
Electrochemical, Electrocatalytic, and Magnetic Properties of Vanadium-Containing Polyoxometalates

 , , and
, , and
Abstract
:1. Introduction
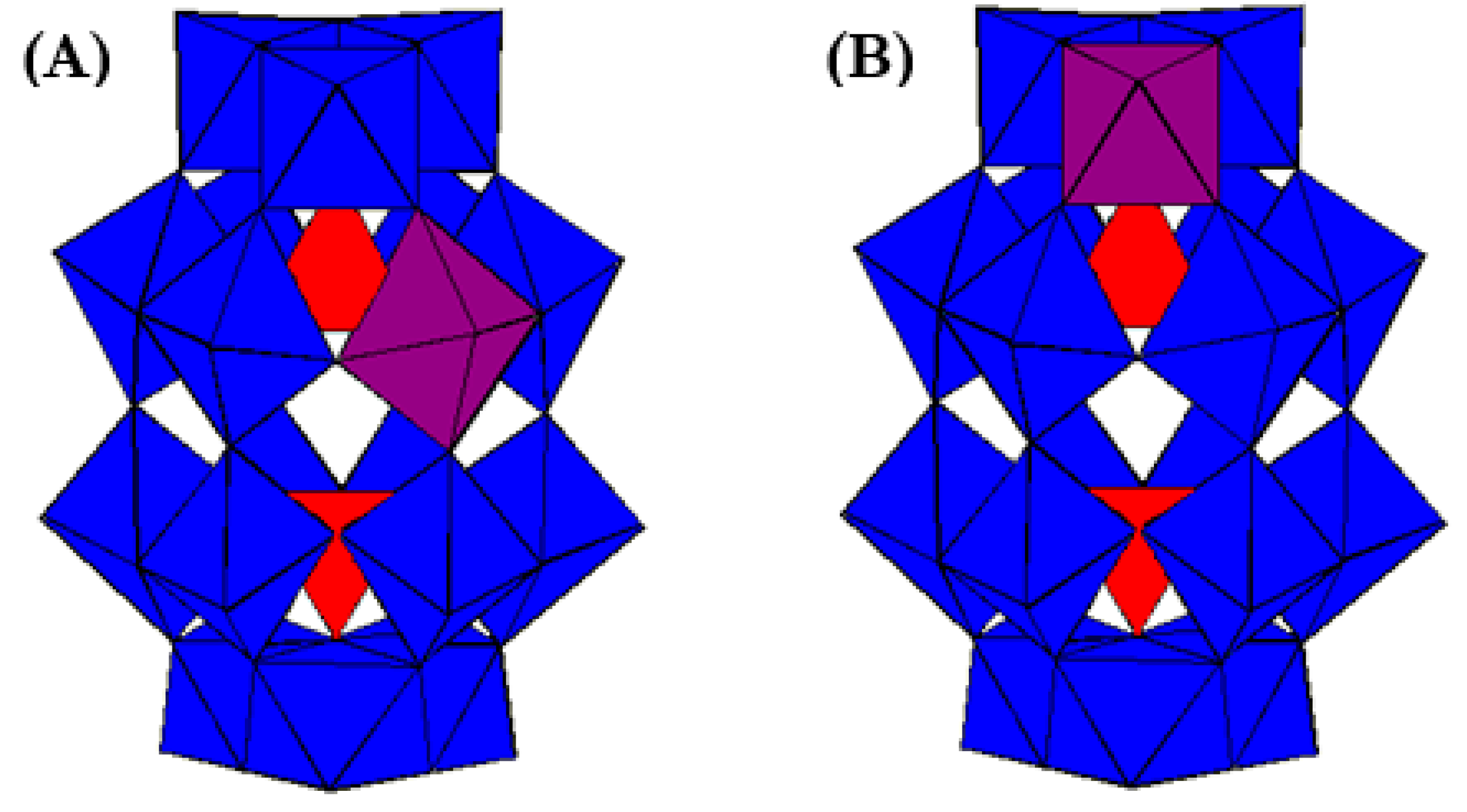
2. Materials and Methods
2.1. Magnetic Properties
2.2. Electrochemistry
3. Results and Discussion
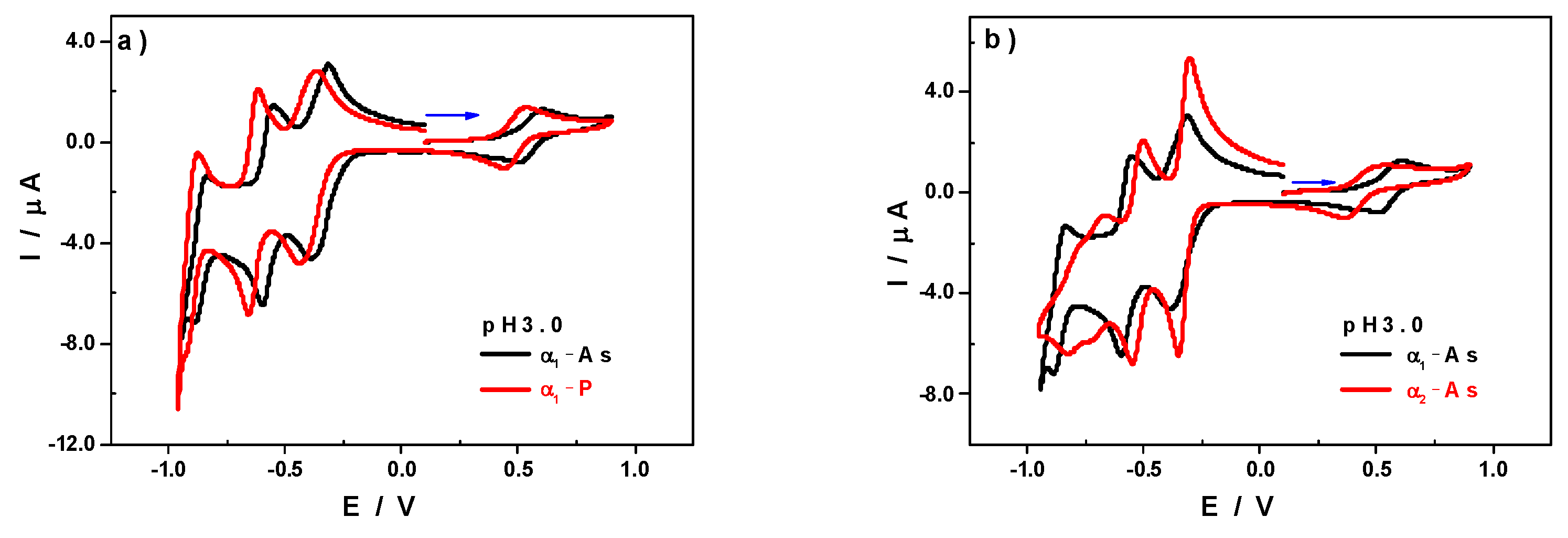
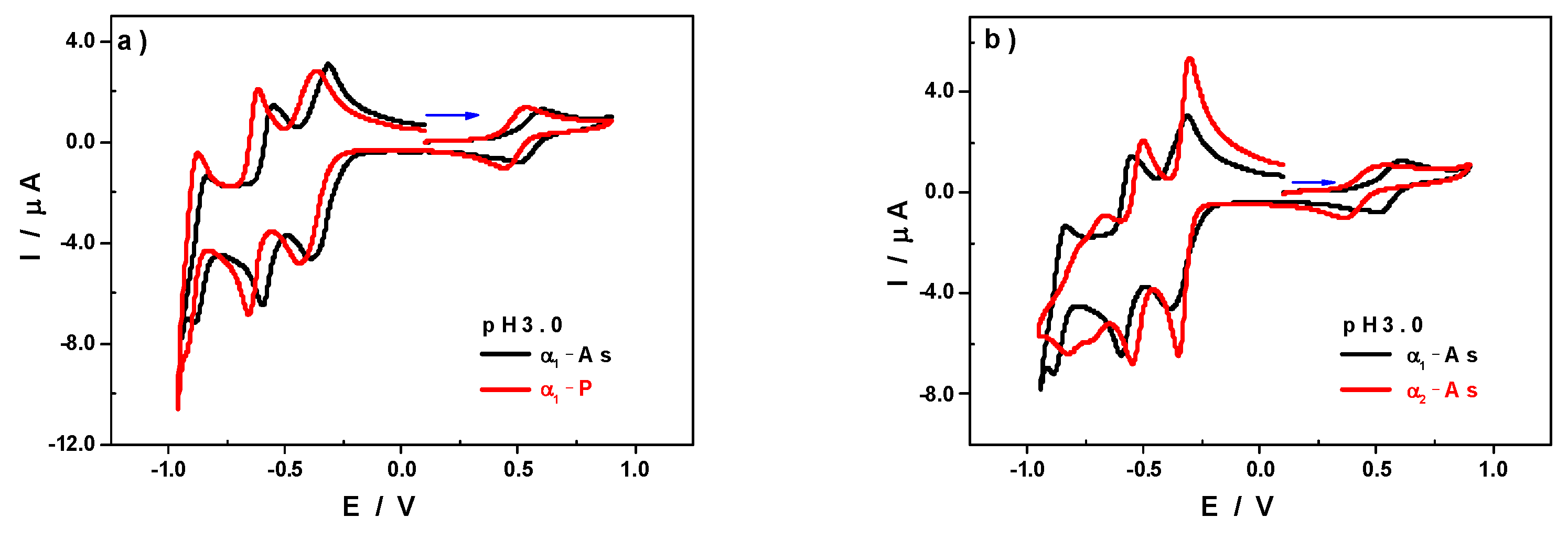
3.1. Cyclic Voltammetry Characterisation at pH 3
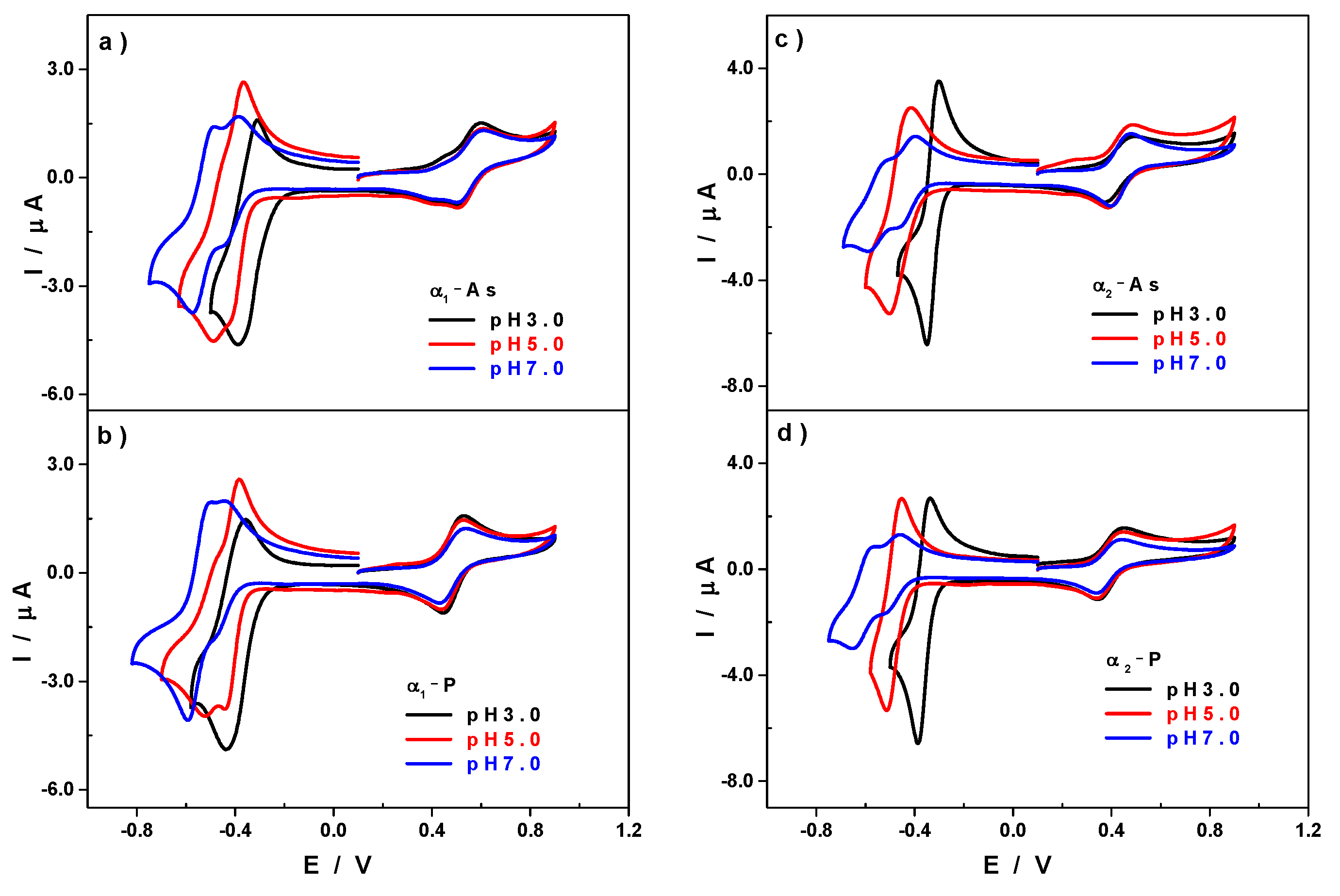
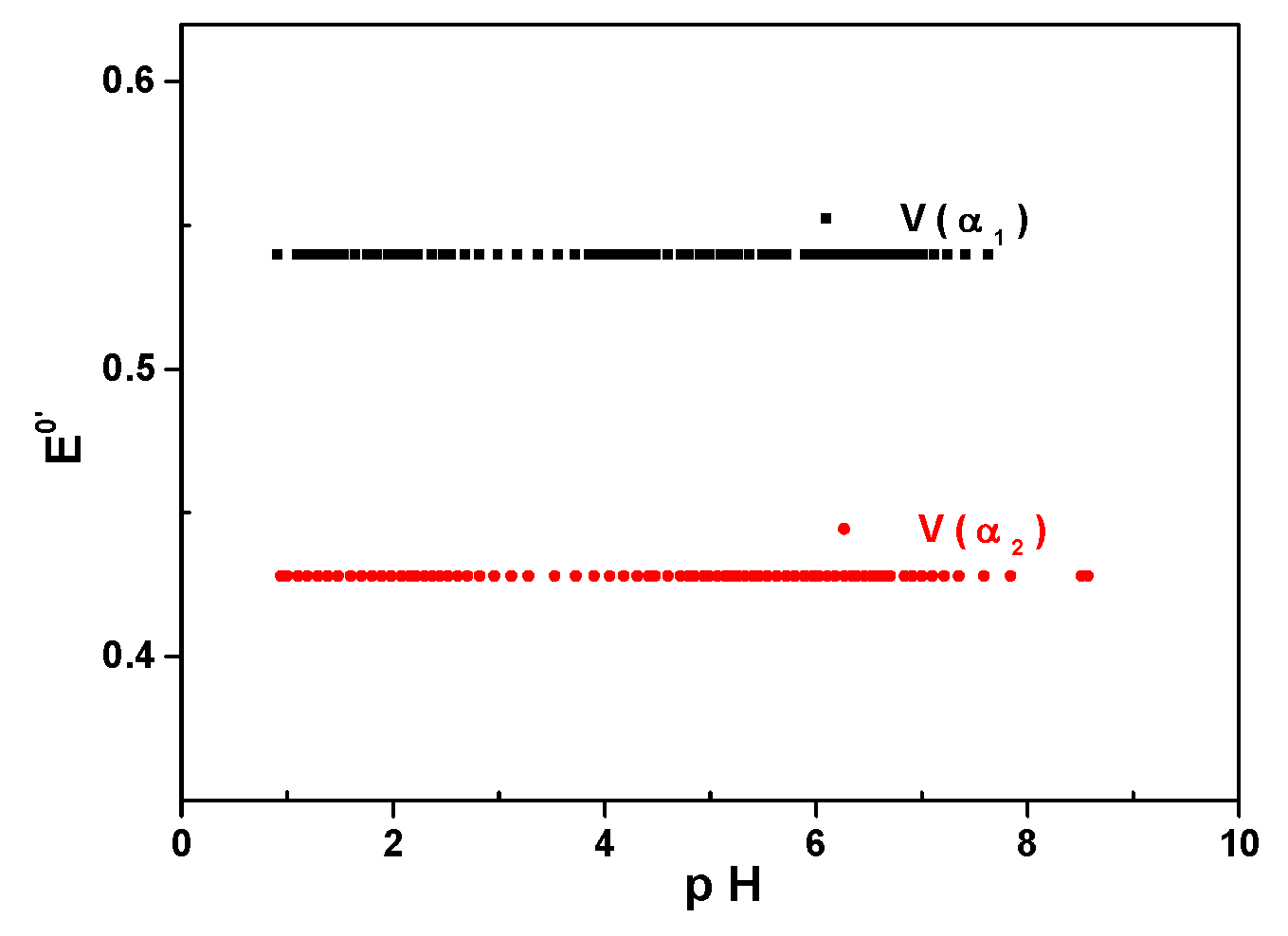
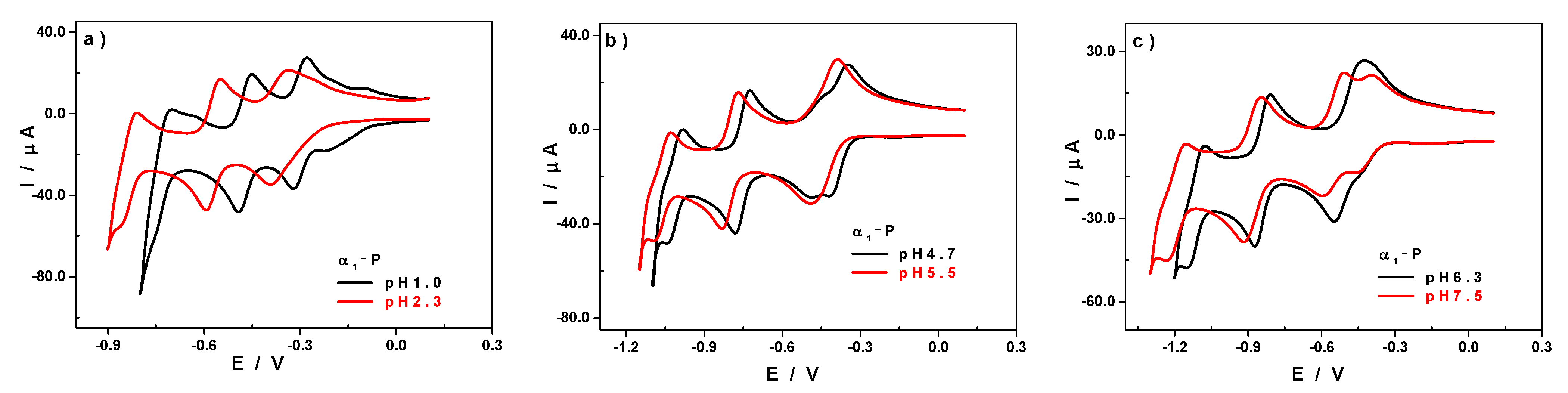
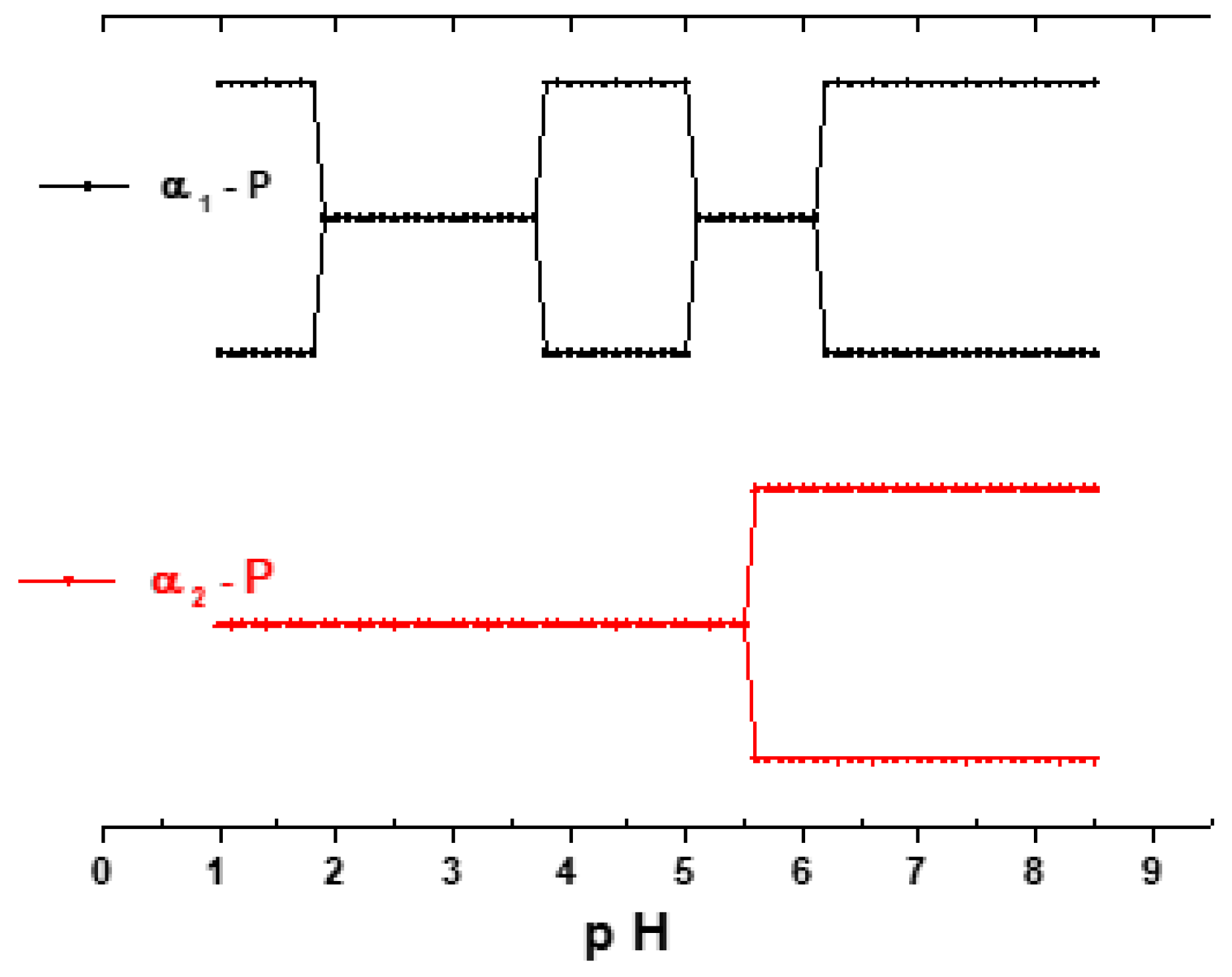
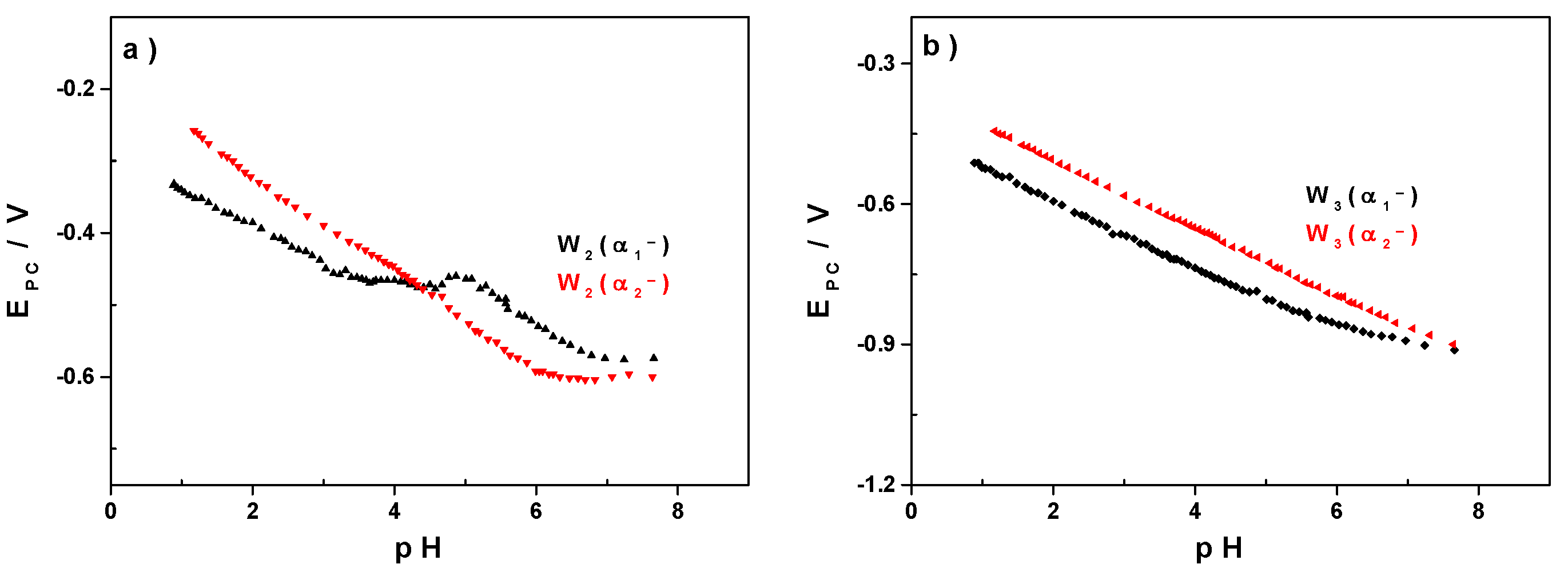
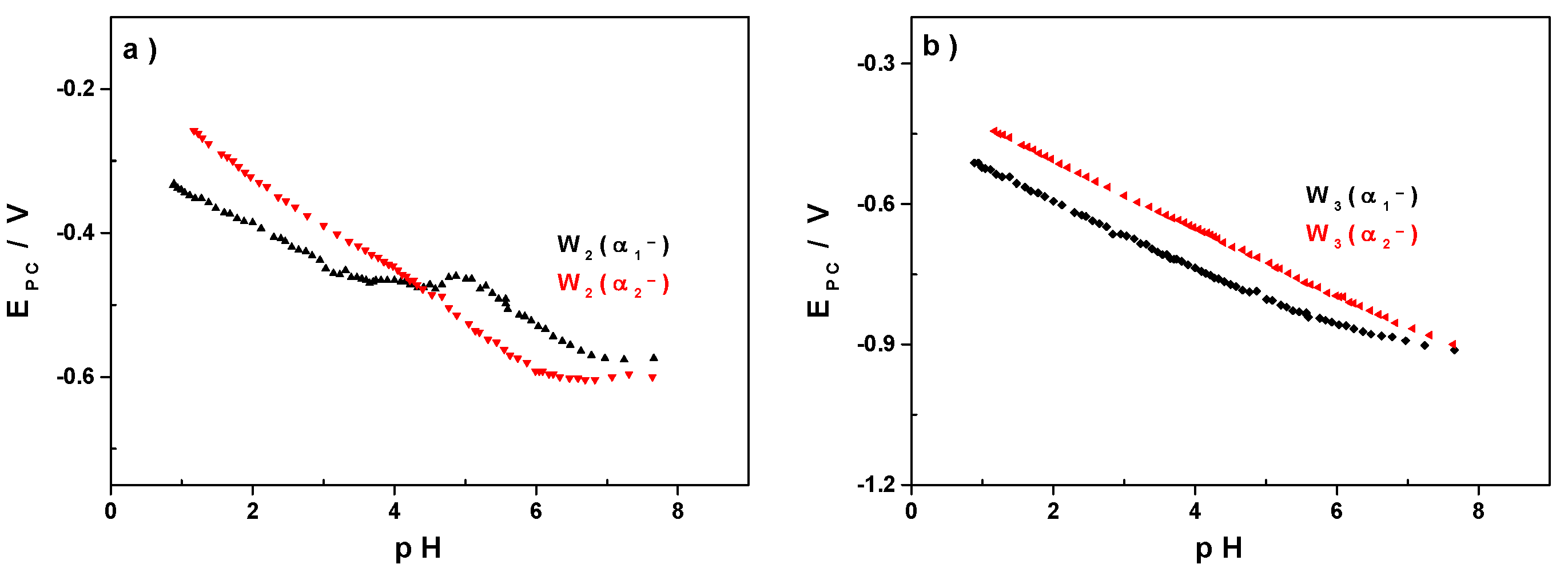
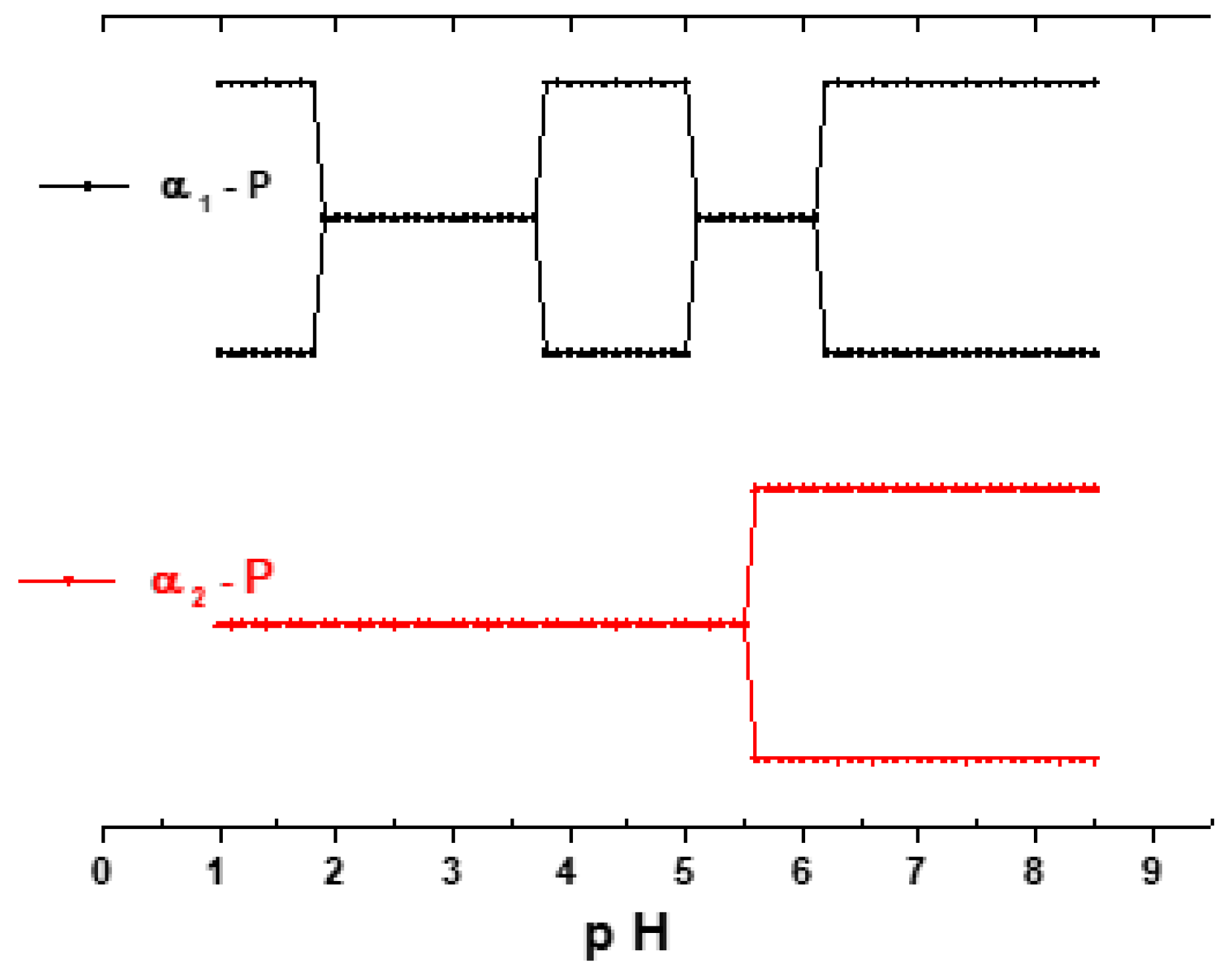
3.2. Influence of the pH
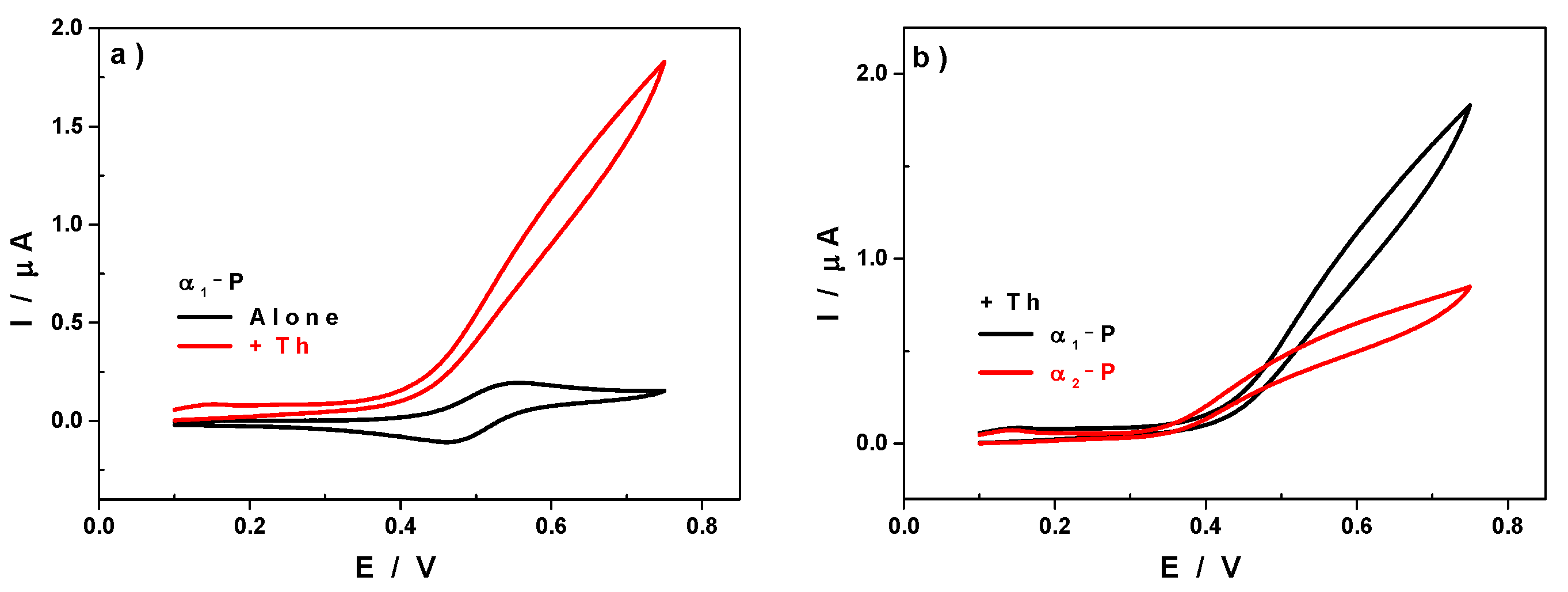
3.3. Electrocatalytic Properties
4. Magnetic Properties
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Pope, M.T.; Müller, A. Polyoxometalate Chemistry: An Old Field with New Dimensions in Several Disciplines. Angew. Chem. Int. Ed. Engl. 1991, 30, 34–48. [Google Scholar] [CrossRef]
- Pope, M. Heteropoly and Isopoly Oxometalates, 1st ed.; Springer: Berlin/Heidelberg, Germany, 1983. [Google Scholar]
- Contant, R.; Thouvenot, R. Hétéropolyanions de type Dawson. 2. Synthèses de polyoxotungstoarsénates lacunaires dérivant de l’octadécatungstodiarsénate. Étude structurale par RMN du tungstène-183 des octadéca(molybdotungstovanado)diarsénates apparentés. Can. J. Chem. 1991, 69, 1498–1506. [Google Scholar] [CrossRef]
- Dawson, B. The structure of the 9(18)-heteropoly anion in potassium 9(18)-tungstophosphate, K6(P2W18O62).14H2O. Acta Crystallogr. 1953, 6, 113–126. [Google Scholar] [CrossRef] [Green Version]
- Lefort, J. Action des acides arsénique et phosphorique sur les tungstates de soude et nouvelle méthode d’analyse des tungstates. Ann. Chim. Phys. 1882, XXV, 200–210. [Google Scholar]
- Souchay, P. Tungstic heteropolyacids. VIII. Phosphotungstates. Ann. Chim 1947, 2, 203–222. [Google Scholar]
- Contant, R.; Thouvenot, R. A reinvestigation of isomerism in the Dawson structure: Syntheses and 183W NMR structural characterization of three new polyoxotungstates [X2W18O62]6− (X = PV, AsV). Inorg. Chim. Acta 1993, 212, 41–50. [Google Scholar] [CrossRef]
- Belghiche, R.; Contant, R.; Lu, Y.W.; Keita, B.; Abbessi, M.; Nadjo, L.; Mahuteau, J. Synthesis and characterization of Fe- or Cu-substituted molybdenum-enriched tungstodiphosphates. Eur. J. Inorg. Chem. 2002, 6, 1410–1414. [Google Scholar] [CrossRef]
- Contant, R.; Richet, M.; Lu, Y.W.; Keita, B.; Nadjo, L. Isomerically pure α1-monosubstituted tungstodiphosphates: Synthesis, characterization and stability in aqueous solutions. Eur. J. Inorg. Chem. 2002, 10, 2587–2593. [Google Scholar] [CrossRef]
- Nishiki, K.; Umehara, N.; Kadota, Y.; Lopez, X.; Poblet, J.M.; Mezui, C.A.; Teillout, A.-L.; Mbomekalle, I.M.; de Oliveira, P.; Miyamoto, M.; et al. Preparation of α1- and α2-isomers of mono-Ru-substituted Dawson-type phosphotungstates with an aqua ligand and comparison of their redox potentials, catalytic activities, and thermal stabilities with Keggin-type derivatives. Dalton Trans. 2016, 45, 3715–3726. [Google Scholar] [CrossRef]
- Keita, B.; Lu, Y.W.; Nadjo, L.; Contant, R.; Abbessi, M.; Canny, J.; Richet, M. Electrochemical and catalytic behavior of Dawson-type complexes derived from [(1),2,3-P2Mo2W15O61]10− and first transition metal ions. J. Electroanal. Chem. 1999, 477, 146–157. [Google Scholar] [CrossRef]
- Contant, R.; Abbessi, M.; Canny, J.; Richet, M.; Keita, B.; Belhouari, A.; Nadjo, L. Synthesis, characterization and electrochemistry of complexes derived from [(1),2,3-P2Mo2W15O61]10− and first-row transition metal ions. Eur. J. Inorg. Chem. 2000, 3, 567–574. [Google Scholar] [CrossRef]
- Abbessi, M.; Contant, R.; Thouvenot, R.; Herve, G. Dawson type heteropolyanions. 1. Multinuclear (phosphorus-31, vanadium-51, tungsten-183) NMR structural investigations of octadeca(molybdotungstovanado)diphosphates .alpha.-1,2,3-[P2MM’2W15O62]n- (M, M’ = Mo, V, W): Syntheses of new related compounds. Inorg. Chem. 1991, 30, 1695–1702. [Google Scholar] [CrossRef]
- Contant, R.; Abbessi, M.; Thouvenot, R.; Hervé, G. Dawson Type Heteropolyanions. 3. Syntheses and 31P, 51V, and 183W NMR Structural Investigation of Octadeca(molybdo−tungsto−vanado)diphosphates Related to the [H2P2W12O48]12- Anion. Inorg. Chem. 2004, 43, 3597–3604. [Google Scholar] [CrossRef]
- Finke, R.G.; Rapko, B.; Saxton, R.J.; Domaille, P.J. Trisubstituted heteropolytungstates as soluble metal oxide analogs. III. Synthesis, characterization, phosphorus-31, silicon-29, vanadium-51, and 1- and 2-D tungsten-183 NMR, deprotonation, and proton mobility studies of organic solvent solute forms of HxSiW9V3O40x-7 and HxP2W15V3O62x-9. J. Am. Chem. Soc. 1986, 108, 2947–2960. [Google Scholar]
- Harmalker, S.P.; Leparulo, M.A.; Pope, M.T. Mixed-valence chemistry of adjacent vanadium centers in heteropolytungstate anions. I. Synthesis and electronic structures of mono-, di-, and trisubstituted derivatives of .alpha.-octadecatungstodiphosphate(6-) ion (.alpha.-[P2W18O62]6-). J. Am. Chem. Soc. 1983, 105, 4286–4292. [Google Scholar] [CrossRef]
- Keita, B.; Girard, F.; Nadjo, L.; Contant, R.; Canny, J.; Richet, M. Metal ion complexes derived from the α1 isomer of (P2W17O61)10−: Comparison with the corresponding α2 species. J. Electroanal. Chem. 1999, 478, 76–82. [Google Scholar] [CrossRef]
- Tommasino, J.B.; Contant, R.; Michaut, J.P.; Roncin, J. Electrochemical characterization of a series of substituted Dawson type tungstophosphates α[P2W18-xMozVyO62]n- (x = y + z; n = 6 + y). Polyhedron 1997, 17, 357–366. [Google Scholar] [CrossRef]
- Finke, R.G.; Droege, M.W. Trivacant heteropolytungstate derivatives. 2. Synthesis, characterization, and tungsten-183 NMR of P4W30M4(H2O)2O11216− (M = Co, Cu, Zn). Inorg. Chem. 1983, 22, 1006–1008. [Google Scholar] [CrossRef]
- Weakley, T.J.R.; Finke, R.G. Single-crystal X-ray structures of the polyoxotungstate salts K8.3Na1.7[Cu4(H2O)2(PW9O34)2].cntdot.24H2O and Na14Cu[Cu4(H2O)2(P2W15O56)2].cntdot.53H2O. Inorg. Chem. 1990, 29, 1235–1241. [Google Scholar] [CrossRef]
- Gomez-Garcia, C.J.; Borras-Almenar, J.J.; Coronado, E.; Ouahab, L. Single-Crystal X-ray Structure and Magnetic Properties of the Polyoxotungstate Complexes Na16[M4(H2O)2(P2W15O56)2].cntdot.nH2O (M = MnII, n = 53; M = NiII, n = 52): An Antiferromagnetic MnII Tetramer and a Ferromagnetic NiII Tetramer. Inorg. Chem. 1994, 33, 4016–4022. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, Q.; Duncan, D.C.; Campana, C.F.; Hill, C.L. Multiiron Polyoxoanions. Syntheses, Characterization, X-ray Crystal Structures, and Catalysis of H2O2-Based Hydrocarbon Oxidations by [FeIII4(H2O)2(P2W15O56)2]12. Inorg. Chem. 1997, 36, 4208–4215. [Google Scholar] [CrossRef]
- Bi, L.-H.; Wang, E.-B.; Peng, J.; Huang, R.-D.; Xu, L.; Hu, C.-W. Crystal Structure and Replacement Reaction of Coordinated Water Molecules of the Heteropoly Compounds of Sandwich-Type Tungstoarsenates. Inorg. Chem. 2000, 39, 671–679. [Google Scholar] [CrossRef]
- Ruhlmann, L.; Canny, J.; Contant, R.; Thouvenot, R. Di- and Tricobalt Dawson Sandwich Complexes: Synthesis, Spectroscopic Characterization, and Electrochemical Behavior of Na18[(NaOH2)2Co2(P2W15O56)2] and Na17[(NaOH2)Co3(H2O)(P2W15O56)2]. Inorg. Chem. 2002, 41, 3811–3819. [Google Scholar] [CrossRef]
- Clemente-Juan, J.M.; Coronado, E.; Gaita-Ariño, A.; Giménez-Saiz, C.; Güdel, H.-U.; Sieber, A.; Bircher, R.; Mutka, H. Magnetic Polyoxometalates: Anisotropic Exchange Interactions in the Moiety of [(NaOH2)Co3(H2O)(P2W15O56)2]17. Inorg. Chem. 2005, 44, 3389–3395. [Google Scholar] [CrossRef] [PubMed]
- Anderson, T.M.; Fang, X.; Mbomekalle, I.M.; Keita, B.; Nadjo, L.; Hardcastle, K.I.; Farsidjani, A.; Hill, C.L. Structural and Electrochemical Studies of Dicupric Wells–Dawson Sandwich-Type Complexes. J. Clust. Sci. 2006, 17, 183–195. [Google Scholar] [CrossRef]
- Ruhlmann, L.; Costa-Coquelard, C.; Canny, J.; Thouvenot, R. Mixed-Metal Dawson Sandwich Complexes: Synthesis, Spectroscopic Characterization and Electrochemical Behaviour of Na16[MIICo3(H2O)2(P2W15O56)2] (M = Mn, Co, Ni, Zn and Cd). Eur. J. Inorg. Chem. 2007, 2007, 1493–1500. [Google Scholar] [CrossRef]
- Mbomekalle, I.M.; Keita, B.; Nadjo, L.; Berthet, P.; Hardcastle, K.I.; Hill, C.L.; Anderson, T.M. Multi-Iron Tungstodiarsenates. Synthesis, Characterization, and Electrocatalytic Studies of αββα-(FeIIIOH2)2FeIII2(As2W15O56)212. Inorg. Chem. 2003, 42, 1163–1169. [Google Scholar] [CrossRef]
- Mbomekalle, I.M.; Keita, B.; Nadjo, L.; Berthet, P.; Neiwert, W.A.; Hill, C.L.; Ritorto, M.D.; Anderson, T.M. Manganous heteropolytungstates. Synthesis and heteroatom effects in Wells-Dawson-derived sandwich complexes. Dalton Trans. 2003, 13, 2646–2650. [Google Scholar] [CrossRef]
- Mbomekalle, I.M.; Mialane, P.; Dolbecq, A.; Marrot, J.; Secheresse, F.; Berthet, P.; Keita, B.; Nadjo, L. Rational Synthesis, Structure, Magnetism and Electrochemistry of Mixed Iron-Nickel-Containing Wells-Dawson-Fragment-Based Sandwich-Type Polyoxometalates. Eur. J. Inorg. Chem. 2009, 34, 5194–5204. [Google Scholar] [CrossRef]
- Bassil, B.S.; Xiang, Y.; Haider, A.; Hurtado, J.; Novitchi, G.; Powell, A.K.; Bossoh, A.M.; Mbomekalle, I.M.; de Oliveira, P.; Kortz, U. Heptanickel(II) double-cubane core in Wells-Dawson heteropolytungstate, [Ni7(OH)6(H2O)6(P2W15O56)2]16. Chem. Commun. 2016, 52, 2601–2604. [Google Scholar] [CrossRef]
- Bassil, B.S.; Bossoh, A.M.; Ibrahim, M.; Bile, B.A.; Mougharbel, A.S.; Lin, Z.; de Oliveira, P.; Mbomekalle, I.-M.; Kortz, U. Synthesis, Structure and Electrochemistry of the Dinickel(II)-Containing 30-Tungsto-4-Phosphate [Ni2Na2(H2O)2(P2W15O56)2]18. Curr. Inorg. Chem. 2017, 7, 21–27. [Google Scholar] [CrossRef]
- Mbomekalle, I.M.; Cao, R.; Hardcastle, K.I.; Hill, C.L.; Ammam, M.; Keita, B.; Nadjo, L.; Anderson, T.M. Synthesis, structural characterization, and electrocatalytic studies of αββα-(ZnIIOH2)2(FeIII)2(X2W15O56)214− (X = P or As). C. R. Chim. 2005, 8, 1077–1086. [Google Scholar] [CrossRef]
- Ayingone Mezui, C.S.; de Oliveira, P.; Teillout, A.-L.; Marrot, J.; Berthet, P.; Lebrini, M.; Mbomekallé, I.M. Synthesis, Structure, and Magnetic Electrochemical Properties of a Family of Tungstoarsenates Containing Just CoII Centers or Both CoII and FeIII Centers. Inorg. Chem. 2017, 56, 1999–2012. [Google Scholar] [CrossRef]
- Floriant, D.; Charyle, S.A.M.; de Pedro, O.; Anne-Lucie, T.; Israel, M.M. Synthesis, Electrochemistry and Electro-Catalytic Properties of The Mixed Copper-Iron-Containing Sandwich-Type Polyoxometalates [(FeIIIOH)2CuII 2 (X2W15O56)2]14− and [(CuIIOH2)2FeIII 2(X2W15O56)2]14− (with X = AsV and PV). Curr. Inorg. Chem. (Discontin.) 2017, 7, 28–38. [Google Scholar]
- Mbomekalle, I.-M.; Bassil, B.S.; Suchopar, A.; Keita, B.; Nadjo, L.; Ammam, M.; Haouas, M.; Taulelle, F.; Kortz, U. Improved Synthesis, Structure, and Solution Characterization of the Cyclic 48-Tungsto-8-Arsenate(V), [H4As8W48O184]36−. J. Cluster Sci. 2014, 25, 277–285. [Google Scholar] [CrossRef]
- Liu, G.; Liu, T.; Mal, S.S.; Kortz, U. Wheel-Shaped Polyoxotungstate [Cu20Cl(OH)24(H2O)12(P8W48O184)]25- Macroanions Form Supramolecular “Blackberry” Structure in Aqueous Solution. J. Am. Chem. Soc. 2006, 128, 10103–10110. [Google Scholar] [CrossRef]
- Ismail, A.H.; Bassil, B.S.; Yassin, G.H.; Keita, B.; Kortz, U. {W48} ring opening: Fe16-containing, Ln4-stabilized 49-tungsto-8-phosphateopen wheel [Fe16O2(OH)23(H2O)9(P8W49O189)Ln4(H2O)20]11−. Chemistry 2012, 18, 6163–6166. [Google Scholar] [CrossRef]
- Mal, S.S.; Kortz, U. The Wheel-Shaped Cu20 Tungstophosphate [Cu20Cl(OH)24(H2O)12(P8W48O184)]25− Ion. Angew. Chem. Int. Ed. 2005, 44, 3777–3780. [Google Scholar] [CrossRef]
- Contant, R.; Teze, A. A new crown heteropolyanion K28Li5H7P8W48O184.92H2O: Synthesis, structure, and properties. Inorg. Chem. 1985, 24, 4610–4614. [Google Scholar] [CrossRef]
- Keita, B.; Mbomekalle, I.-M.; Nadjo, L.; de Oliveira, P.; Ranjbari, A.; Contant, R. Vanadium-substituted Dawson-type polyoxometalates as versatile electrocatalysts. C. R. Chim. 2005, 8, 1057–1066. [Google Scholar] [CrossRef]
- Mbomekalle, I.M.; Keita, B.; Nadjo, L.; Contant, R.; Belai, N.; Pope, M.T. Rationalization and improvement of the syntheses of two octadecatungstoarsenates: The novel α-K7[H4AsW18O62]·18H2O and the well known symmetrical α-K6[As2W18O62]·14H2O. Inorg. Chim. Acta 2003, 342, 219–228. [Google Scholar] [CrossRef]
- Mbomekalle, I.-M.; Lu, Y.W.; Keita, B.; Nadjo, L. Simple, high yield and reagent-saving synthesis of pure α-K6P2W18O62 14H2O. Inorg. Chem. Commun. 2003, 7, 86–90. [Google Scholar] [CrossRef]
- Sadakane, M.; Steckhan, E. Electrochemical Properties of Polyoxometalates as Electrocatalysts. Chem. Rev. 1998, 98, 219–238. [Google Scholar] [CrossRef]
- Ueda, T. Electrochemistry of Polyoxometalates: From Fundamental Aspects to Applications. ChemElectroChem 2018, 5, 823–838. [Google Scholar] [CrossRef]
- Keita, B.; Nadjo, L. Electrochemistry of Isopoly and Heteropoly Oxometalates. In Encyclopedia of Electrochemistry; Bard, A.J., Stratmann, M., Eds.; Wiley-VCH: Weinheim, Germany, 2005; Volume 7. [Google Scholar]
- Vila, N.; Aparicio, P.A.; Secheresse, F.; Poblet, J.M.; Lopez, X.; Mbomekalle, I.M. Electrochemical behavior of alpha1/alpha2-[Fe(H2O)P2W17O61](7-) isomers in solution: Experimental and DFT studies. Inorg. Chem. 2012, 51, 6129–6138. [Google Scholar] [CrossRef]
- Keita, B.; Mbomekalle, I.M.; Nadjo, L.; Contant, R. Compared behaviours of Dawson-type tungstodiarsenates and -diphosphates. Eur. J. Inorg. Chem. 2002, 2, 473–479. [Google Scholar] [CrossRef]
- Keita, B.; Mbomekalle, I.M.; de Oliveira, P.; Ranjbari, A.; Justum, Y.; Nadjo, L.; Pompon, D. Reactions of V-substituted polyoxometalates with L-cysteine. J. Cluster Sci. 2006, 17, 221–233. [Google Scholar] [CrossRef]
- Mbomekallé, I.-M.; López, X.; Poblet, J.M.; Sécheresse, F.; Keita, B.; Nadjo, L. Influence of the Heteroatom Size on the Redox Potentials of Selected Polyoxoanions. Inorg. Chem. 2010, 49, 7001–7006. [Google Scholar] [CrossRef] [PubMed]
- Pope, M.T.; Varga, G.M. Heteropoly Blues. I. Reduction Stoichiometries and Reduction Potentials of Some 12-Tungstates. Inorg. Chem. 1966, 5, 1249–1254. [Google Scholar] [CrossRef]
- Nakajima, K.; Eda, K.; Himeno, S. Effect of the Central Oxoanion Size on the Voltammetric Properties of Keggin-Type [XW12O40]n− (n = 2−6) Complexes. Inorg. Chem. 2010, 49, 5212–5215. [Google Scholar] [CrossRef]
- Keita, B.; Lu, Y.W.; Nadjo, L.; Contant, R. Influence of charge distribution on the properties of Keggin- and Dawson-type heteropolyanions. Eur. J. Inorg. Chem. 2000, 12, 2463–2471. [Google Scholar] [CrossRef]
- Keita, B.; Jean, Y.; Levy, B.; Nadjo, L.; Contant, R. Toward a qualitative understanding of the initial electron transfer site in Dawson-type heteropolyanions. New J. Chem. 2002, 26, 1314–1319. [Google Scholar] [CrossRef]
- Kahn, O. Molecular Magnetism; VCH: New York, NY, USA, 1993. [Google Scholar]
- Mialane, P.; Marrot, J.; Rivière, E.; Nebout, J.; Hervé, G. Structural Characterization and Magnetic Properties of Sandwich-Type Tungstoarsenate Complexes. Study of a Mixed-Valent VIV2/VV Heteropolyanion. Inorg. Chem. 2001, 40, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Miras, H.N.; Raptis, R.G.; Lalioti, N.; Sigalas, M.P.; Baran, P.; Kabanos, T.A. A novel series of vanadium-sulfite polyoxometalates: Synthesis, structural, and physical studies. Chemistry 2005, 11, 2295–2306. [Google Scholar] [CrossRef] [PubMed]

 ) and α2-PV5+ (
) and α2-PV5+ (  ) at 2 K compared to the theoretical curve for S=1/2.
) and α2-PV5+ ( ) at 2 K compared to the theoretical curve for S=1/2.
) at 2 K compared to the theoretical curve for S=1/2.
) and α2-PV5+ ( ) at 2 K compared to the theoretical curve for S=1/2.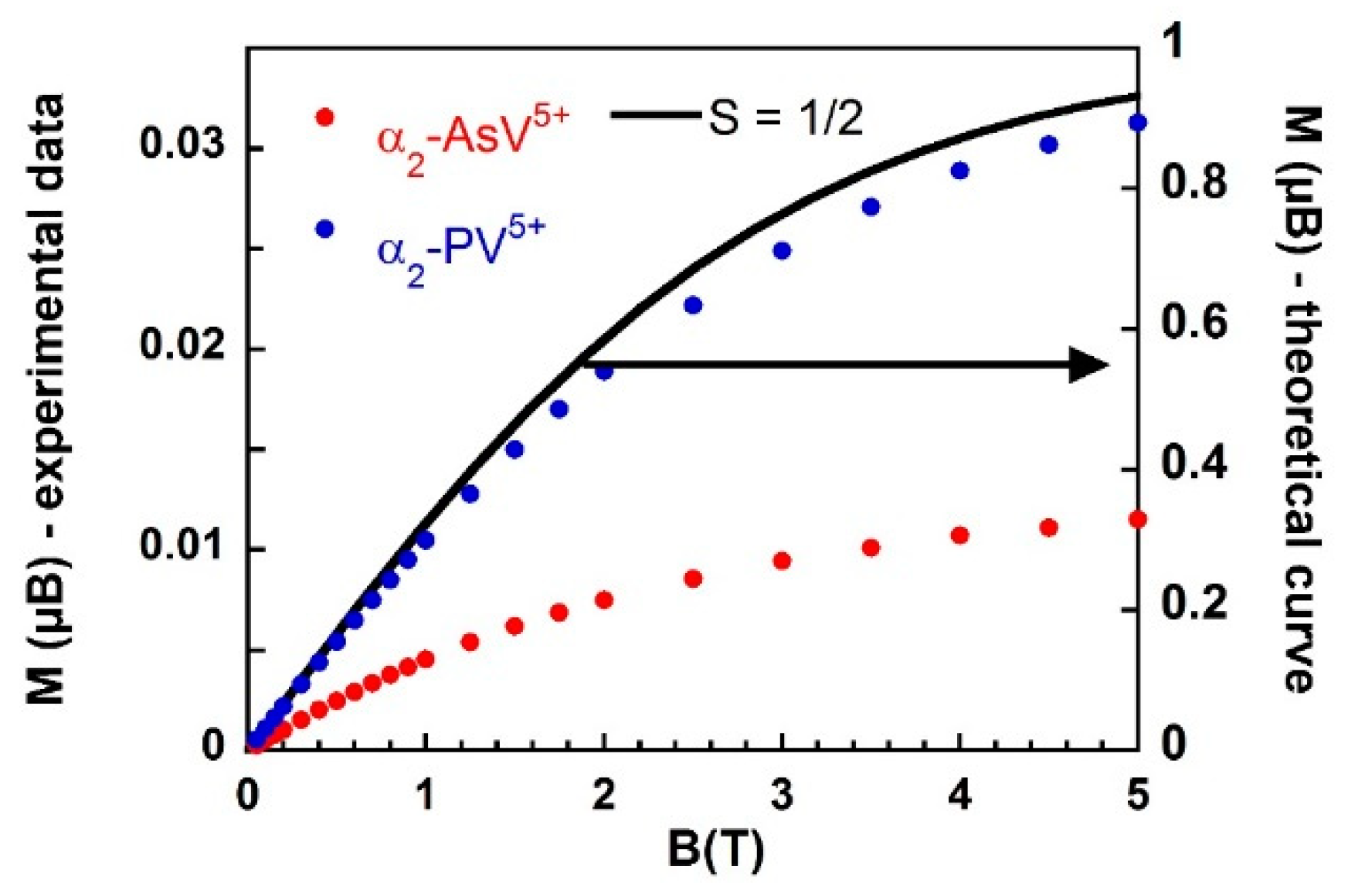 ) and α2-PV5+ ( ) under a magnetic field of 1 T.
) and α2-PV5+ ( ) under a magnetic field of 1 T.
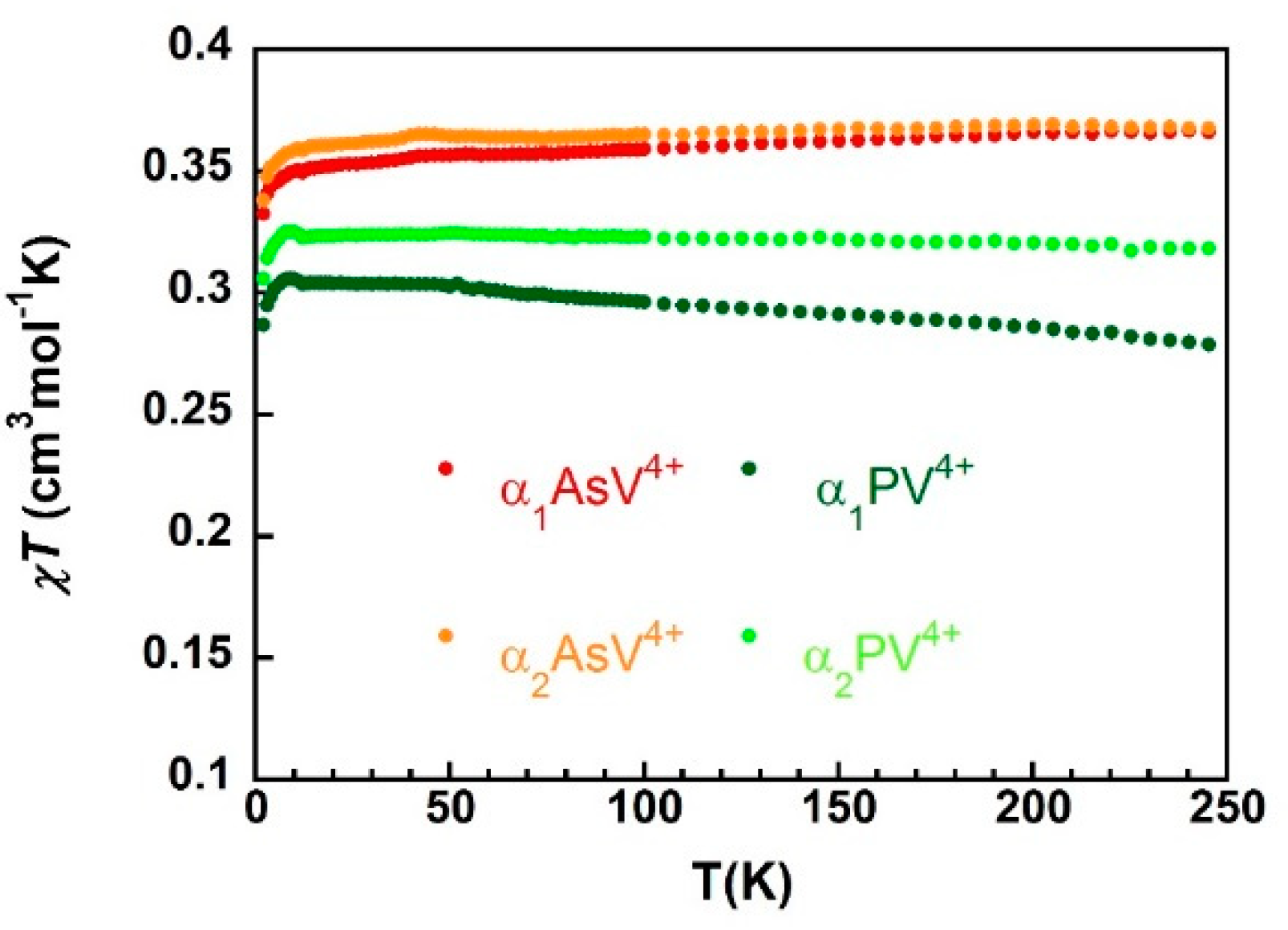
 ), α2-As V4+ (
), α2-As V4+ (  ), α1-PV4+ (
), α1-PV4+ (  ) and α2-PV4+ (
) and α2-PV4+ (  ).
), α2-As V4+ ( ), α1-PV4+ ( ) and α2-PV4+ ( ).
).
), α2-As V4+ ( ), α1-PV4+ ( ) and α2-PV4+ ( ).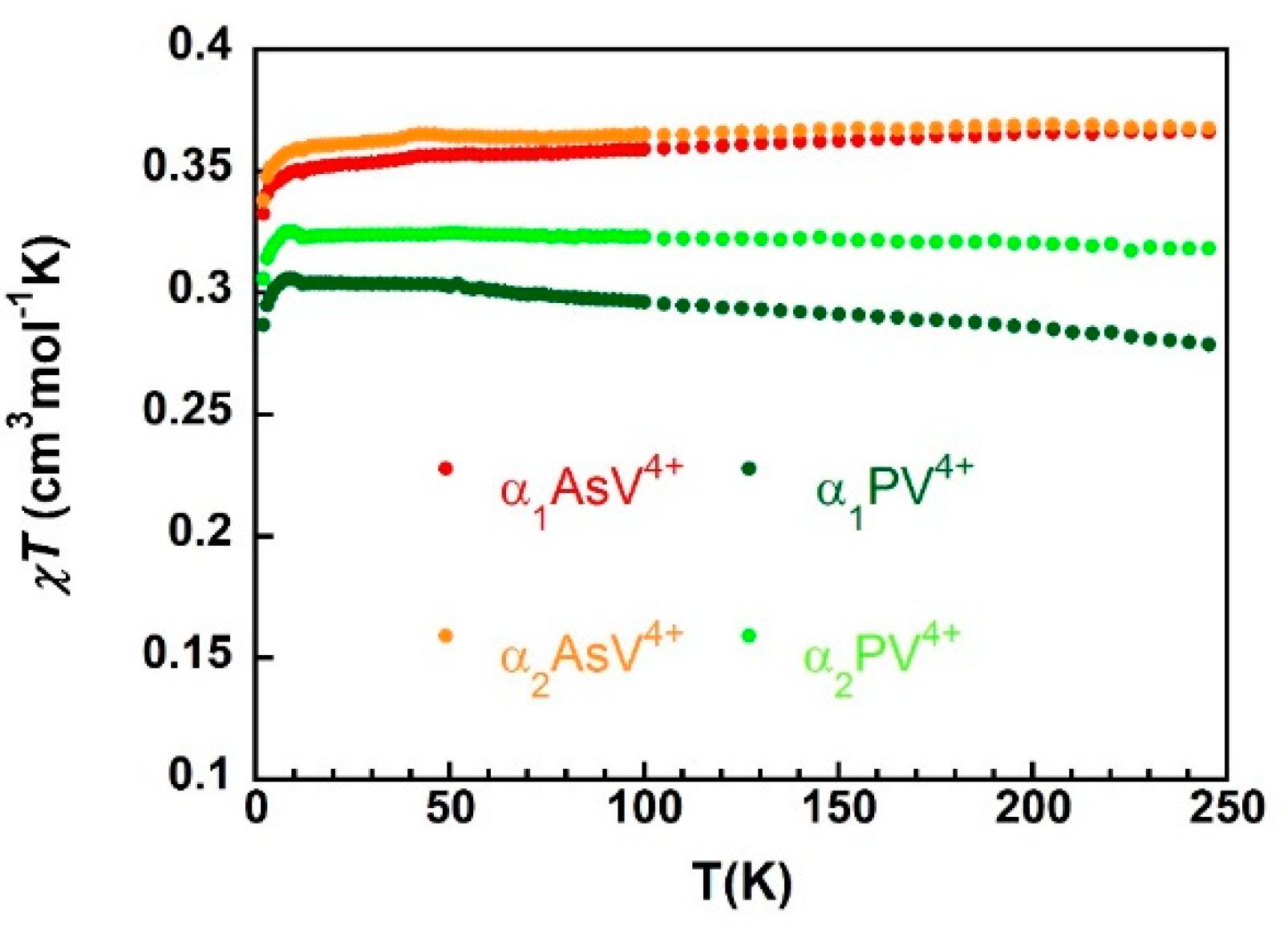 ), 4 K ( ), and 2 K (
), 4 K ( ), and 2 K (  ); Brillouin function (-) for a S = 1/2 ion. The g values are given in Table 3.
), 4 K ( ), and 2 K ( ); Brillouin function (-) for a S = 1/2 ion. The g values are given in Table 3.
); Brillouin function (-) for a S = 1/2 ion. The g values are given in Table 3.
), 4 K ( ), and 2 K ( ); Brillouin function (-) for a S = 1/2 ion. The g values are given in Table 3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| V | W1 | W2 | W3 | W4 | α1-As | |
| Ea | 0.610 | −0.314 | −0.552 | −0.836 | - | |
| Ec | 0.514 | −0.386 | −0.596 | −0.884 | - | |
| E0′ = (Ea + Ec)/2 | 0.562 | −0.350 | −0.574 | −0.860 | - | |
| # electrons | 1 | 2 | 2 | 2 | - | |
| V | W1 | W2 | W3 | W4 | α2-As | |
| Ea | 0.512 | −0.304 | −0.504 | −0.664 | −0.776 | |
| Ec | 0.366 | −0.350 | −0.550 | −0.728 | −0.826 | |
| E0′ = (Ea + Ec)/2 | 0.439 | −0.327 | −0.527 | −0.696 | −0.801 | |
| # electrons | 1 | 2 | 2 | 1 | 1 | |
| V | W1 | W2 | W3 | W4 | α1-P | |
| Ea | 0.532 | −0.366 | −0.616 | −0.874 | - | |
| Ec | 0.446 | −0.436 | −0.656 | −0.922 | - | |
| E0′ = (Ea + Ec)/2 | 0.489 | −0.401 | −0.636 | −0.898 | - | |
| # electrons | 1 | 2 | 2 | 2 | - | |
| V | W1 | W2 | W3 | W4 | α2-P | |
| Ea | 0.450 | −0.338 | −0.550 | −0.736 | −0.840 | |
| Ec | 0.346 | −0.386 | −0.586 | −0.774 | −0.876 | |
| E0′ = (Ea + Ec)/2 | 0.398 | −0.362 | −0.568 | −0.755 | −0.858 | |
| # electrons | 1 | 2 | 2 | 1 | 1 |
| α1-As | α2-As | α1-P | α2-P | |
|---|---|---|---|---|
| I0 (μA) | 0.17 | 0.16 | 0.15 | 0.15 |
| Icat (μA) | 1.86 | 1.34 | 1.83 | 0.85 |
| CAT (%) | 994 | 738 | 1120 | 467 |
| ONSET (V vs. SCE) | 0.42 | 0.35 | 0.42 | 0.37 |
| S = 1/2 (S.O.) | α1-AsV4+ | α2-AsV4+ | α1-PV4+ | α2-PV4+ | ||
|---|---|---|---|---|---|---|
| χT vs. T | C (cm3.mol−1) | 0.375 | 0.352 | 0.362 | 0.307 | 0.325 |
| χTIP (cm3.mol−1) | 60 × 10−6 | 29 × 10−6 | ||||
| χdia (cm3.mol−1) | −112 × 10−6 | −25 × 10−6 | ||||
| M vs. B | g | 2.0 | 1.91 | 1.93 | 1.75 | 1.82 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manga, J.D.; Teillout, A.-L.; Rivière, E.; de Oliveira, P.; Mbomekallé, I.M. Electrochemical, Electrocatalytic, and Magnetic Properties of Vanadium-Containing Polyoxometalates. Magnetochemistry 2021, 7, 157. https://doi.org/10.3390/magnetochemistry7120157
Manga JD, Teillout A-L, Rivière E, de Oliveira P, Mbomekallé IM. Electrochemical, Electrocatalytic, and Magnetic Properties of Vanadium-Containing Polyoxometalates. Magnetochemistry. 2021; 7(12):157. https://doi.org/10.3390/magnetochemistry7120157
Chicago/Turabian StyleManga, Joseph Dika, Anne-Lucie Teillout, Eric Rivière, Pedro de Oliveira, and Israël Martyr Mbomekallé. 2021. "Electrochemical, Electrocatalytic, and Magnetic Properties of Vanadium-Containing Polyoxometalates" Magnetochemistry 7, no. 12: 157. https://doi.org/10.3390/magnetochemistry7120157
APA StyleManga, J. D., Teillout, A.-L., Rivière, E., de Oliveira, P., & Mbomekallé, I. M. (2021). Electrochemical, Electrocatalytic, and Magnetic Properties of Vanadium-Containing Polyoxometalates. Magnetochemistry, 7(12), 157. https://doi.org/10.3390/magnetochemistry7120157