 2019, 2, 6513–6527. [Google Scholar] [CrossRef]
- Jin, Y.; Kneusels, N.-J.; Marbella, L.E.; Castillo-Martínez, E.; Magusin, P.C.M.M.; Weatherup, R.S.; Jónsson, E.; Liu, T.; Paul, S.; Grey, C.P. Understanding Fluoroethylene Carbonate and Vinylene Carbonate Based Electrolytes for Si Anodes in Lithium Ion Batteries with NMR Spectroscopy. J. Am. Chem. Soc. 2018, 140, 9854–9867. [Google Scholar] [CrossRef] [Green Version]
- Pathak, A.D.; Samanta, K.; Sahu, K.K.; Pati, S. Mechanistic insight into the performance enhancement of Si anode of a lithium-ion battery with a fluoroethylene carbonate electrolyte additive. J. Appl. Electrochem. 2021, 51, 143–154. [Google Scholar] [CrossRef]
- Zhang, T.; Gao, J.; Zhang, H.; Yang, L.; Wu, Y.; Wu, H. Preparation and electrochemical properties of core-shell Si/SiO nanocomposite as anode material for lithium ion batteries. Electrochem. Commun. 2007, 9, 886–890. [Google Scholar] [CrossRef]
- Wu, J.; Ma, F.; Liu, X.; Fan, X.; Shen, L.; Wu, Z.; Ding, X.; Han, X.; Deng, Y.; Hu, W.; et al. Recent Progress in Advanced Characterization Methods for Silicon-Based Lithium-Ion Batteries. Small Methods 2019, 3, 425–445. [Google Scholar] [CrossRef]
- Philippe, B.; Dedryvère, R.; Allouche, J.; Lindgren, F.; Gorgoi, M.; Rensmo, H.; Gonbeau, D.; Edstroem, K. Nanosilicon Electrodes for Lithium-Ion Batteries: Interfacial Mechanisms Studied by Hard and Soft X-ray Photoelectron Spectroscopy. Chem. Mater. 2012, 24, 1107–1115. [Google Scholar] [CrossRef]
- Chen, T.; Wu, J.; Zhang, Q.; Su, X. Recent advancement of SiOx based anodes for lithium-ion batteries. J. Power Sources 2017, 363, 126–144. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, Q.; Zhao, Y.; He, R.; Xu, M.; Feng, S.; Li, S.; Zhou, L.; Mai, L. Silicon oxides: A promising family of anode materials for lithium-ion batteries. Chem. Soc. Rev. 2019, 48, 285–309. [Google Scholar] [CrossRef]
- Ge, G.; Li, G.; Wang, X.; Chen, X.; Fu, L.; Liu, X.; Mao, E.; Liu, J.; Yang, X.; Qian, C.; et al. Manipulating Oxidation of Silicon with Fresh Surface Enabling Stable Battery Anode. Nano Lett. 2021, 21, 3127–3133. [Google Scholar] [CrossRef] [PubMed]
- Sivonxay, E.; Aykol, M.; Persson, K.A. The lithiation process and Li diffusion in amorphous SiO2 and Si from first-principles. Electrochim. Acta 2020, 331, 135344. [Google Scholar] [CrossRef]
- Wetjen, M.; Pritzl, D.; Jung, R.; Solchenbach, S.; Ghadimi, R.; Gasteiger, H. Differentiating the Degradation Phenomena in Silicon-Graphite Electrodes for Lithium-Ion Batteries. J. Electrochem. Soc. 2017, 164, A2840–A2852. [Google Scholar] [CrossRef]
- Schmitz, R.W.; Murmann, P.; Schmitz, R.; Müller, R.; Krämer, L.; Kasnatscheew, J.; Isken, P.; Niehoff, P.; Nowak, S.; Röschenthaler, G.-V.; et al. Investigations on novel electrolytes, solvents and SEI additives for use in lithium-ion batteries: Systematic electrochemical characterization and detailed analysis by spectroscopic methods. Prog. Solid State Chem. 2014, 42, 65–84. [Google Scholar] [CrossRef]
- Zheng, J.; Yin, J.; Zhang, D.; Li, G.; Bock, D.C.; Tang, T.; Zhao, Q.; Liu, X.; Warren, A.; Deng, Y.; et al. Spontaneous and field-induced crystallographic reorientation of metal electrodeposits at battery anodes. Sci. Adv. 2020, 6, eabb1122. [Google Scholar] [CrossRef]
- Liu, D.; Shadike, Z.; Lin, R.; Qian, K.; Li, H.; Li, K.; Wang, S.; Yu, Q.; Liu, M.; Ganapathy, S.; et al. Review of Recent Development of In Situ/Operando Characterization Techniques for Lithium Battery Research. Adv. Mater. 2019, 31, e1806620. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.; Iturrondobeitia, A.; Kasper, M.; Ghanbari, N.; Aguesse, F.; Bekaert, E.; Daniel, L.; Genies, S.; Gordon, I.J.; Löble, M.W.; et al. Review—Post-Mortem Analysis of Aged Lithium-Ion Batteries: Disassembly Methodology and Physico-Chemical Analysis Techniques. J. Electrochem. Soc. 2016, 163, A2149–A2164. [Google Scholar] [CrossRef]
- Richter, K.; Waldmann, T.; Kasper, M.; Pfeifer, C.; Memm, M.; Axmann, P.; Wohlfahrt-Mehrens, M. Surface Film Formation and Dissolution in Si/C Anodes of Li-Ion Batteries: A Glow Discharge Optical Emission Spectroscopy Depth Profiling Study. J. Phys. Chem. C 2019, 123, 18795–18803. [Google Scholar] [CrossRef]
- Oudenhoven, J.F.M.; Labohm, F.; Mulder, M.; Niessen, R.A.H.; Mulder, F.M.; Notten, P.H.L. In Situ Neutron Depth Profiling: A Powerful Method to Probe Lithium Transport in Micro-Batteries. Adv. Mater. 2011, 23, 4103–4106. [Google Scholar] [CrossRef]
- Möller, S.; Höschen, D.; Kurth, S.; Esser, G.; Hiller, A.; Scholtysik, C.; Dellen, C.; Linsmeier, C. A New High-Throughput Focused MeV Ion-Beam Analysis Setup. Instruments 2021, 5, 10. [Google Scholar] [CrossRef]
- Habrioux, A.; Surblé, S.; Berger, P.; Khodja, H.; D’Affroux, A.; Mailley, S.; Gutel, T.; Patoux, S. Nuclear microanalysis of lithium dispersion in LiFePO4 based cathode materials for Li-ion batteries. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2012, 290, 13–18. [Google Scholar] [CrossRef]
- Yamazaki, A.; Orikasa, Y.; Chen, K.; Uchimoto, Y.; Kamiya, T.; Koka, M.; Satoh, T.; Mima, K.; Kato, Y.; Fujita, K. In-situ measurement of the lithium distribution in Li-ion batteries using micro-IBA techniques. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2016, 371, 298–302. [Google Scholar] [CrossRef] [Green Version]
- Uhlenbruck, S.; Dellen, C.; Möller, S.; Lobe, S.; Tsai, C.-L.; Finsterbusch, M.; Bram, M.; Guillon, O. Reactions of garnet-based solid-state lithium electrolytes with water—A depth-resolved study. Solid State Ion. 2018, 320, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Suzuki, K.; Yoshino, K.; Taminato, S.; Satoh, T.; Finsterbusch, M.; Kamiya, T.; Yamazaki, A.; Kato, Y.; Fujita, K.; et al. Ex-situ Analysis of Lithium Distribution in a Sulfide-based All-solid-state Lithium Battery by Particle-induced X-ray and Gamma-ray Emission Measurements. Electrochemistry 2020, 88, 45–49. [Google Scholar] [CrossRef] [Green Version]
- Möller, S.; Satoh, T.; Ishii, Y.; Teßmer, B.; Guerdelli, R.; Kamiya, T.; Fujita, K.; Suzuki, K.; Kato, Y.; Wiemhöfer, H.-D.; et al. Absolute Local Quantification of Li as Function of State-of-Charge in All-Solid-State Li Batteries via 2D MeV Ion-Beam Analysis. Batteries 2021, 7, 41. [Google Scholar] [CrossRef]
- Mathayan, V.; Moro, M.V.; Morita, K.; Tsuchiya, B.; Ye, R.; Baba, M.; Primetzhofer, D. In-operando observation of Li depth distribution and Li transport in thin film Li ion batteries. Appl. Phys. Lett. 2020, 117, 023902. [Google Scholar] [CrossRef]
- Mayer, M. RESOLNRA User’s Guide; Max-Planck-Institut für Plasmaphysik: Garching, Germany, 2017; p. 38. [Google Scholar]
- Chia-Shou, L.; Wan-Shou, H.; Min, W.; Jen-Chang, C. Cross-section measurements for the reaction in the proton energy range 1.0–2.6 MeV. Nucl. Phys. A 1977, 275, 93–99. [Google Scholar] [CrossRef]
- Bashkin, S.; Richards, H.T. Proton Bombardment of the Lithium Isotopes. Phys. Rev. 1951, 84, 1124–1129. [Google Scholar] [CrossRef]
- Paneta, V.; Kafkarkou, A.; Kokkoris, M.; Lagoyannis, A. Differential cross-section measurements for the 7Li(p,p0)7Li, 7Li(p,p1)7Li, 7Li(p,α0)4He, 19F(p,p0)19F, 19F(p,α0)16O and 19F(p,α1,2)16O reactions. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2012, 288, 53–59. [Google Scholar] [CrossRef]
- Paul, P.; Lieb, K. The excited state at 25 MeV in Be8. Nucl. Phys. 1964, 53, 465–476. [Google Scholar] [CrossRef]
- ASTM International. ASTM Standard E521-96. Standard Practice for Neutron Radiation Damage Simulation by Charged-Particle Irradiation; ASTM International: West Conshohocken, PA, USA, 2009. [Google Scholar]
- Stoller, R.E.; Toloczko, M.B.; Was, G.S.; Certain, A.G.; Dwaraknath, S.; Garner, F.A. On the use of SRIM for computing radiation damage exposure. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2016, 310, 75–80. [Google Scholar] [CrossRef]
- Möller, S.; Krug, R.; Rayaprolu, R.; Kuhn, B.; Joußen, E.; Kreter, A. Deuterium retention in tungsten and reduced activation steels after 3 MeV proton irradiation. Nucl. Mater. Energy 2020, 23, 100742. [Google Scholar] [CrossRef]
- Möller, S. Accelerator Technology: Applications in Science, Medicine, and Industry; Springer: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Mayer, M. SIMNRA User’s Guide; Max-Planck-Institut für Plasmaphysik Garching bei München: Munich, Germany, 1997; p. 67. [Google Scholar]
- Gurbich, A.F. SigmaCalc recent development and present status of the evaluated cross-sections for IBA. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2016, 371, 27–32. [Google Scholar] [CrossRef]
- Silva, T.; Rodrigues, C.; Mayer, M.; Moro, M.; Trindade, G.; Aguirre, F.; Added, N.; A Rizzutto, M.; Tabacniks, M. MultiSIMNRA: A computational tool for self-consistent ion beam analysis using SIMNRA. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. At. 2016, 371, 86–89. [Google Scholar] [CrossRef] [Green Version]
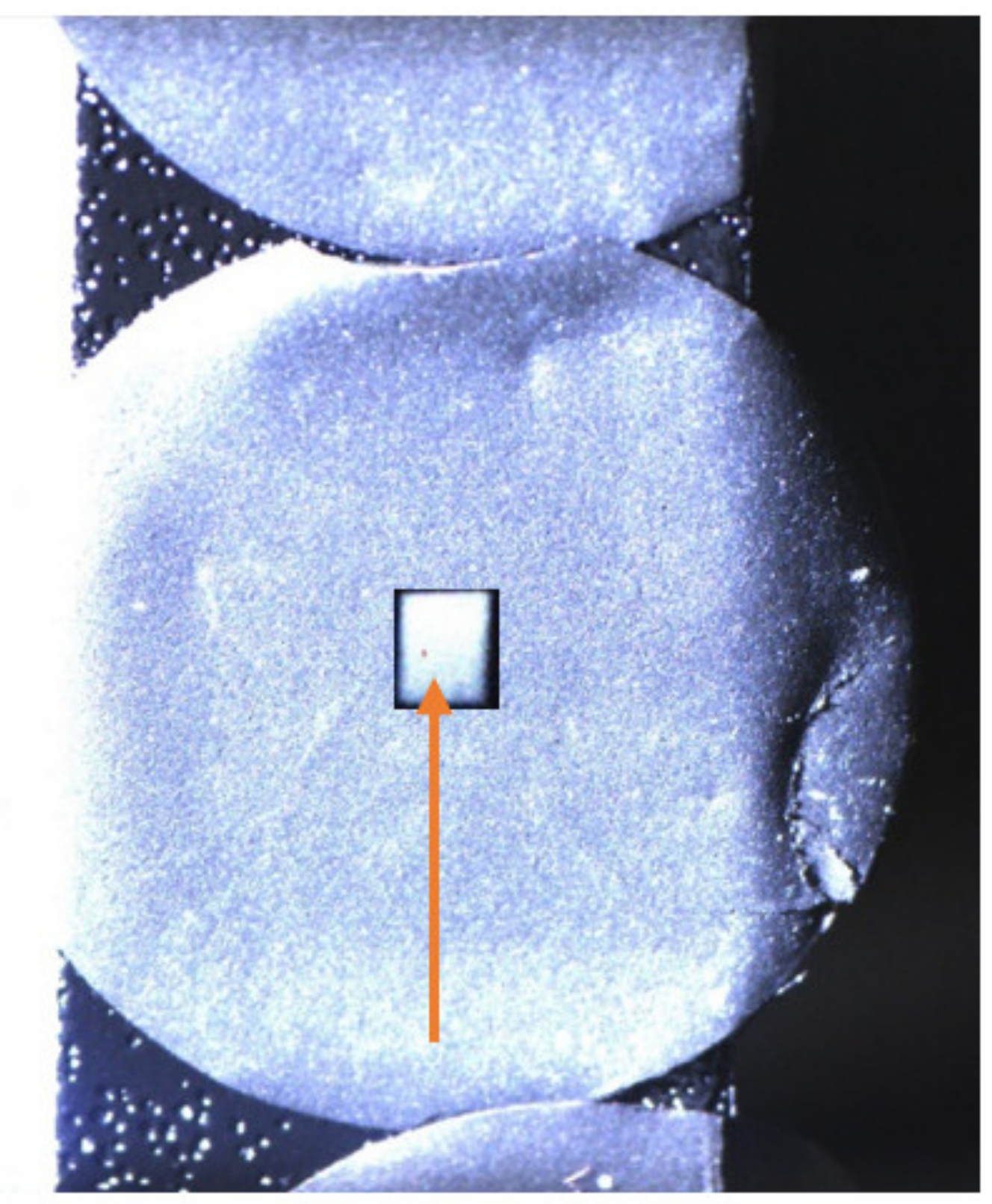
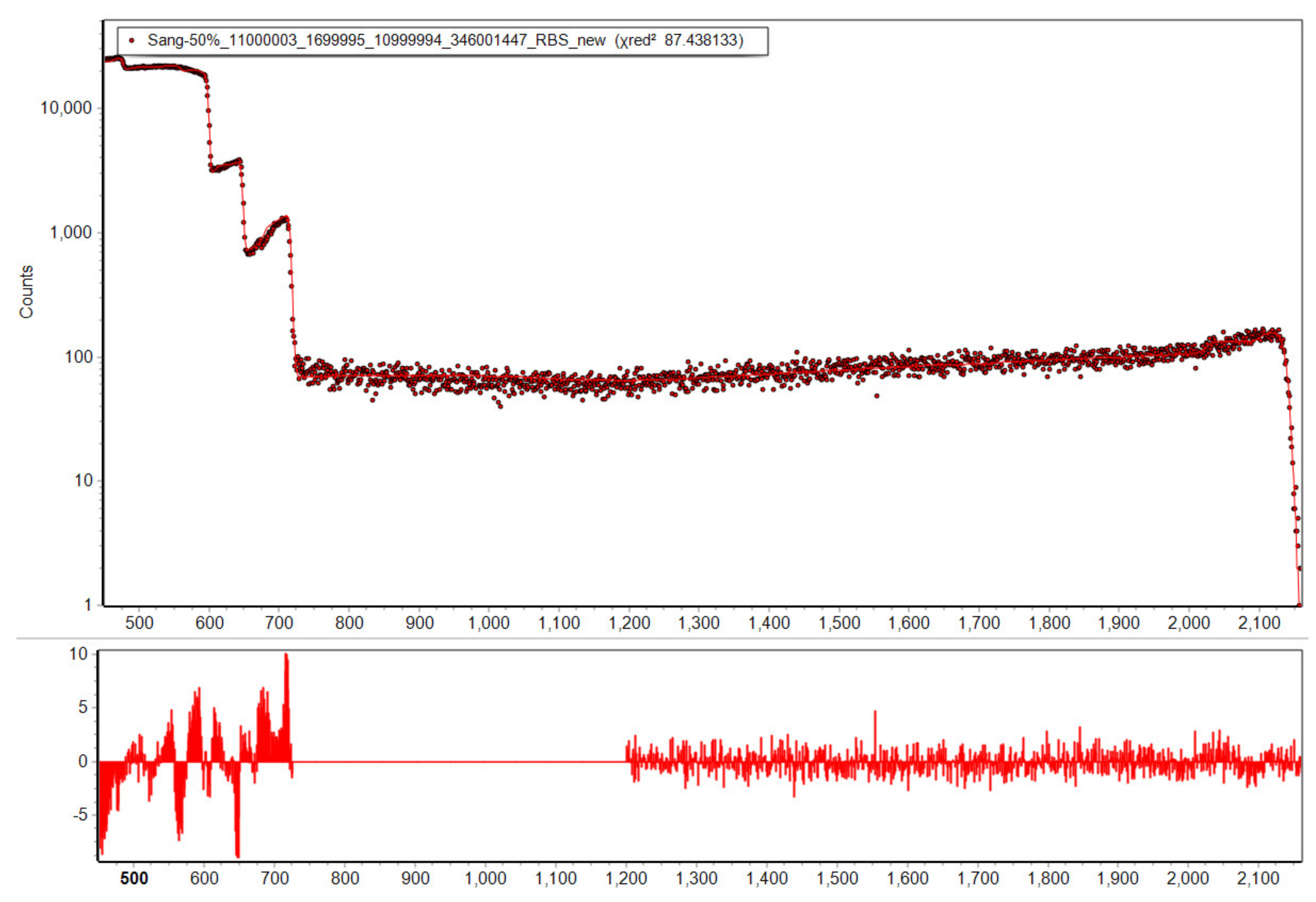

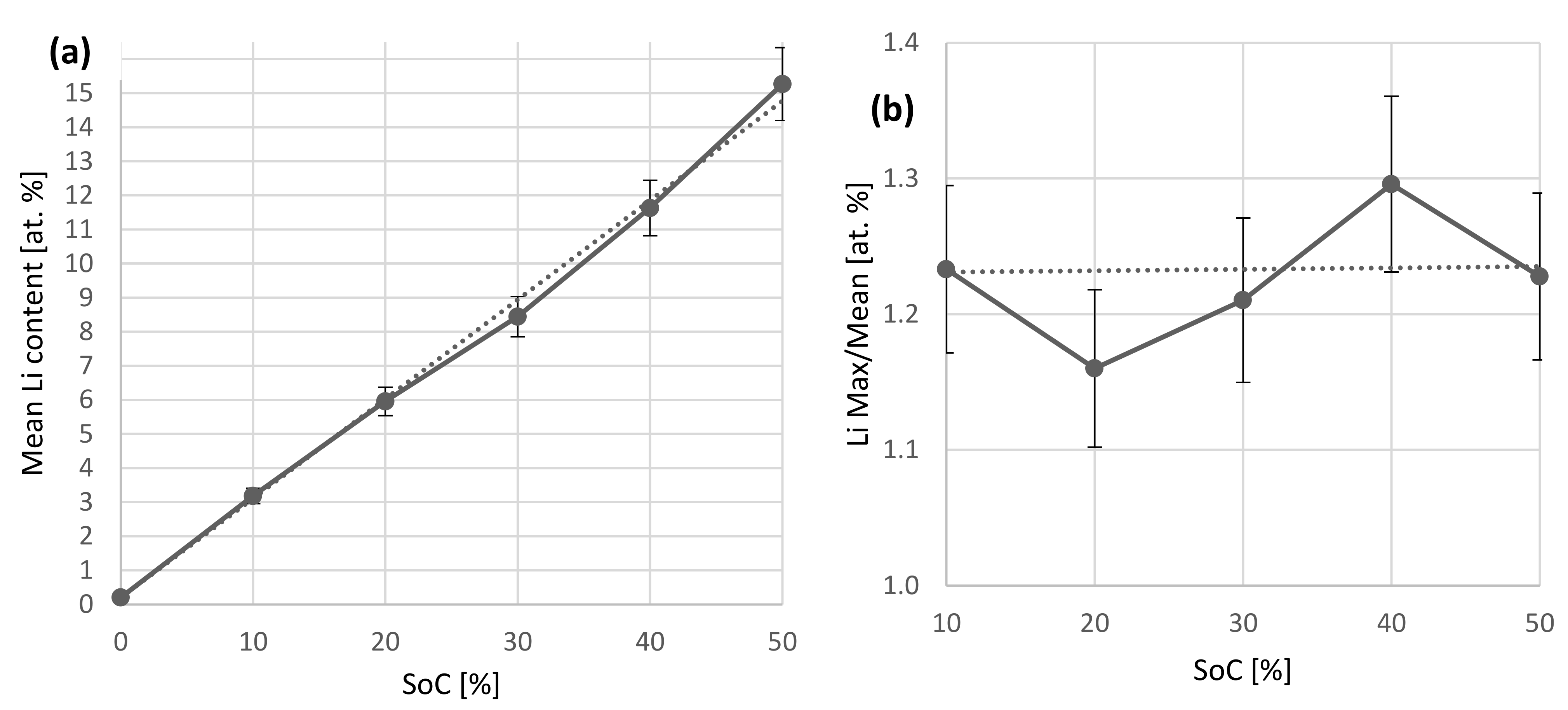
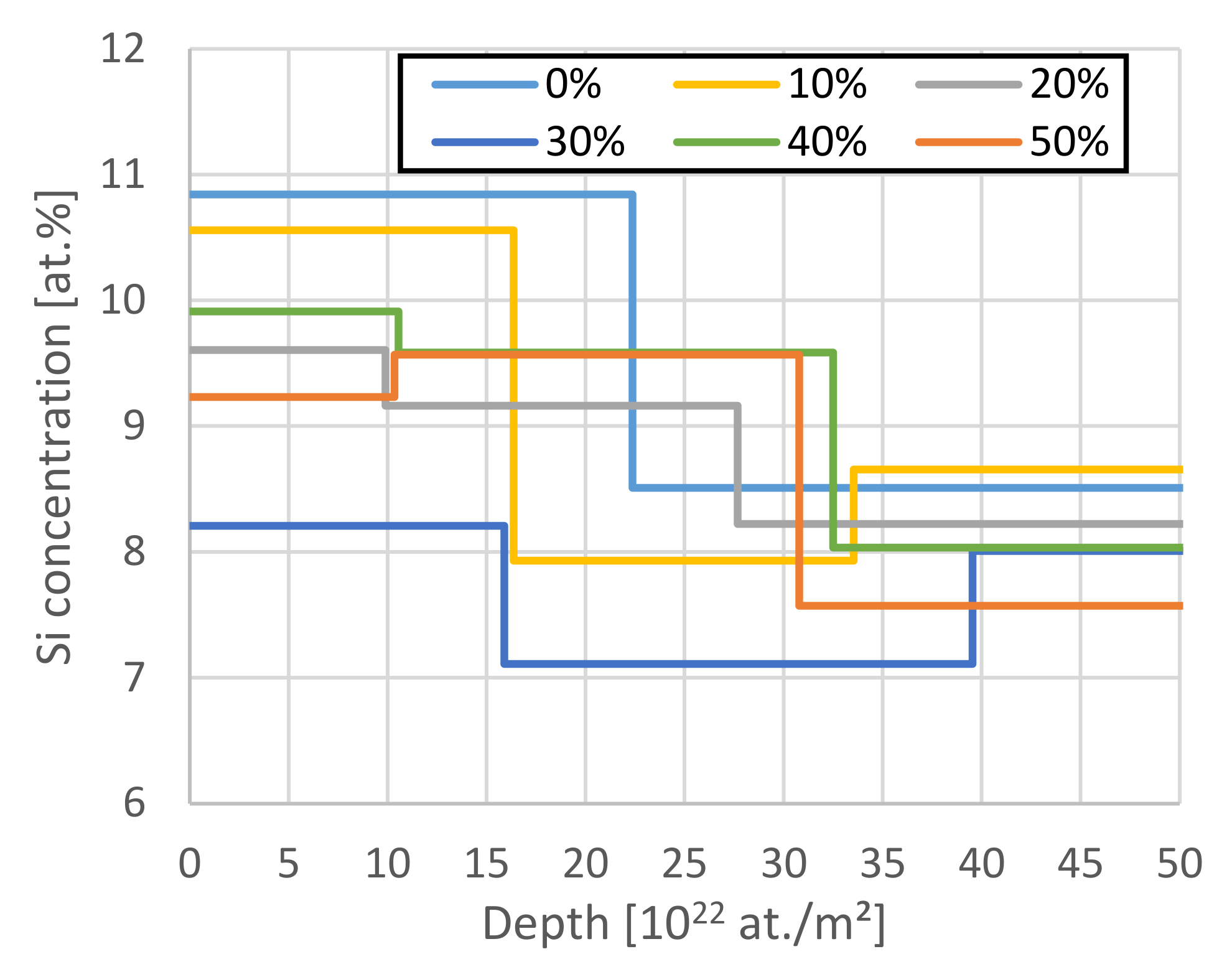



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction | 7Li(p,α0)4He | 7Li(d,α0)5He | 7Li(3He,α0)6Li | 7Li(d,p0)8Li | 7Li(3He,p0)9Be | 6Li(3He,p0)8Be | 6Li(p, 3He)4He | 6Li(3He, α0)5Li | 6Li(d, α0)4He |
|---|---|---|---|---|---|---|---|---|---|
| Q-value [MeV] | 17.34 | 14.23 | 13.33 | −0.19 | 11.2 | 16.79 | 4.02 | 14.91 | 22.37 |
| Range [1022 at/m2] | 280 | 230 | 55 | 110 | 70 | 50 | 50 | 50 | 350 |
| Resolution [1022 at/m2] | 1.70 | 1.70 | 2.15 | 2.30 | 3.50 | 7.70 | 0.65 | 2.60 | 2.75 |
| Cross-section | Paneta [45] | n.a. >1.6 MeV | Not available | Paul [46] | Not available | Not available | Chia-Shou Lin [43] | Not available | Only <2 MeV |
| Lithiation Degree/SoC (%) | Anode Capacity (mAh/cm2) | Lithiation Charged (mAh) | Lithiation Charged (1019 Atoms/cm2) | Process Time (h) | IBA Detected Li (1019 Atoms/cm2) |
|---|---|---|---|---|---|
| 0 | 3.78 | 0 | 0.00 | 0 | 0.057 |
| 10 | 3.78 | 0.38 | 0.85 | 0.5 | 0.892 |
| 20 | 3.78 | 0.76 | 1.70 | 1 | 1.668 |
| 30 | 3.78 | 1.13 | 2.55 | 1.5 | 2.364 |
| 40 | 3.78 | 1.51 | 3.40 | 2 | 3.257 |
| 50 | 3.78 | 1.89 | 4.25 | 2.5 | 4.274 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Möller, S.; Joo, H.; Rasinski, M.; Mann, M.; Figgemeier, E.; Finsterbusch, M. Quantitative Lithiation Depth Profiling in Silicon Containing Anodes Investigated by Ion Beam Analysis. Batteries 2022, 8, 14. https://doi.org/10.3390/batteries8020014
Möller S, Joo H, Rasinski M, Mann M, Figgemeier E, Finsterbusch M. Quantitative Lithiation Depth Profiling in Silicon Containing Anodes Investigated by Ion Beam Analysis. Batteries. 2022; 8(2):14. https://doi.org/10.3390/batteries8020014
Chicago/Turabian StyleMöller, Sören, Hyunsang Joo, Marcin Rasinski, Markus Mann, Egbert Figgemeier, and Martin Finsterbusch. 2022. "Quantitative Lithiation Depth Profiling in Silicon Containing Anodes Investigated by Ion Beam Analysis" Batteries 8, no. 2: 14. https://doi.org/10.3390/batteries8020014
APA StyleMöller, S., Joo, H., Rasinski, M., Mann, M., Figgemeier, E., & Finsterbusch, M. (2022). Quantitative Lithiation Depth Profiling in Silicon Containing Anodes Investigated by Ion Beam Analysis. Batteries, 8(2), 14. https://doi.org/10.3390/batteries8020014