1. Introduction
Over recent decades, lithium-ion batteries (LIBs) have been widely adopted as secondary energy storage devices, particularly in electric vehicles and portable electronics [
1]. However, constraints related to energy density and the escalating costs of LIB technology have driven efforts to explore alternative battery systems [
2]. Due to the widespread availability of K in the Earth’s crust, potassium-ion batteries (KIBs) have emerged as a promising alternative. [
3]. Despite this advantage, KIBs suffer from low gravimetric and volumetric energy densities, primarily due to the large ionic size and high atomic weight of K ions. Current studies on KIBs cathode materials has focused on Prussian blue analogs, layered metal oxides, polyanionic compounds, and organic materials, yet none have achieved adequate capacity for practical applications. Se, known for its high electrical conductivity (1 × 10
−3 S m
−1), theoretical capacity of 675 mA h g
−1, and volumetric capacity of 3253 mA h cm
−3, has emerged as a potential cathode material for advanced KIBs [
4]. The K–Se battery operates according to the reaction Se + 2M⁺ + 2e
− ↔ M
2Se (where M = Li, Na, K), offering a notable theoretical gravimetric capacity, making it a promising contender for next-generation energy storage solutions.
However, utilizing Se as a cathode material in K–Se batteries introduces critical challenges. The substantial large ionic radius of K⁺ ions leads to sluggish ion transport [
5]. Additionally, the shuttle effect, arising from the dissolution of polyselenide intermediates during redox reactions, decreases Coulombic efficiency and shortens cycle life [
6]. The solid-state reaction between K and Se also leads to considerable volumetric expansion during charge/discharge cycles, causing mechanical degradation of the active material [
7]. Several approaches have been proposed to address these issues. Yu et al. demonstrated that suppressing the formation of soluble polyselenide intermediates in carbonate-based electrolytes effectively alleviates the shuttle effect [
7]. Despite such advances, Se cathodes still face challenges such as limited active material utilization, poor rate performance, and rapid capacity decay.
Unmodified commercial carbon materials such as Super P and Ketjen Black have been widely utilized as Se-loading hosts due to their availability and cost-effectiveness [
8]. However, their non-uniform particle size distribution and small particle dimensions limit their performance [
9]. Non-uniform particle sizes hinder the uniform distribution of Se within the internal pores of the host, leading to its accumulation in interparticle voids and reducing its utilization efficiency within the electrode structure [
9]. This non-uniform dispersion limits the interaction between the active material and electrolyte, leading to a decline in total electrode capacity. Additionally, small particle sizes increase surface energy and promote particle aggregation [
9], obstructing the pore structure and exacerbating non-uniform Se loading. Consequently, the aggregation and surface accumulation of Se degrade electrochemical performance and lead to rapid capacity fading. The combination of non-uniform and small particle sizes further restricts selenium from fully penetrating the internal pores of the carbon host, causing it to accumulate primarily on the surface or within interparticle voids [
10]. Consequently, Se utilization is significantly diminished, severely limiting the energy density and efficiency of active materials in the battery. To overcome these limitations, extensive research has been conducted on host materials with a well-defined pore distribution. Yu et al. demonstrated that a uniform pore structure prevents the localized accumulation of Se and facilitates its homogeneous dispersion within the host material [
11]. This approach enhances Se loading efficiency and utilization while improving interactions between the active material and electrolyte. Additionally, it contributes to increased electrode capacity and mitigates performance degradation caused by Se aggregation and surface accumulation [
10].
However, aggregation issues stemming from non-uniform and small particle sizes cannot be fully resolved solely through pore design. Achieving a uniform particle size in carbon hosts is critical for improving performance. Uniformly synthesized flower-like microstructures constructed from two-dimensional (2D) building blocks offer several advantages for Se-based batteries [
12]. The high surface-to-volume ratio of these microstructures significantly increases the number of electrochemical reaction sites, thereby enhancing active material utilization and reaction kinetics. Moreover, the three-dimensional (3D) architecture formed by the assembly of 2D building blocks improves electrolyte accessibility and optimizes ion diffusion pathways [
12], resulting in enhanced Se utilization and long-term cycling stability. Additionally, the uniformly synthesized flower-like structure effectively mitigates aggregation issues caused by nonuniform and small particle sizes.
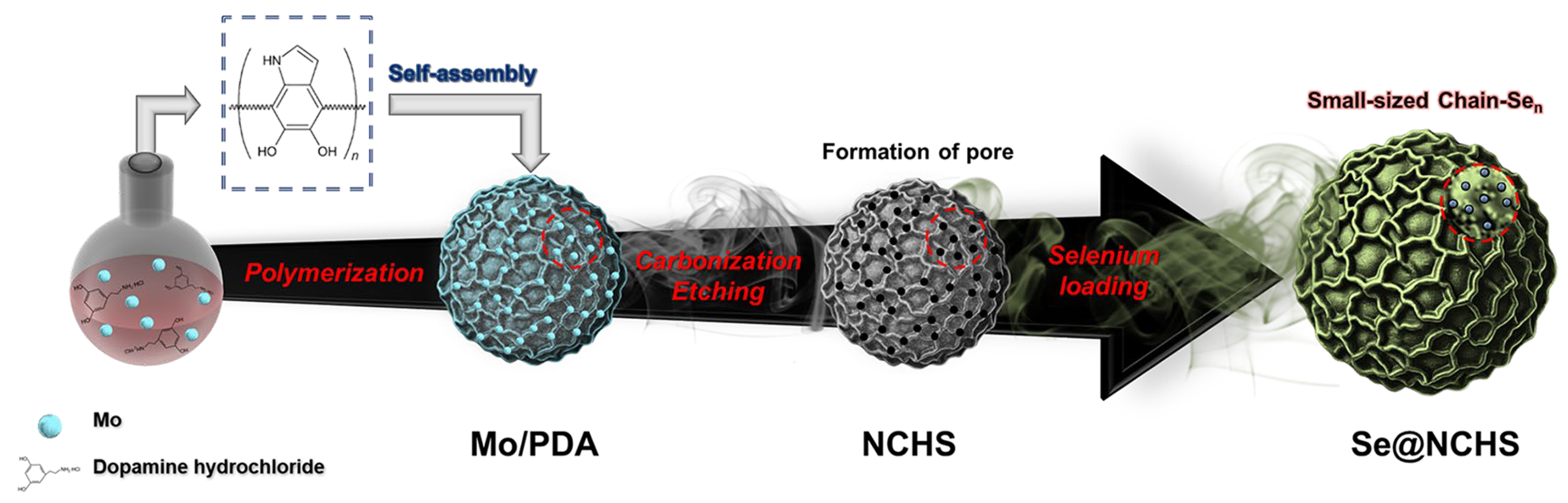
In this study, Se-loaded N-doped carbon spheres (Se@NCHS) with a uniform flower-like hierarchical architecture were developed as a cathode material for K–Se batteries. The uniform microstructure and precisely distributed pores formed along the periphery of the flower-like architecture effectively address the key challenges in Se-based batteries, significantly enhancing electrochemical performance. The uniformly distributed pores formed through the Mo etching process along the periphery of the flower-like structure minimize Se aggregation and surface accumulation while promoting efficient electrolyte infiltration and ion transport [
11,
13]. Furthermore, the interconnected framework constructed from 2D building blocks optimizes electrolyte access and ion diffusion pathways, ensuring long-term cycling stability. The flower-like microstructure with a uniform particle size mitigates aggregation issues caused by nonuniform and small particle sizes, ensuring stable and efficient performance. N doping, naturally introduced during synthesis, strengthens chemical bonding between Se and the carbon host, suppresses side reactions, and accelerates reaction kinetics, thereby improving Coulombic efficiency [
14]. By combining structural and chemical modifications, the Se@NCHS electrode exhibits superior Se utilization, prolonged cycling stability, and enhanced electrochemical properties, making it a promising candidate for next-generation K–Se batteries.
3. Results and Discussion
Figure 1 illustrates the synthesis process of Se@NCHS. Mo@PDA spheres were initially synthesized as uniform particles with an average size of approximately 1.3 μm, as observed in SEM images (
Figure S1a,b). During synthesis, dopamine underwent polymerization, and 2D building blocks self-assembled into spheres with a unique flower-shaped morphology. The Mo@PDA spheres were then carbonized at 750 °C under an argon atmosphere, resulting in the formation of MoO
x@NC. MoO
x@NC particles, approximately 1.4 μm in size, exhibited no significant structural deformation of the carbon observed post-carbonization (
Figure S1c,d).
After etching with aqua regia, the obtained NCHS retained its structure and size (
Figure 2a,b). TEM images (
Figure 2c,d) revealed a flower-shaped morphology with a large central core, indicating that the petal-like external structure had a higher pore density compared to the inner core. This unique flower-shaped morphology enhances the electrolyte contact area, thereby improving electrochemical performance [
11,
14]. High-resolution TEM (HR-TEM) images (
Figure 2e,f) confirmed that no distinct lattice fringes were present within the carbon matrix of NCHS, while the selected area electron diffraction (SAED) pattern (
Figure 2g) exhibited no lattice characteristics corresponding to metallic Mo. These findings confirm the complete removal of MoO
x, resulting in a porous amorphous carbon matrix. Elemental mapping via energy-dispersive X-ray spectroscopy (EDS) (
Figure 2h) demonstrated that the carbon spheres primarily consisted of C and N, verifying the successful formation of N-doped carbon material. The NCHS material features a flower-shaped microstructure with uniformly distributed pores and nitrogen-induced defects, which facilitate additional ion diffusion channels and significantly enhance rate performance [
15]. Furthermore, the flower-shaped design improves electrolyte accessibility and expands the reaction surface area, thereby maximizing electrochemical efficiency [
11,
14]. Additionally, the hierarchical architecture, developed through aqua regia etching, ensures a well-organized porous network across the flower-like surface, further optimizing charge transport [
13]. These structural benefits collectively result in superior electrochemical performance compared to conventional carbon hosts [
16].
Se@NCHS was synthesized via a melt-diffusion method, effectively embedding Se into the well-distributed pores and the flower-like architecture of NCHS. SEM images (
Figure 3a,b) revealed that the Se@NCHS, approximately 1.2 μm in size, retained its structure without significant deformation after Se loading. Notably, no residual Se powder was observed on the outer surface of the carbon after Se loading, indicating the successful encapsulation of Se within the NCHS structure. HR-TEM images, along with SAED patterns, showed no distinct lattice fringes (
Figure 3c–e) or lattice structures corresponding to Se, confirming the preservation of the amorphous carbon structure and atomic-scale confinement of Se within the pores of NCHS. The elemental mapping via energy-dispersive X-ray spectroscopy (EDX) of Se@NCHS (
Figure 3f) revealed a homogeneous dispersion of C, N, and Se. The unique flower-shaped structure formed by the assembly of 2D building blocks provides a larger contact area than spherical carbon structures [
11,
14]. This increased surface area allowed for the uniform distribution of Se within the carbon matrix and created additional adsorption within the pores, contributing to the observed results. This structure enabled high surface Se loading, with no unincorporated or overgrown Se observed, and showed that more Se was loaded on the surface than within the internal pores. This distribution is attributed to the hierarchical structure of the NCHS, which was synthesized via aqua regia etching and is characterized by uniformly distributed pores across its flower-shaped surface. This porous architecture enabled Se to efficiently infiltrate the internal pores, thereby improving its utilization. Additionally, nitrogen within the Se@NCHS introduces polar sites with a strong affinity for charged Se species, effectively suppressing undesirable side reactions in the electrolyte [
14]. These abundant active sites facilitated K⁺ ion transport, enhanced electronic conductivity, promoted the conversion of Se species, accelerated reaction kinetics, and significantly improved redox capacity [
14].
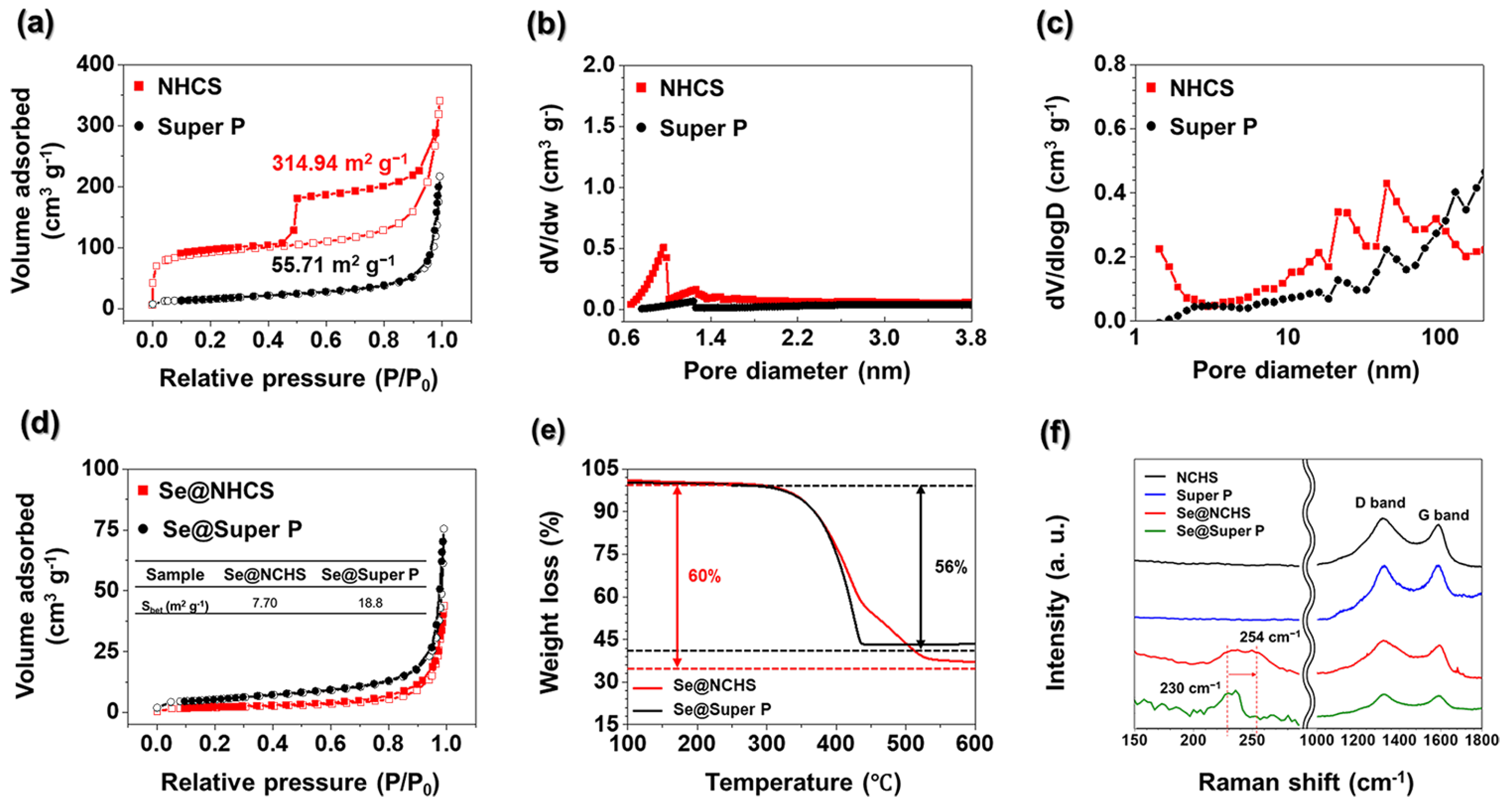
The N
2 absorption–desorption isotherms of NCHS, Super P, Se@NCHS, and Se@Super P were analyzed to assess the surface properties of the synthesized materials, as illustrated in
Figure 4a. The adsorption-desorption characteristics of NCHS exhibited a type III isotherm, demonstrating substantial nitrogen absorption at low pressure levels and a distinct hysteresis loop at elevated pressures, validating its porous structure [
16]. A steep increase in nitrogen adsorption at low relative pressures (P/P
0 < 0.01) indicated the presence of micropores. The BET surface area of NCHS was measured at 341.94 m
2 g
−1, which was substantially larger than that of Super P (55.71 m
2 g
−1). The micropore distribution, determined using the HK method, is presented in
Figure 4b. The NCHS sample exhibited a distinct micropore peak at 0.9 nm, whereas Super P displayed a relatively low micropore concentration. The micropore volumes of NCHS and Super P, determined through t-plot analysis, were 0.10 cm
3 g
−1 and 0.003 cm
3 g
−1, respectively. The structural characteristics of these materials are outlined in
Table S1.
The BJH pore size distribution curves for NCHS and Super P samples are shown in
Figure 4c. Unlike Super P, NCHS has a hierarchical structure comprising mesopores and micropores. The mesopores formed by etching metallic Mo help mitigate volumetric changes in Se [
13], enhance capacity retention, and provide stable cycling performance. Furthermore, the micropores generated by N-doping-induced defects are essential for facilitating the formation of chain-like Se
n, thereby improving electrochemical activity [
15]. After Se loading, the N
2 adsorption–desorption isotherms (
Figure 4d) show a significant reduction in surface areas for Se@NCHS and Se@Super P, measured at 7.69 and 18.78 cm
3 g
−1, respectively. This reduction corresponds to a notable decrease in pore volume. The substantial decrease in the surface area of NCHS suggests that Se effectively penetrated the internal pores, blocking them and thereby reducing the available surface area. In contrast, the lower reduction rate observed in Super P indicates that Se did not fully infiltrate its internal pores, with residual Se remaining outside the pore structure [
11,
15].
TGA was performed to determine Se loading (
Figure 4e). The Se content in Se@NCHS was measured at 60 wt.%, indicating that all the Se successfully infiltrated the internal pores during impregnation. The delayed weight loss at higher temperatures suggests that Se was deeply embedded within the inner pores, requiring additional time for sublimation or evaporation [
11].
In contrast, Se@Super P exhibited a Se content of 56 wt.%, indicating that the loaded 60 wt% Se did not fully penetrate the internal pores of Super P. Unlike Se@NCHS, Se@Super P exhibited rapid weight loss at lower temperatures, which can be attributed to aggregation issues caused by non-uniform and smaller particle sizes [
8]. These limitations restricted the infiltration of Se into the internal pores, resulting in its accumulation on the surface or within interparticle voids.
Raman spectroscopy was employed to analyze the structural properties of Se and carbon hosts in the samples
(Figure 4f). The Raman spectra of Se@Super P and Se@NCHS offered key insights into how the carbon matrix affects the structural evolution of Se. For Se@Super P, a distinct peak appeared at 230 cm
−1, corresponding to the stretching vibration mode of cyclic Se
8 molecules [
17,
18]. Conversely, the Se@NCHS sample exhibited a peak shift from 230 cm
−1 to 254 cm
−1, signifying a partial transformation of cyclic Se
8 into chain-like Se
n structures [
12]. This shift implies that the microporous structure of NCHS plays a crucial role in facilitating this structural transition during the Se melting-diffusion process. Importantly, the presence of the 254 cm
−1 peak confirms that the well-developed micropores in NCHS effectively suppress the stabilization of cyclic Se
8 while promoting its structural conversion into chain-like Se
n [
18]. Since chain-like Se
n demonstrates superior electrochemical properties compared to cyclic Se
8, this transformation is a key factor in enhancing Se utilization for energy storage applications. Additionally, the structural features of the carbon hosts were examined through the D-band (1321 cm
−1) and G-band (1592 cm
−1) peaks, which correspond to disordered sp
3-hybridized carbon and graphitic sp
2-hybridized carbon, respectively [
19]. The
ID/
IG ratio for NCHS was significantly higher than that of Super P, suggesting that KOH activation generated additional structural defects [
20]. These defects are beneficial as strengthen the interaction between the carbon matrix and Se, thereby enhancing the confinement and stabilization of Se within the micropores [
21]. Interestingly, after Se loading, the
ID/
IG values for both the Se@NCHS and Se@Super P samples remained unchanged compared to those of their pristine carbon hosts. This finding verifies that the Se melt-diffusion process preserves the structural stability of the carbon framework, further highlighting the durability of NCHS as a host material for Se [
21].
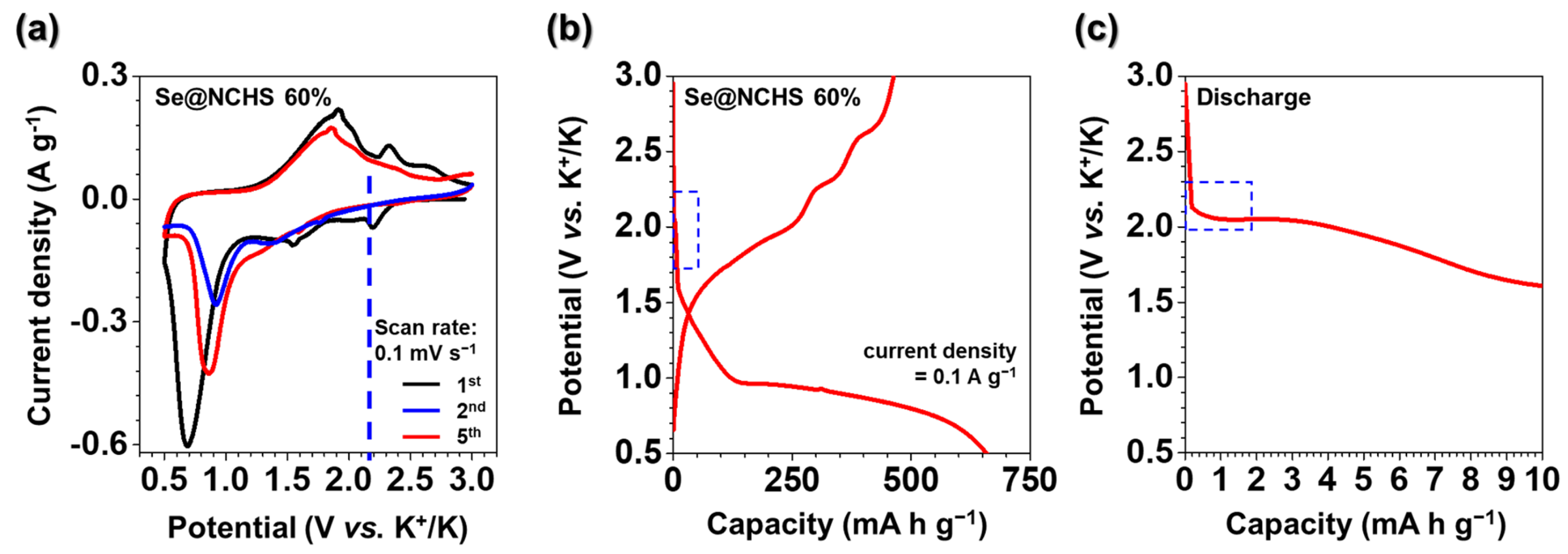
CV tests were performed to examine the charge-discharge mechanism of Se@NCHS as a cathode material for K–Se batteries (
Figure 5). The measurements were conducted at a scan rate of 0.1 mV s
−1 within a voltage window of 0.5–3.0 V (vs. K⁺/K) (
Figure 5a). This voltage range does not include the 0.01–0.5 V region, where carbon interacts with K⁺ ions, confirming that carbon primarily functions to enhance conductivity and maintain structural integrity [
22]. During the first CV scan of Se@NCHS, three cathodic peaks were observed at 0.7, 1.54, and 2.12 V. The 0.7 V peak significantly diminished in subsequent cycles, most likely due to irreversible capacity loss caused by electrolyte decomposition and the formation of a solid electrolyte interphase (SEI) layer [
23]. In contrast, anodic peaks appeared at 1.89, 2.01, and 2.31 V, corresponding to the reversible transition from K
2Se to Se [
23]. The first charge-discharge profile of Se@NCHS (
Figure 5b) exhibits discharge and charge capacities of 658.0 and 463.0 mA h g
−1, respectively, resulting in a coulombic efficiency of 70.4%. The initial decline in coulombic efficiency is mainly due to the irreversible formation of the SEI layer and side reactions involving K⁺ ions with functional groups on the carbon surface. [
24]. According to prior research [
18], suggest that Se reduction in carbonate-based electrolytes follows a stepwise mechanism: Se → K
2Se
2 → K
2Se. Consequently, the cathodic peaks at 1.54 and 2.12 V in the first CV scan of Se@NCHS correspond to the formation of K
2Se (Se → K
2Se
2 → K
2Se) [
18]. An expanded view of the first discharge plateau for Se@NCHS (
Figure 5c) shows that the initial plateau at 2.12 V aligns with the first CV cycle, indicating that the electrochemical reactions are consistent, particularly in the stepwise reduction process of Se [
18,
21]. This finding confirms that Se is effectively incorporated into the NCHS matrix, demonstrating that the conversion reactions of Se proceeded efficiently within the electrode structure, enabling a high utilization rate of the active material.
The cycling durability of Se@NCHS and Se@Super P electrodes at a current density of 0.5 C over 500 cycles is presented in
Figure 6a. While the Se@Super P electrode exhibited negligible capacity, the Se@NCHS sample retained a discharge capacity of 199.4 mA h g
−1. This notable variation is mainly due to the enhanced utilization of chain-like Se (Seₙ), enabled by the hierarchical structure of NCHS [
25]. The minimal capacity retention in Se@Super P can be explained by the voltage window (0.5–3.0 V) selected for K–Se batteries, which is consistent with the range used in CV tests and lies outside the potassium ion interaction range (0.1–0.5 V). Additionally, the coulombic efficiency stabilized at approximately 99% by the 50th cycle and remained consistent in the following cycles (indicated by a black arrow). To examine the carbon host’s contribution to cathode capacity, galvanostatic measurements were conducted on NCHS without Se. As shown in the first charge/discharge profile of NCHS (
Figure S2a), a substantial decline in capacity occurred after the initial cycle, resulting in a coulombic efficiency of 64.12%, primarily caused by irreversible side reactions. The cycling performance data (
Figure S2b) indicate that the capacity of NCHS dropped to nearly zero under conditions identical to Se@NCHS, verifying that the carbon matrix has minimal influence on the overall capacity of K–Se batteries.
The rate capability of the Se@NCHS and Se@Super P electrodes at various current densities is depicted in
Figure 6b. The capacity decline observed during the initial 0.1 C is primarily associated with the stabilization of the electrode-electrolyte interface and structural rearrangements within the electrode. To ensure the accuracy and reproducibility of the data, the discharge capacity of the Se@NCHS electrode was assessed at current densities of 0.2, 0.5, 1.0, 1.5, 2.0, 2.5, and 3.0 C, yielding respective values of 478, 351, 313, 277, 250, 226, 204, and 188 mA h g
−1. When the current density reverted to 0.5 C, the capacity retention remained high, reaching 282 mA h g
−1. In contrast, the Se@Super P electrode exhibited a sharp decline in capacity, dropping from 53 to 1 mA h g
−1 as the current density increased from 0.1 to 3.0 C. These results confirm that the enhanced electrochemical performance of Se@NCHS electrode stems from the synergistic effects of its structural architecture and morphological attributes. The hierarchical framework of the Se@NCHS electrode effectively accommodates Se volume fluctuations during cycling and facilitates the formation of chain-like Se (Se
n), thereby significantly improving Se utilization [
18,
21]. Furthermore, its distinct flower-shaped architecture provides a greater electrolyte contact area compared to spherical carbon, effectively shortening ion diffusion pathways [
11].
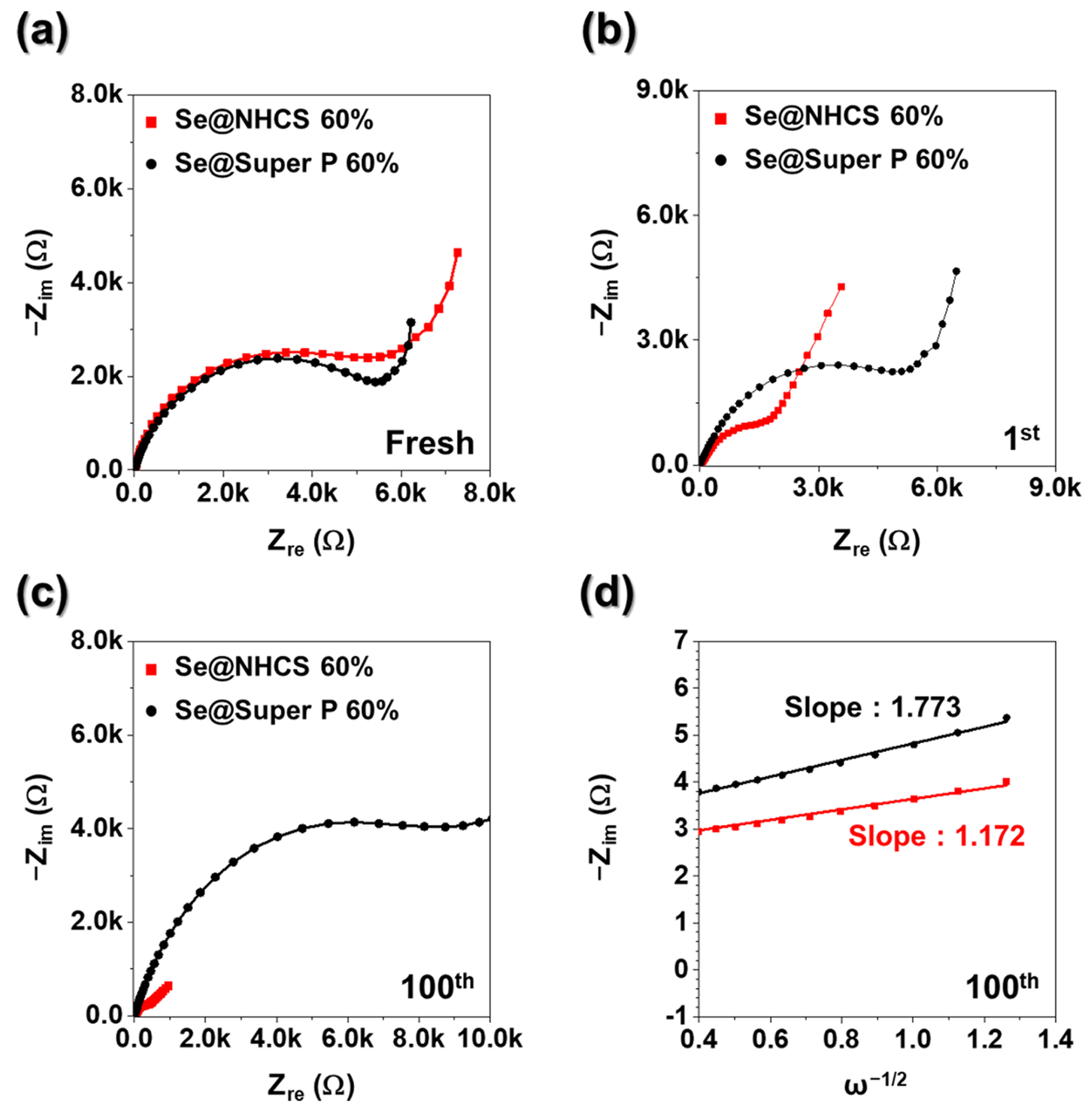
To examine the influence of the hierarchical architecture and morphology on electrode resistance variations during cycling, ex situ EIS measurements were performed on Se@NCHS and Se@Super P electrodes. In the Nyquist plot, the semicircle radius represents the impedance of the electrode, with a larger semicircle indicating higher resistance [
26]. In the fresh state, the Se@Super P electrode (
Figure 7a) exhibited a smaller semicircle compared to Se@NCHS, suggesting that the high electrical conductivity of Super P nanoparticles facilitated more efficient electron and ion transport during the initial EIS analysis [
26]. Over the course of cycling, the semicircle radius of the Se@NCHS electrode progressively decreased, while that of the Se@Super P electrode exhibited an increased in R
ct after cycling (
Figure 7a–c). Prior studies indicate that Rct tends to decrease as electronic conductivity increases, primarily due to the intrinsically high conductivity of carbon-based materials [
27]. The Se@NCHS electrode demonstrated a continuous reduction in R
ct over successive cycles, which can be attributed to enhanced electrolyte penetration and shortened ion diffusion paths, facilitated by its distinct flower-like structure of NCHS. These findings highlight that the hierarchical structure of the Se@NCHS electrode effectively mitigates volume expansion during charge/discharge cycles, thereby preserving structural integrity and stabilizing electrode interfaces.
Figure 7d presents the correlation between phase angle (ω
−1/2) and real impedance (Z′) for both electrodes after the 100 cycles in the low-frequency region. The Se@NCHS electrode displayed a lower slope than Se@Super P electrode, indicating a higher K⁺ diffusion rate. This enhancement is attributed to the uniform dispersion of active materials within the electrode, facilitated by its hierarchical structure, which effectively minimizes interfacial resistance at the electrode-electrolyte interface.
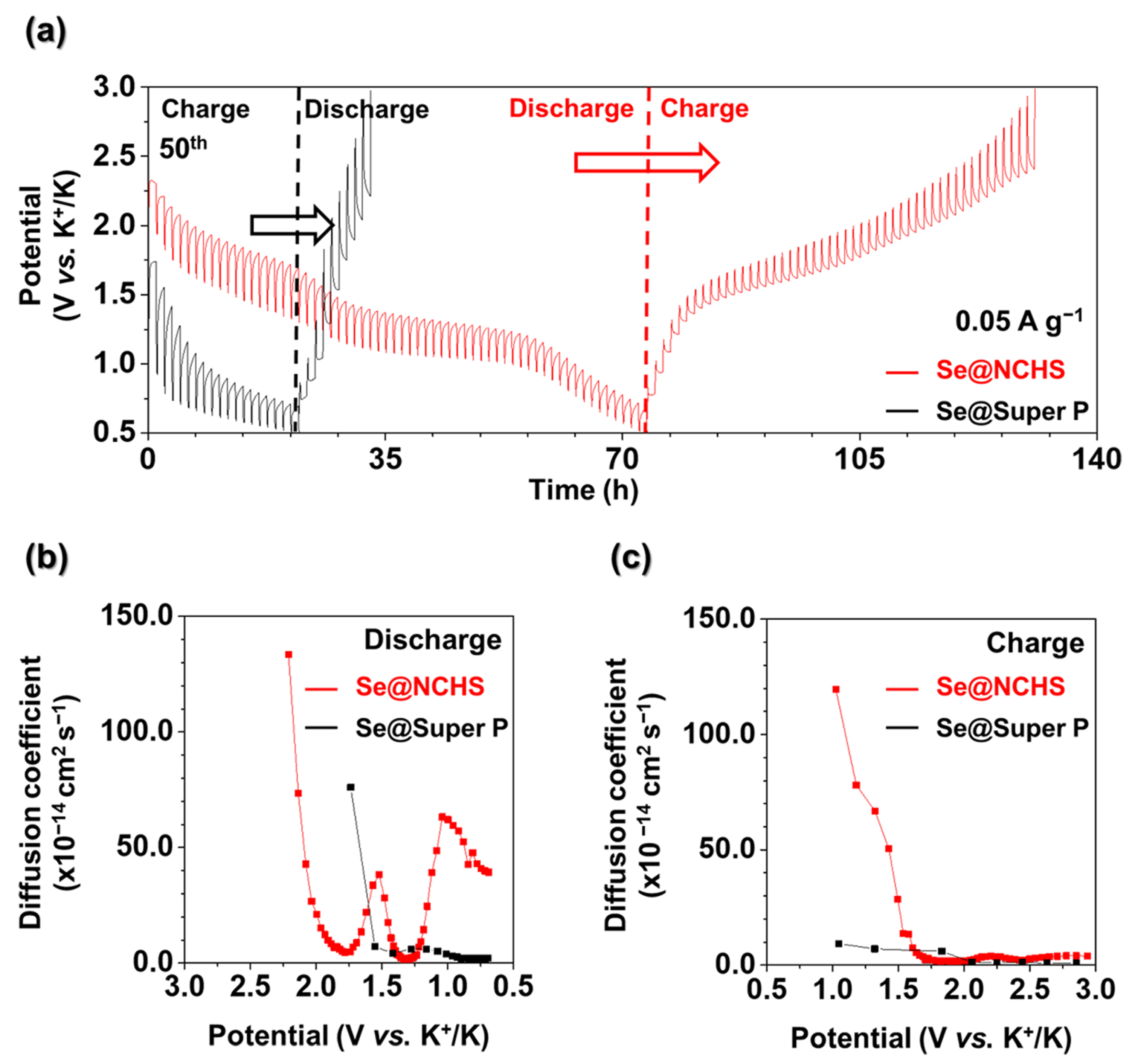
Consequently, the decrease in impedance detected in EIS measurements during cycling can be associated with the mitigation of volume expansion and improved ion transport. These findings indicate that the hierarchical structure of NCHS, defined by its uniform pore distribution and precisely controlled particle size, is essential for reducing resistance and enhancing long-term electrochemical performance. To further explore the effect of pore architecture on ion movement, galvanostatic intermittent titration technique (GITT) tests were performed (
Figure 8). Moreover, the potassium-ion diffusion coefficient (D
K) for Se@NCHS and Se@Super P was derived using Fick’s second law to assess ion diffusion behavior. In GITT, a longer reaction time indicates deeper ion penetration into the electrode, allowing for a more complete reaction with the entire active material and facilitating uniform utilization of active sites. The longer reaction time observed for Se@NCHS compared to Se@Super P suggests higher structural stability, enabling the electrode to maintain a greater capacity during charge-discharge cycles. This structural robustness was credited to the hierarchical structure of the Se@NCHS electrode, which effectively accommodates volume variations, thereby enhancing overall electrochemical performance. Furthermore, Se@NCHS exhibited significantly higher ion diffusion rates during both charge and discharge processes than Se@Super P. This improvement was attributed to the hierarchical structure, which provided additional ion pathways through the uniform pore distribution created by etching and nitrogen-doping-induced defects, thereby enabling faster ion diffusion. Additionally, the unique flower-shaped morphology formed by the assembly of 2D building blocks increased the contact area with the electrolyte relative to spherical carbon spheres, effectively shortening ion diffusion pathways [
14,
15]. The variation in the diffusion coefficient during the discharge of Se@NCHS can be attributed to the electrochemical reaction of Se, in which the transformation from cyclo-Se
8 to chain-like Se
n affects the diffusion coefficient. This phase transition alters the diffusion pathway, resulting in significant fluctuations in diffusion coefficients within a specific voltage range. In contrast, no significant variation in the diffusion coefficient was observed for Se@Super P, due to the non-uniform and small particle sizes of Super P, which limit Se penetration into the internal pores and result in Se accumulation on the surface, minimizing obstruction of the diffusion pathway. These findings provide strong evidence that the hierarchical structure, uniform pore distribution, and particle size of NCHS contribute to enhanced electrochemical performance.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}