Triggering of Valence Tautomeric Transitions in Dioxolene-Based Cobalt Complexes Influenced by Ligand Substituents, Co-ligands, and Anions



Abstract
:
1. Introduction
2. Materials and Methods
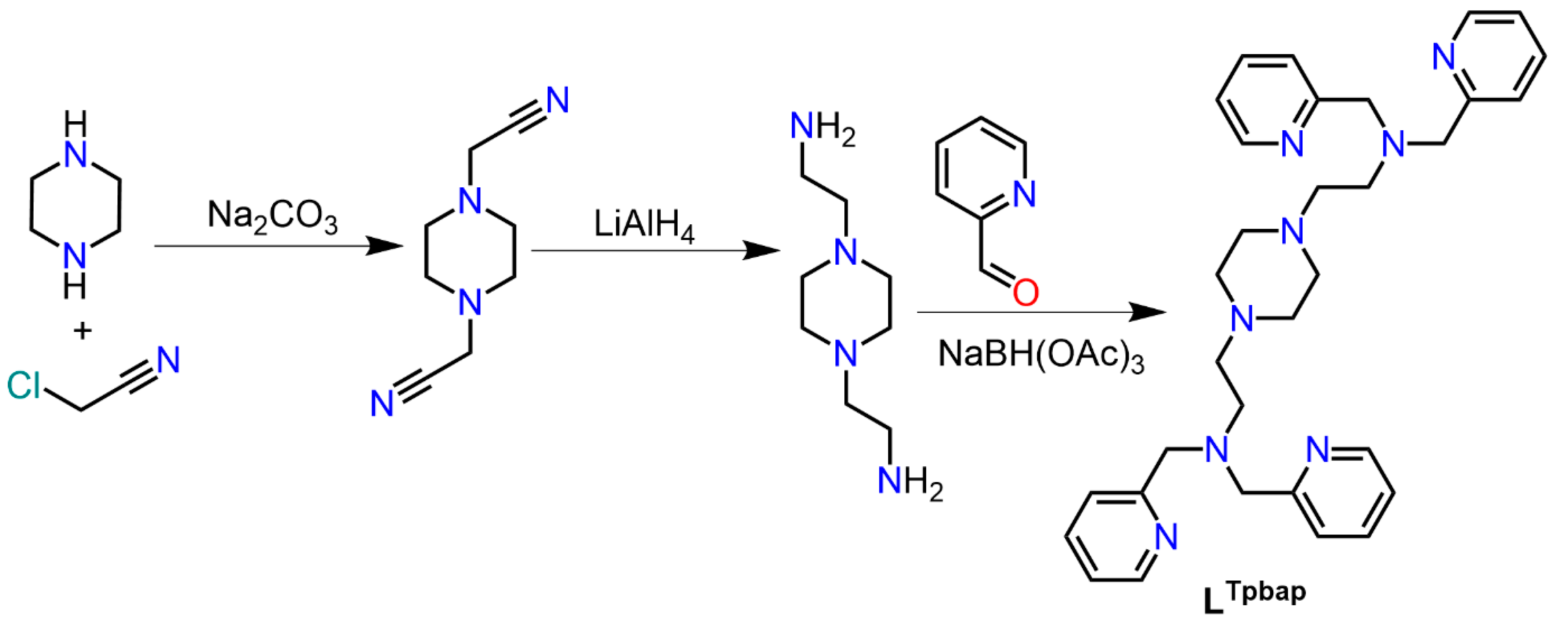
2.1. Ligand Synthesis
2.1.1. N,N′-Bis(cyanomethyl)piperazine
2.1.2. N,N′-Bis(2-aminoethyl)piperazine
2.1.3. 1,1′-(piperazine-1,4-diyl)bis(N,N-bis(pyridin-2-ylmethyl)methanamine)(Ltpbap)
2.2. Complex Synthesis
2.2.1. [Co2(OH2)(piv)4(Hpiv)4] (P1)
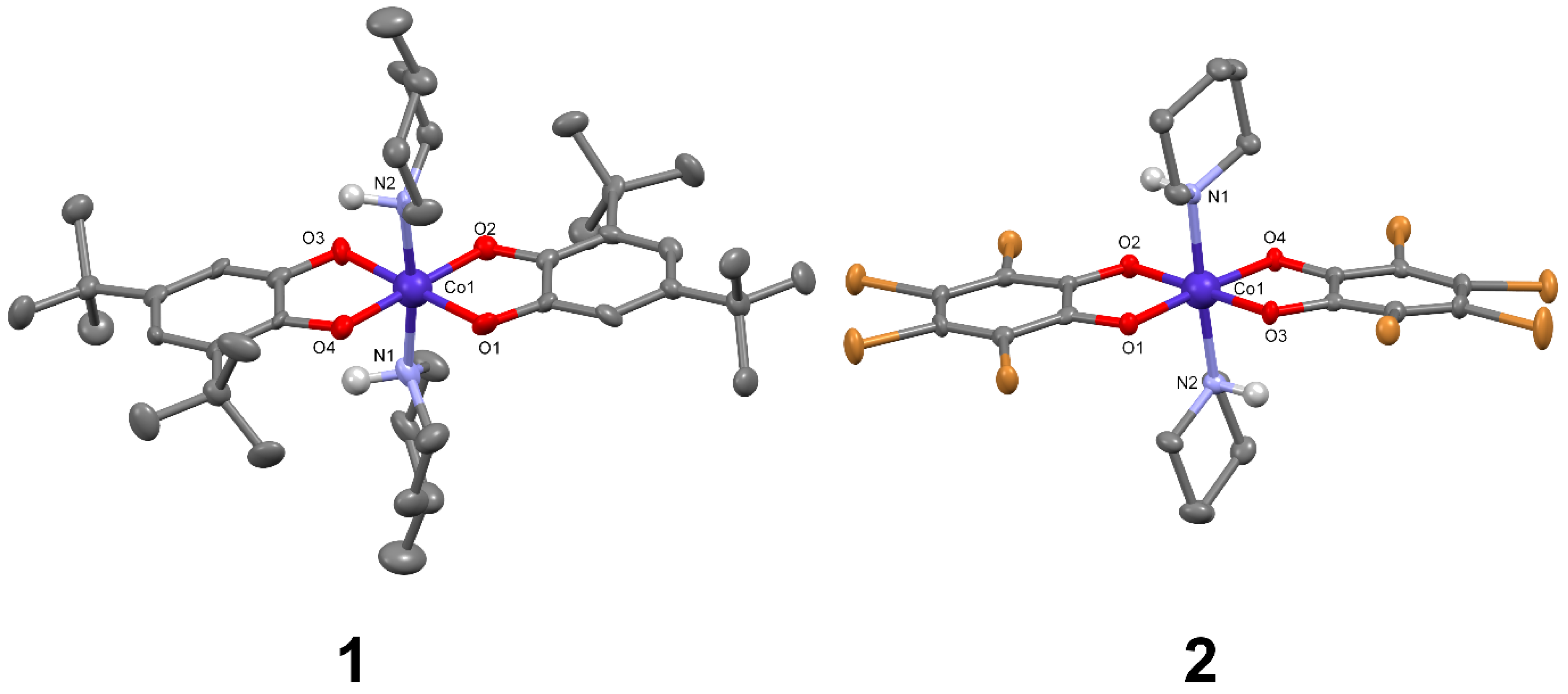
2.2.2. [Co(3,5-dbsq)(3,5-dbcat)(4-Mepip)2] (1)
2.2.3. (Hpip)[Co(tbcat)2(pip)2]CH3CN (2)
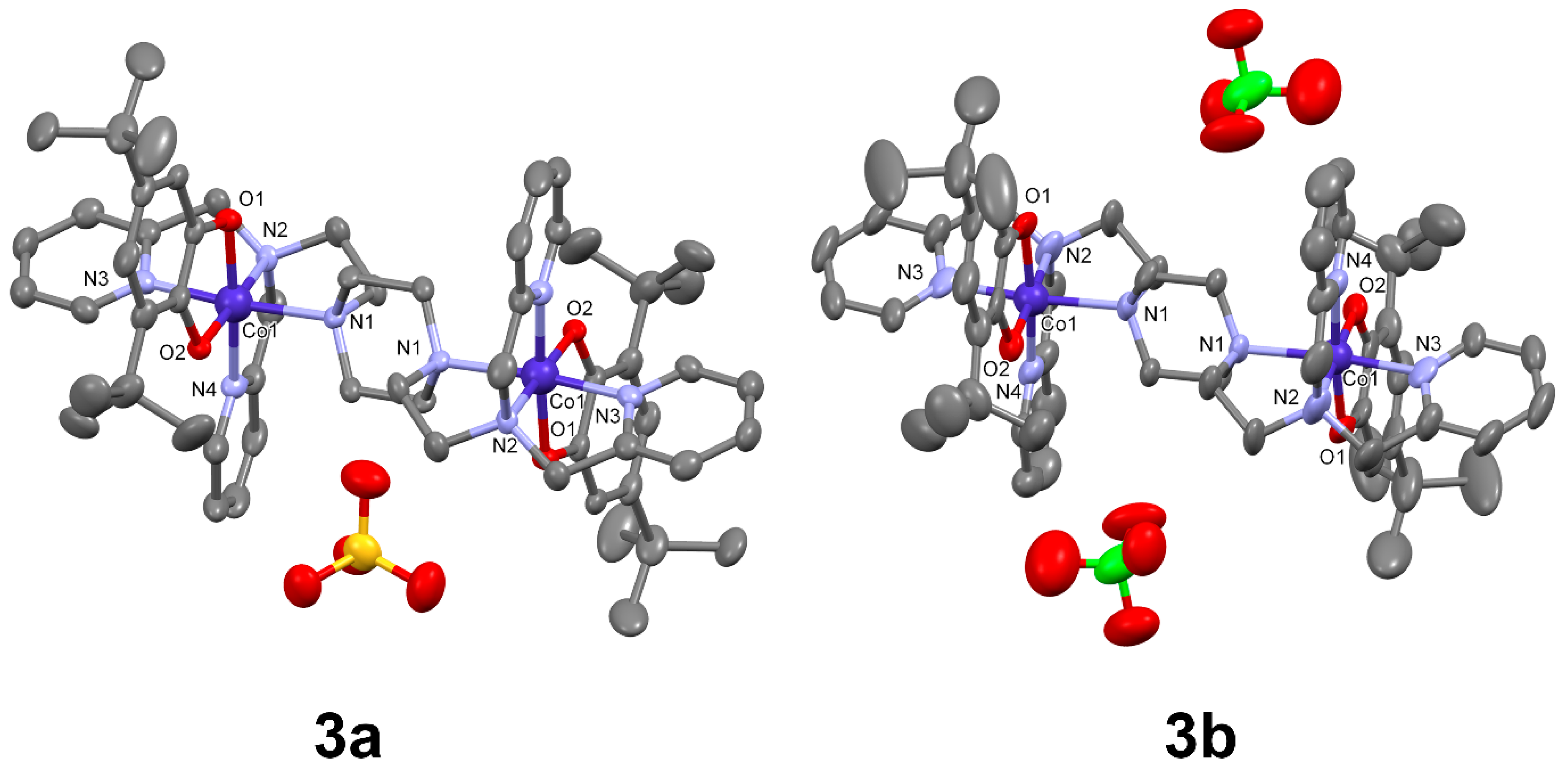
2.2.4. [Co2(Ltpbap)(3,5-dbcat)2](SO4)·5.5MeOH·2H2O (3a)
2.2.5. [Co2(Ltpbap)(3,5-dbcat)2](ClO4)2·1.5 H2O (3b)
3. Results and Discussion
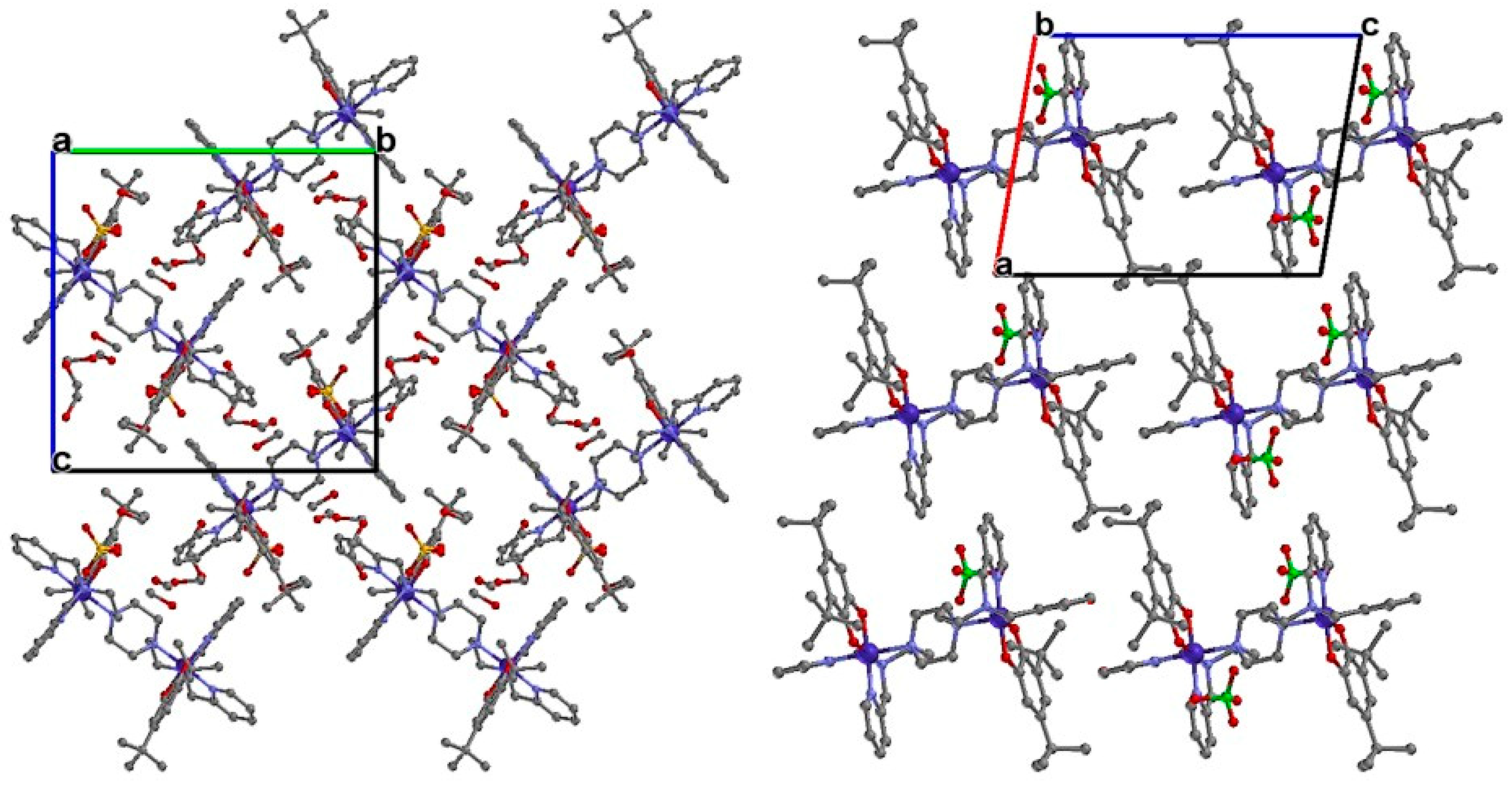
3.1. Crystal Structures
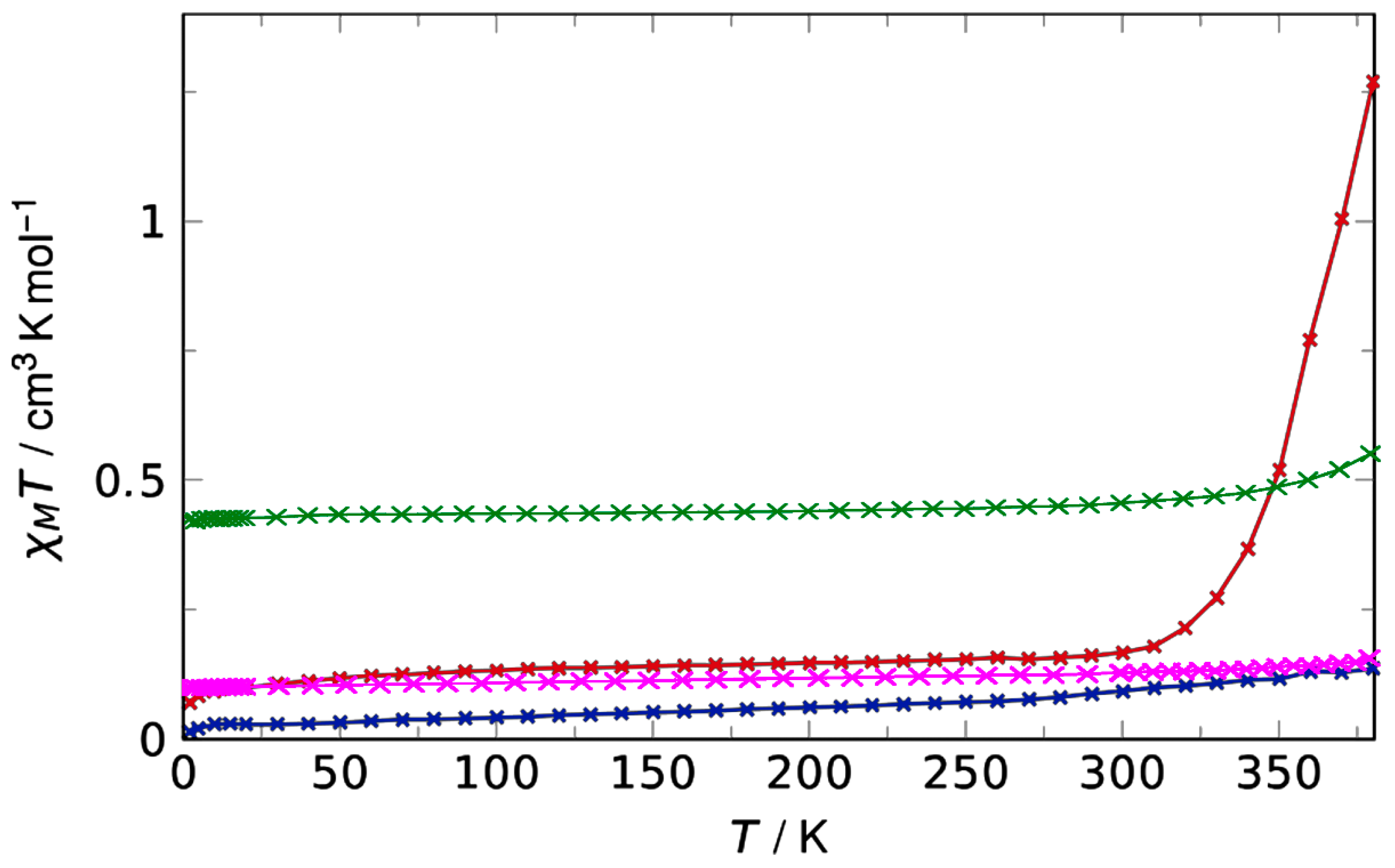
3.2. Magnetic Characterisation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bousseksou, A.; Molnár, G.; Salmon, L.; Nicolazzi, W. Molecular spin crossover phenomenon: Recent achievements and prospects. Chem. Soc. Rev. 2011, 40, 3313–3335. [Google Scholar] [CrossRef]
- Bousseksou, A.; Molnár, G.; Real, J.A.; Tanaka, K. Spin crossover and photomagnetism in dinuclear iron(II) compounds. Coord. Chem. Rev. 2007, 251, 1822–1833. [Google Scholar] [CrossRef]
- Enriquez-Cabrera, A.; Rapakousiou, A.; Piedrahita Bello, M.; Molnár, G.; Salmon, L.; Bousseksou, A. Spin crossover polymer composites, polymers and related soft materials. Coord. Chem. Rev. 2020, 419, 213396. [Google Scholar] [CrossRef]
- Gaspar, A.B.; Seredyuk, M. Spin crossover in soft matter. Coord. Chem. Rev. 2014, 268, 41–58. [Google Scholar] [CrossRef]
- Atmani, C.; El Hajj, F.; Benmansour, S.; Marchivie, M.; Triki, S.; Conan, F.; Patinec, V.; Handel, H.; Dupouy, G.; Gómez-García, C.J. Guidelines to design new spin crossover materials. Coord. Chem. Rev. 2010, 254, 1559–1569. [Google Scholar] [CrossRef]
- Sundaresan, S.; Eppelsheimer, J.; Carrella, L.M.; Rentschler, E. Three Novel Thiazole-Arm Containing 1, 3, 4-Oxadiazole-Based [HS-HS] Fe (II) Dinuclear Complexes. Crystals 2022, 12, 404. [Google Scholar] [CrossRef]
- Halcrow, M.A. Structure:Function Relationships in Molecular Spin-Crossover Complexes. Chem. Soc. Rev. 2011, 40, 4119. [Google Scholar] [CrossRef]
- Halcrow, M.A. (Ed.) Spin-Crossover Materials: Properties and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2013; p. 568. [Google Scholar]
- Hogue, R.W.; Singh, S.; Brooker, S. Spin crossover in discrete polynuclear iron(II) complexes. Chem. Soc. Rev. 2018, 47, 7303–7338 and inside front cover. [Google Scholar] [CrossRef]
- Harding, D.J.; Harding, P.; Phonsri, W. Spin crossover in iron(III) complexes. Coord. Chem. Rev. 2016, 313, 38–61. [Google Scholar] [CrossRef]
- Sundaresan, S.; Kitchen, J.A.; Brooker, S. Hydrophobic tail length in spin crossover active iron(II) complexes predictably tunes T1/2 in solution and enables surface immobilization. Inorg. Chem. Front. 2020, 7, 2050–2059. [Google Scholar] [CrossRef]
- Sundaresan, S.; Kühne, I.A.; Evesson, C.; Harris, M.M.; Fitzpatrick, A.J.; Ahmed, A.; Müller-Bunz, H.; Morgan, G.G. Compressed Jahn-Teller octahedra and spin quintet-triplet switching in coordinatively elastic manganese(III) complexes. Polyhedron 2021, 208, 115386. [Google Scholar] [CrossRef]
- Boskovic, C. Valence Tautomeric Transitions in Cobalt-dioxolene Complexes. In Spin-Crossover Materials: Properties and Applications, 1st ed.; Halcrow, M.A., Ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013; pp. 202–224. [Google Scholar]
- Gütlich, P.; Dei, A. Valence Tautomeric Interconversion in Transition Metal 1,2-Benzoquinone Complexes. Angew. Chem. Int. Ed. 1997, 36, 2734–2736. [Google Scholar] [CrossRef]
- Shaikh, N.; Goswami, S.; Panja, A.; Wang, X.-Y.; Gao, S.; Butcher, R.J.; Banerjee, P. New Route to the Mixed Valence Semiquinone-Catecholate Based Mononuclear FeIII and Catecholate Based Dinuclear MnIII Complexes: First Experimental Evidence of Valence Tautomerism in an Iron Complex. Inorg. Chem. 2004, 43, 5908–5918. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Maruyama, H.; Sato, O. Valence Tautomeric Transitions with Thermal Hysteresis around Room Temperature and Photoinduced Effects Observed in a Cobalt−Tetraoxolene Complex. J. Am. Chem. Soc. 2006, 128, 1790–1791. [Google Scholar] [CrossRef]
- Tezgerevska, T.; Alley, K.G.; Boskovic, C. Valence tautomerism in metal complexes: Stimulated and reversible intramolecular electron transfer between metal centers and organic ligands. Coord. Chem. Rev. 2014, 268, 23–40. [Google Scholar] [CrossRef]
- Affronte, M.; Beni, A.; Dei, A.; Sorace, L. Valence Tautomerism Interconversion Triggers Transition to Stable Charge Distribution in Solid Polymeric Cobalt-polyoxolene Complexes. Dalton Trans. 2007, 45, 5253–5259. [Google Scholar] [CrossRef]
- Dapporto, P.; Dei, A.; Poneti, G.; Sorace, L. Complete Direct and Reverse Optically Induced Valence Tautomeric Interconversion in a Cobalt–Dioxolene Complex. Chem. Eur. J. 2008, 14, 10915–10918. [Google Scholar] [CrossRef]
- Nadurata, V.L.; Hay, M.A.; Janetzki, J.T.; Gransbury, G.K.; Boskovic, C. Rich redox-activity and solvatochromism in a family of heteroleptic cobalt complexes. Dalton Trans. 2021, 50, 16631–16646. [Google Scholar] [CrossRef]
- Poneti, G.; Mannini, M.; Sorace, L.; Sainctavit, P.; Arrio, M.-A.; Otero, E.; Criginski Cezar, J.; Dei, A. Soft-X-ray-Induced Redox Isomerism in a Cobalt Dioxolene Complex. Angew. Chem. Int. Ed. 2010, 49, 1954–1957. [Google Scholar] [CrossRef]
- Alley, K.G.; Poneti, G.; Aitken, J.B.; Hocking, R.K.; Moubaraki, B.; Murray, K.S.; Abrahams, B.F.; Harris, H.H.; Sorace, L.; Boskovic, C. A Two-Step Valence Tautomeric Transition in a Dinuclear Cobalt Complex. Inorg. Chem. 2012, 51, 3944–3946. [Google Scholar] [CrossRef]
- Alley, K.G.; Poneti, G.; Robinson, P.S.D.; Nafady, A.; Moubaraki, B.; Aitken, J.B.; Drew, S.C.; Ritchie, C.; Abrahams, B.F.; Hocking, R.K.; et al. Redox Activity and Two-Step Valence Tautomerism in a Family of Dinuclear Cobalt Complexes with a Spiroconjugated Bis(dioxolene) Ligand. J. Am. Chem. Soc. 2013, 135, 8304–8323. [Google Scholar] [CrossRef]
- Evangelio, E.; Ruiz-Molina, D. Valence Tautomerism: New Challenges for Electroactive Ligands. Eur. J. Inorg. Chem. 2005, 2005, 2957–2971. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXTL-Plus. Graphical Interface for Crystal Structure Solution and Refinement; Bruker Analytical X-ray Instruments Inc.: Madison, WI, USA, 1998. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. A short history of SHELX. Acta Cryst. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- O’Connor, C.J. Magnetochemistry—Advances in Theory and Experimentation. Prog. Inorg. Chem. 1982, 29, 203–283. [Google Scholar]
- Aromí, G.; Batsanov, A.S.; Christian, P.; Helliwell, M.; Parkin, A.; Parsons, S.; Smith, A.A.; Timco, G.A.; Winpenny, R.E.P. Synthetic and Structural Studies of Cobalt–Pivalate Complexes. Chem. Eur. J. 2003, 9, 5142–5161. [Google Scholar] [CrossRef]
- Jung, O.-S.; Jo, D.H.; Lee, Y.-A.; Conklin, B.J.; Pierpont, C.G. Bistability and Molecular Switching for Semiquinone and Catechol Complexes of Cobalt. Studies on Redox Isomerism for the Bis(pyridine) Ether Series Co(py2X)(3,6-DBQ)2, X = O, S, Se, and Te. Inorg. Chem. 1997, 36, 19–24. [Google Scholar] [CrossRef]
- Liang, H.; Na, M.Y.; Chun, S.I.; Kwon, S.S.; Lee, Y.-A.; Jung, O.-S. Dinuclear Valence Tautomeric 1,2-Semiquinonato/Catecholatocobalt Complexes Containing 1,1,4,7,10,10-Hexamethyltriethylenetetramine. Bull. Chem. Soc. Jpn. 2007, 80, 916–921. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mono-Nuclear | 1 | 2 | Di-Nuclear | 3a | 3b |
|---|---|---|---|---|---|
| Metal-Donor | Bond Length (Å) | Bond Length (Å) | Metal-Donor | Bond Length (Å) | Bond Length (Å) |
| Co1-O1 | 1.901 (9) | 1.909 (3) | Co1-O1 | 1.878 (4) | 1.892 (6) |
| Co1-O2 | 1.905 (9) | 1.901 (3) | Co1-O2 | 1.888 (4) | 1.884 (5) |
| Co1-O3 | 1.885 (9) | 1.907 (3) | Co1-N1 | 2.063 (5) | 2.056 (5) |
| Co1-O4 | 1.884 (9) | 1.893 (3) | Co1-N2 | 1.954 (4) | 1.940 (6) |
| Co1-N1 | 1.969 (9) | 2.014 (4) | Co1-N3 | 1.936 (5) | 1.956 (6) |
| Co1-N2 | 1.978 (9) | 2.019 (4) | Co1-N4 | 1.925 (5) | 1.908 (7) |
| Compound | 1 | 2 |
| Empirical formula | C40H66CoN2O4 | C29H37Br8CoN4O4 |
| Formula weight/g mol−1 | 697.87 | 1203.83 |
| Crystal size/mm | 0.15 × 0.13 × 0.05 | 0.19 × 0.12 × 0.03 |
| Crystal system | Orthorhombic | Monoclinic |
| Space group | Pca21 | P21 |
| CCDC No. | 2202223 | 2195460 |
| Unit cell dimensions | ||
| a/Å | 18.7294(3) | 10.3888(2) |
| b/Å | 10.7376(2) | 17.3101(3) |
| c/Å | 19.4878(4) | 10.5887(2) |
| α/° | 90 | 90 |
| β/° | 90 | 99.6854(9) |
| γ/° | 90 | 90 |
| Volume/Å | 3919.2(14) | 1877.04(6) |
| Z | 4 | 2 |
| ρcalc./g cm−1 | 1.183 | 2.130 |
| μ/mm−1 | 0.478 | 9.012 |
| F(000) | 1516.0 | 1156.0 |
| Temperature/K | 173(2) | 173(2) |
| Radiation | Mo-Kα | Mo-Kα |
| Index ranges | −22 < h < 20 | −15 < h < 16 |
| −12 < k <12 | −26 < k < 26 | |
| −12 < l < 23 | −16 < l < 15 | |
| Collected reflections | 14473 | 54646 |
| Independent reflections | 5030 | 14399 |
| Rint | 0.1705 | 0.0673 |
| Rsigma | 0.2964 | 0.08060 |
| Data/restraints/parameters | 5030/105/439 | 14399/23/429 |
| Goodness-of-fit on F2 | 0.622 | 0.800 |
| Final R1 [I ≥ 2σ(I)] | 0.0469 | 0.0319 |
| Final wR2 [I ≥ 2σ(I)] | 0.0666 | 0.0496 |
| Final R1 [alldata] | 0.1652 | 0.0577 |
| Final wR2 [alldata] | 0.0961 | 0.0529 |
| Compound | 3a | 3b |
| Empirical formula | C69H116Co2N8O17S | C60H80Cl2Co2N8O12 |
| Formula weight/g mol−1 | 1479.61 | 1294.08 |
| Crystal size/mm | 0.46 × 0.34 × 0.3 | 0.19 × 0.15 × 0.02 |
| Crystal system | Monoclinic | Triclinic |
| Space group | C2/c | P |
| CCDC No. | 2170727 | 2170728 |
| Unit cell dimensions | ||
| a/Å | 26.051(3) | 11.071(2) |
| b/Å | 17.448(2) | 11.332(18) |
| c/Å | 17.215(2) | 15.543(3) |
| α/° | 90 | 107.932(6) |
| β/° | 94.177(3) | 97.683(7) |
| γ/° | 90 | 94.664(7) |
| Volume/Å | 7804.4(18) | 1823.1(5) |
| Z | 4 | 1 |
| ρcalc./g cm−1 | 1.259 | 1.179 |
| μ/mm−1 | 0.519 | 0.585 |
| F(000) | 3168.0 | 680.0 |
| Temperature/K | 173(2) | 173(2) |
| Radiation | Mo-Kα | Mo-Kα |
| Index ranges | −34 < h < 34 | −13 < h < 14 |
| 0 < k <22 | −14 < k < 14 | |
| 0 < l < 22 | −20 < l < 20 | |
| Collected reflections | 13257 | 18429 |
| Independent reflections | 13257 | 8470 |
| Rint | 0.0207 | 0.1584 |
| Rsigma | 0.1309 | 0.3032 |
| Data/ restraints/ parameters | 13257/38/455 | 8740/79/379 |
| Goodness-of-fit on F2 | 1.041 | 0.941 |
| Final R1 [I ≥ 2σ(I)] | 0.0884 | 0.1072 |
| Final wR2 [I ≥ 2σ(I)] | 0.2066 | 0.2082 |
| Final R1 [alldata] | 0.1786 | 0.2736 |
| Final wR2 [alldata] | 0.2753 | 0.2692 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sundaresan, S.; Diehl, M.; Carrella, L.M.; Rentschler, E. Triggering of Valence Tautomeric Transitions in Dioxolene-Based Cobalt Complexes Influenced by Ligand Substituents, Co-ligands, and Anions. Magnetochemistry 2022, 8, 109. https://doi.org/10.3390/magnetochemistry8090109
Sundaresan S, Diehl M, Carrella LM, Rentschler E. Triggering of Valence Tautomeric Transitions in Dioxolene-Based Cobalt Complexes Influenced by Ligand Substituents, Co-ligands, and Anions. Magnetochemistry. 2022; 8(9):109. https://doi.org/10.3390/magnetochemistry8090109
Chicago/Turabian StyleSundaresan, Sriram, Marcel Diehl, Luca M. Carrella, and Eva Rentschler. 2022. "Triggering of Valence Tautomeric Transitions in Dioxolene-Based Cobalt Complexes Influenced by Ligand Substituents, Co-ligands, and Anions" Magnetochemistry 8, no. 9: 109. https://doi.org/10.3390/magnetochemistry8090109
APA StyleSundaresan, S., Diehl, M., Carrella, L. M., & Rentschler, E. (2022). Triggering of Valence Tautomeric Transitions in Dioxolene-Based Cobalt Complexes Influenced by Ligand Substituents, Co-ligands, and Anions. Magnetochemistry, 8(9), 109. https://doi.org/10.3390/magnetochemistry8090109