Diversity of Coordination Modes in a Flexible Ditopic Ligand Containing 2-Pyridyl, Carbonyl and Hydrazone Functionalities: Mononuclear and Dinuclear Cobalt(III) Complexes, and Tetranuclear Copper(II) and Nickel(II) Clusters †


 ,
,  ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Synthetic Comments
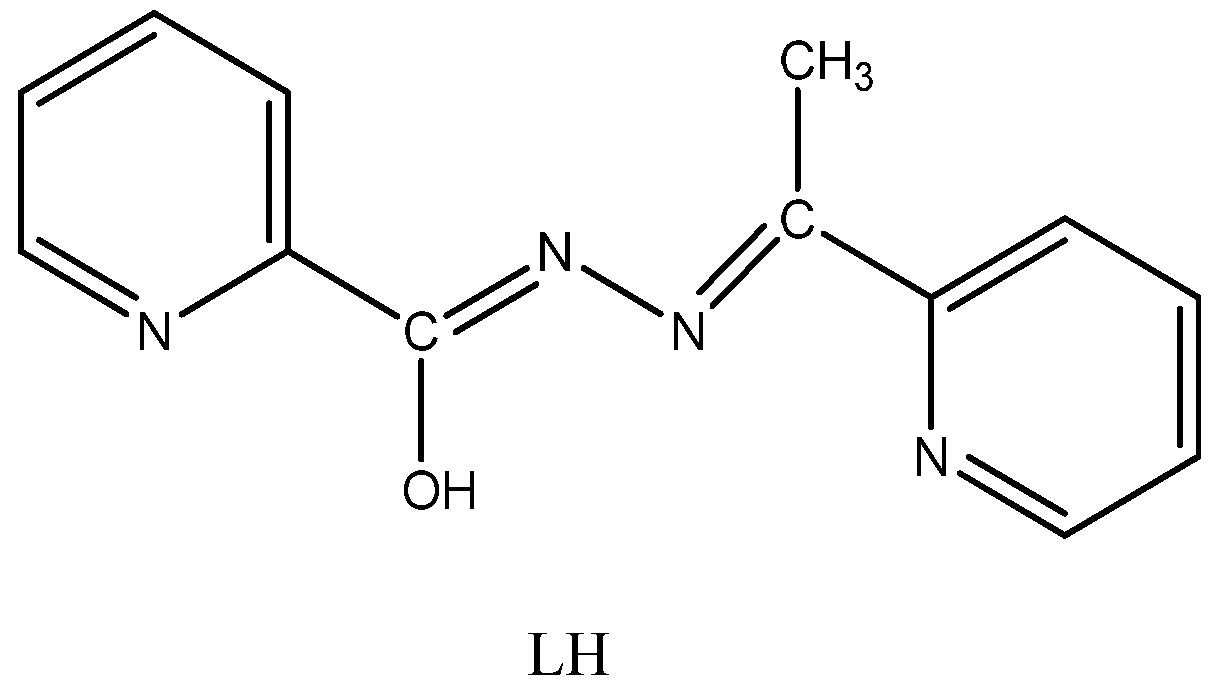
2.2. Spectroscopic Characterization in Brief
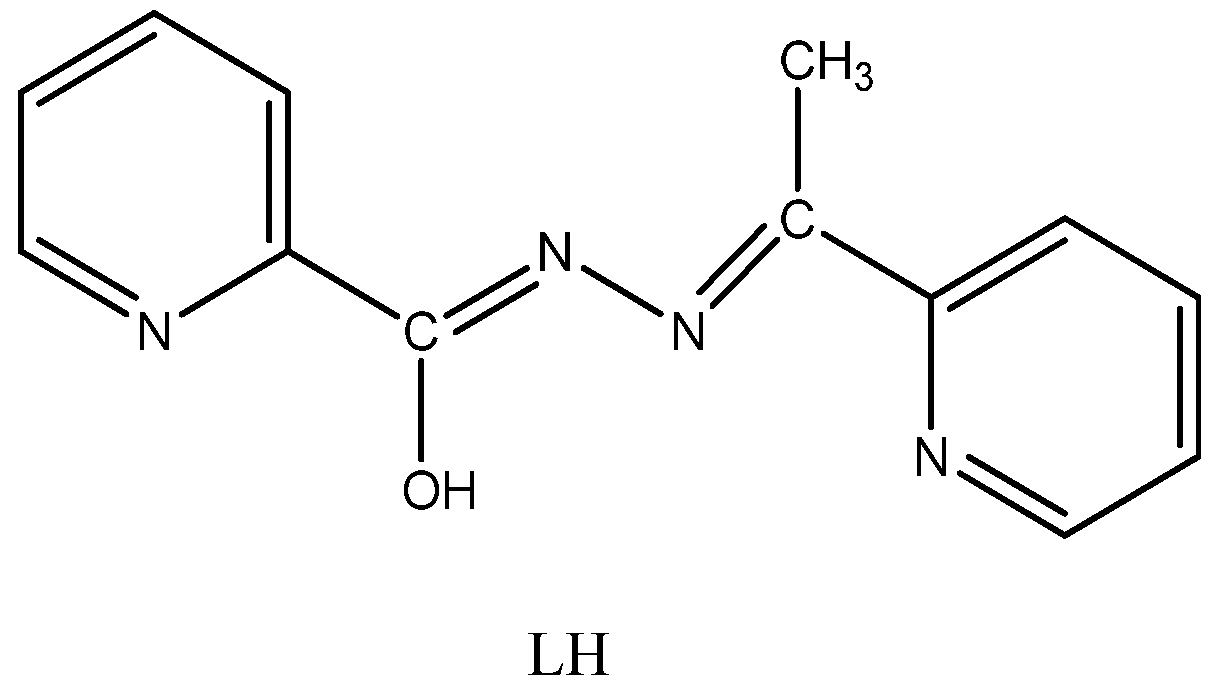
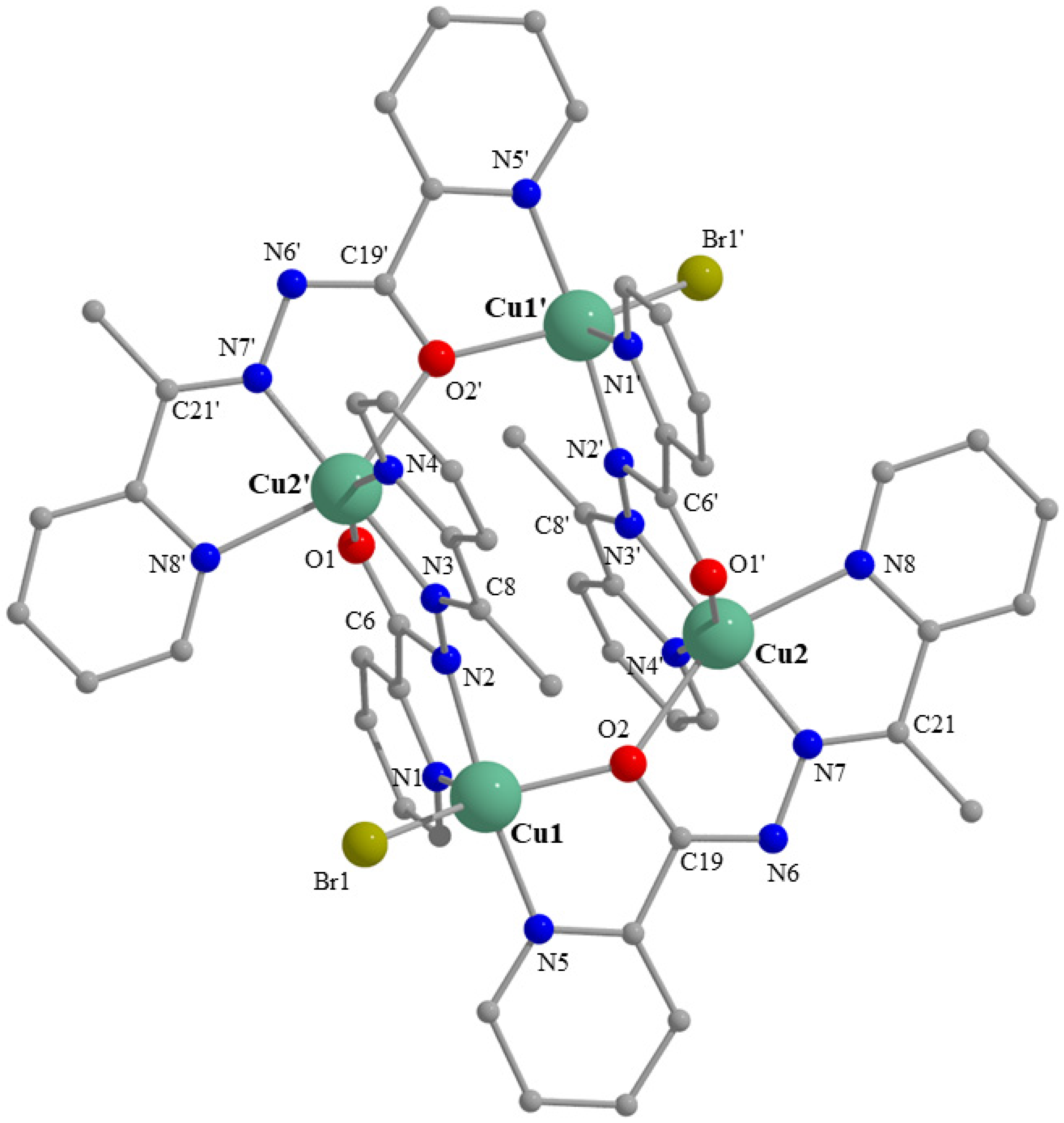
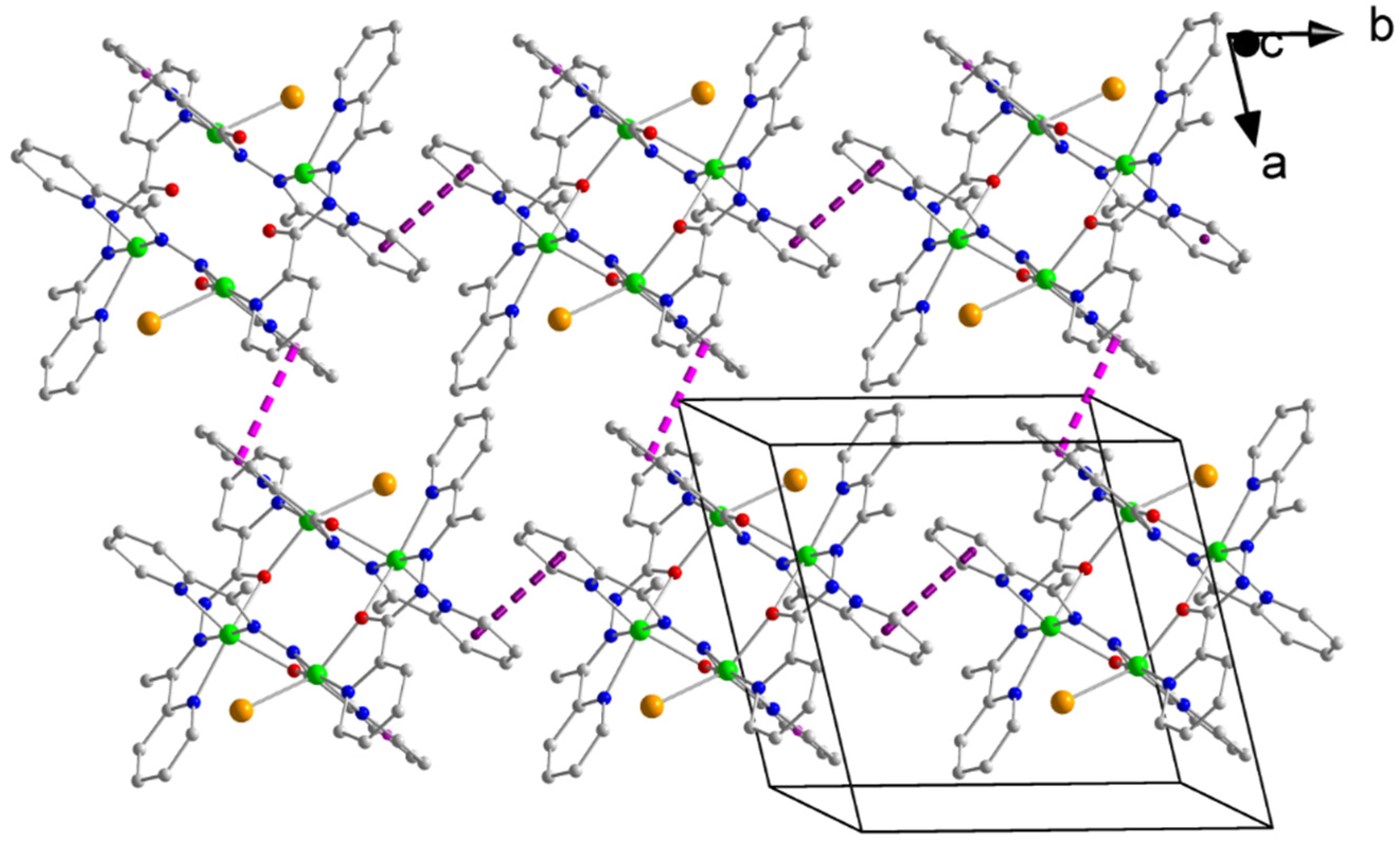
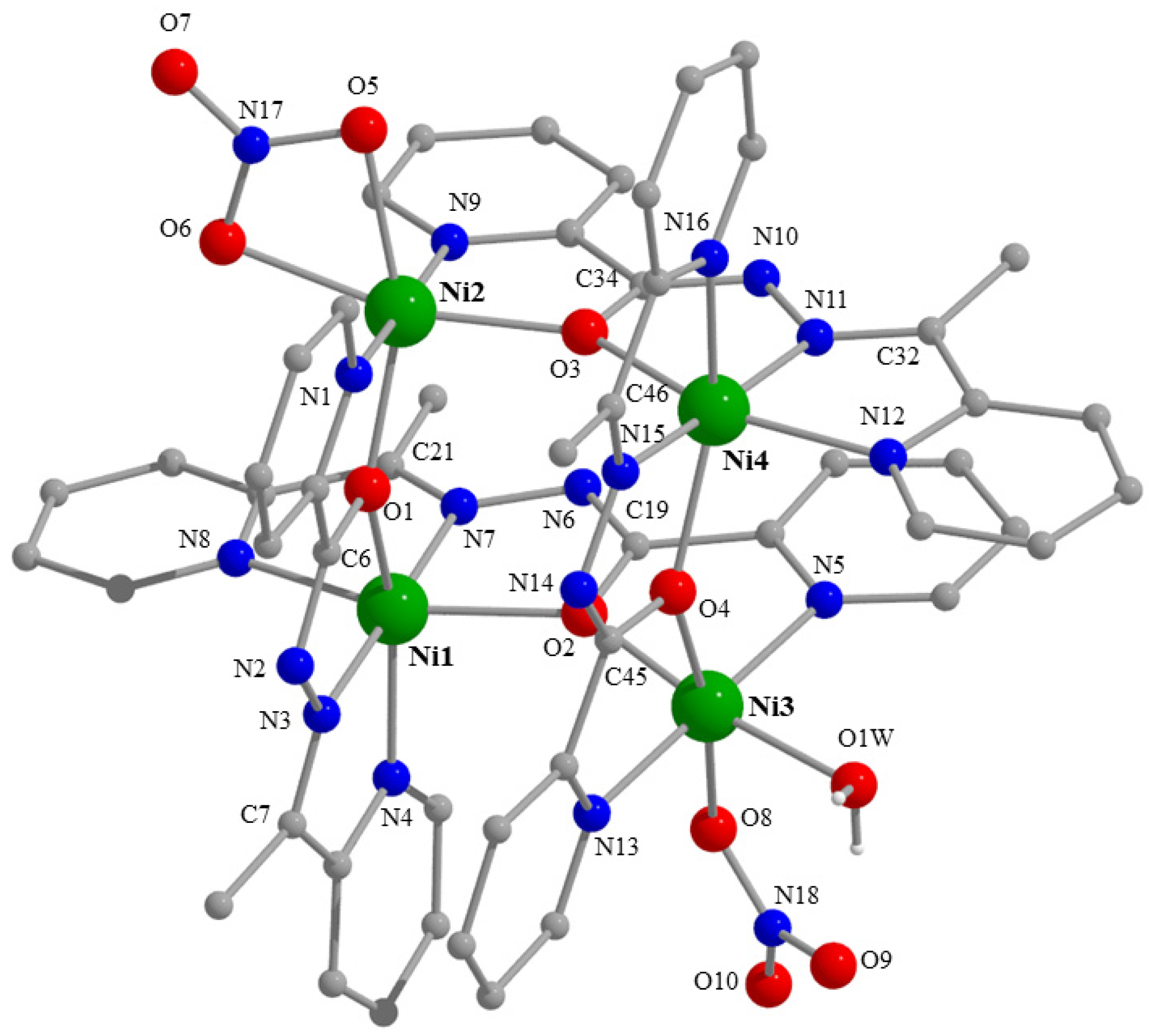
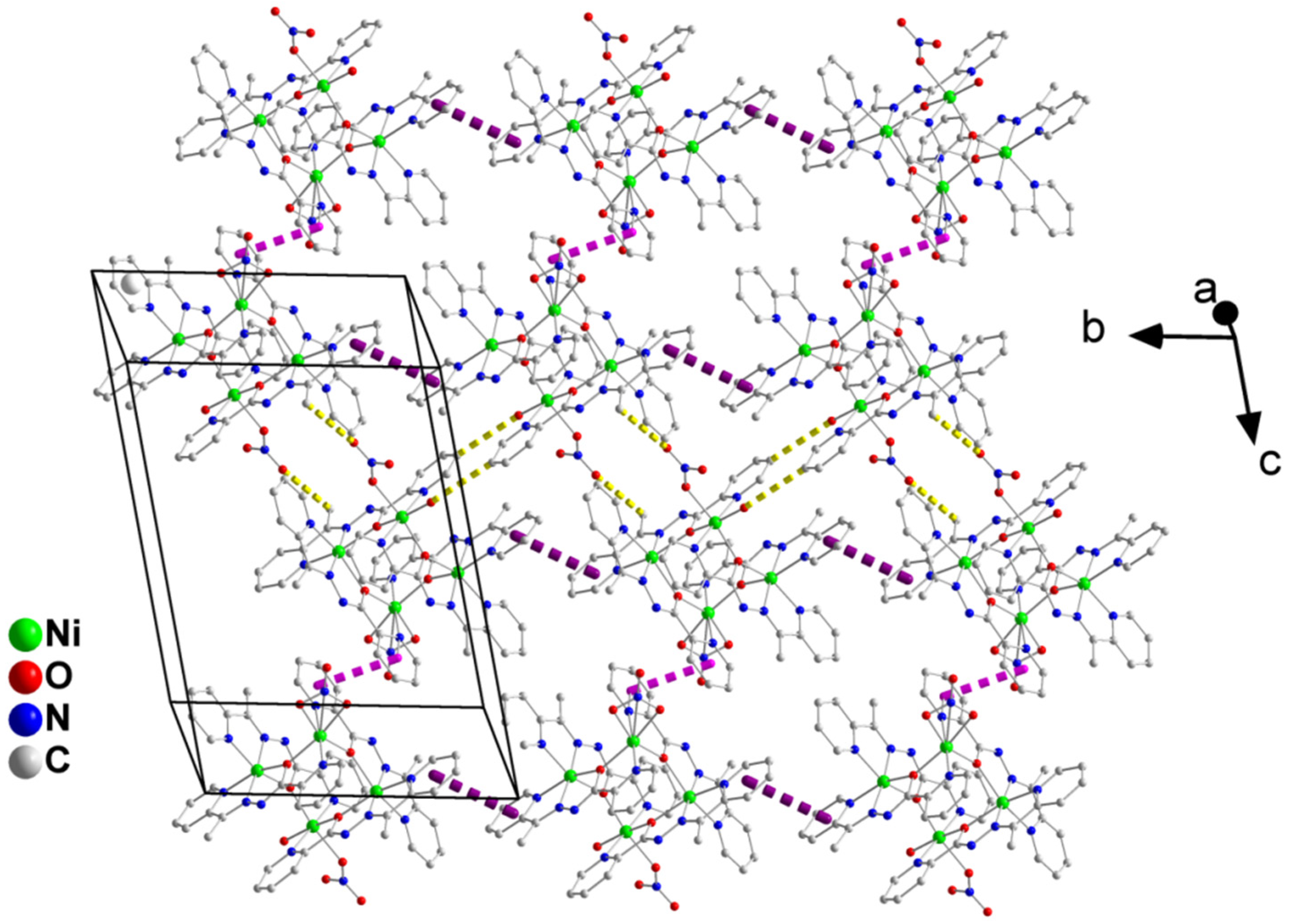
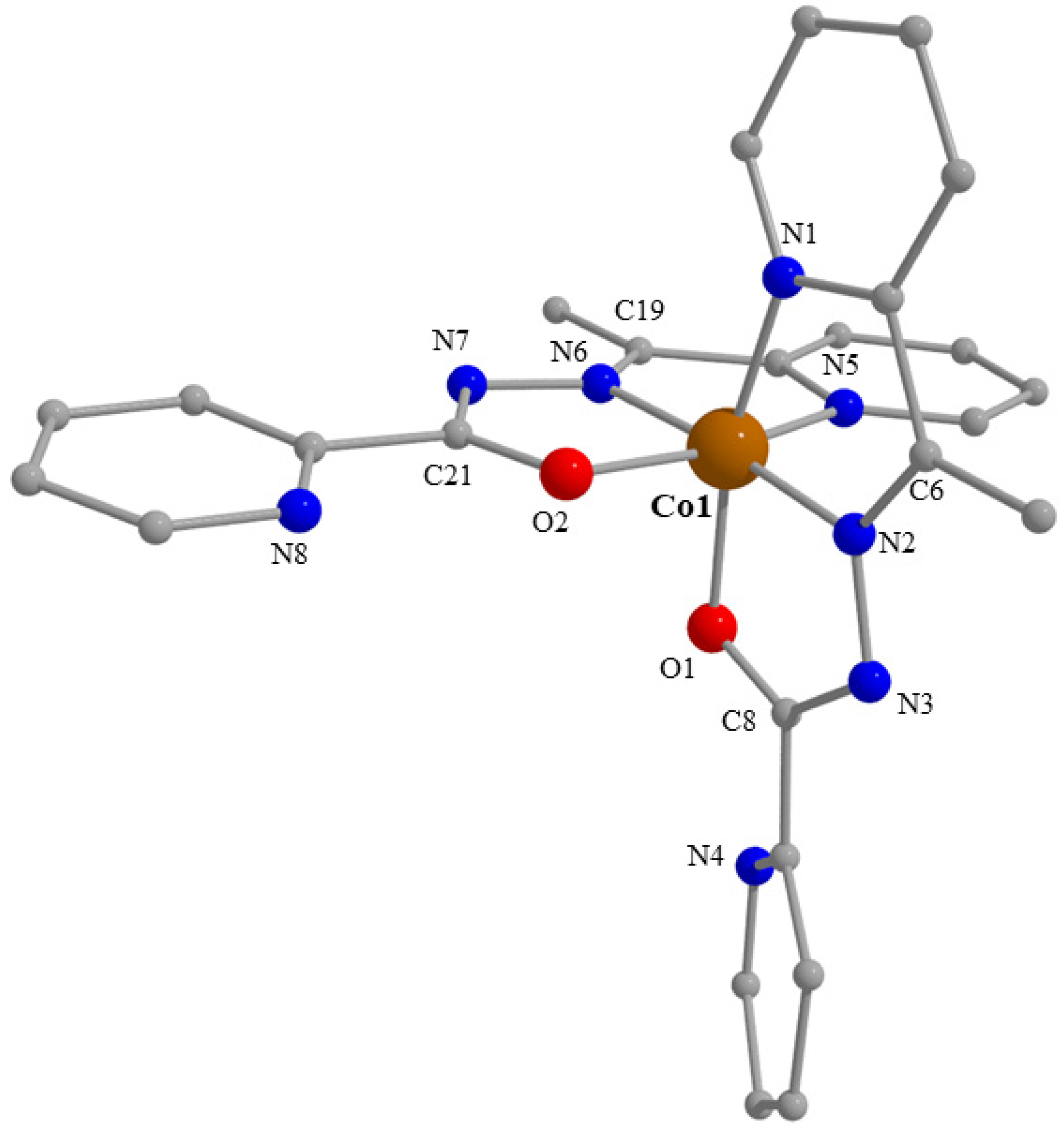
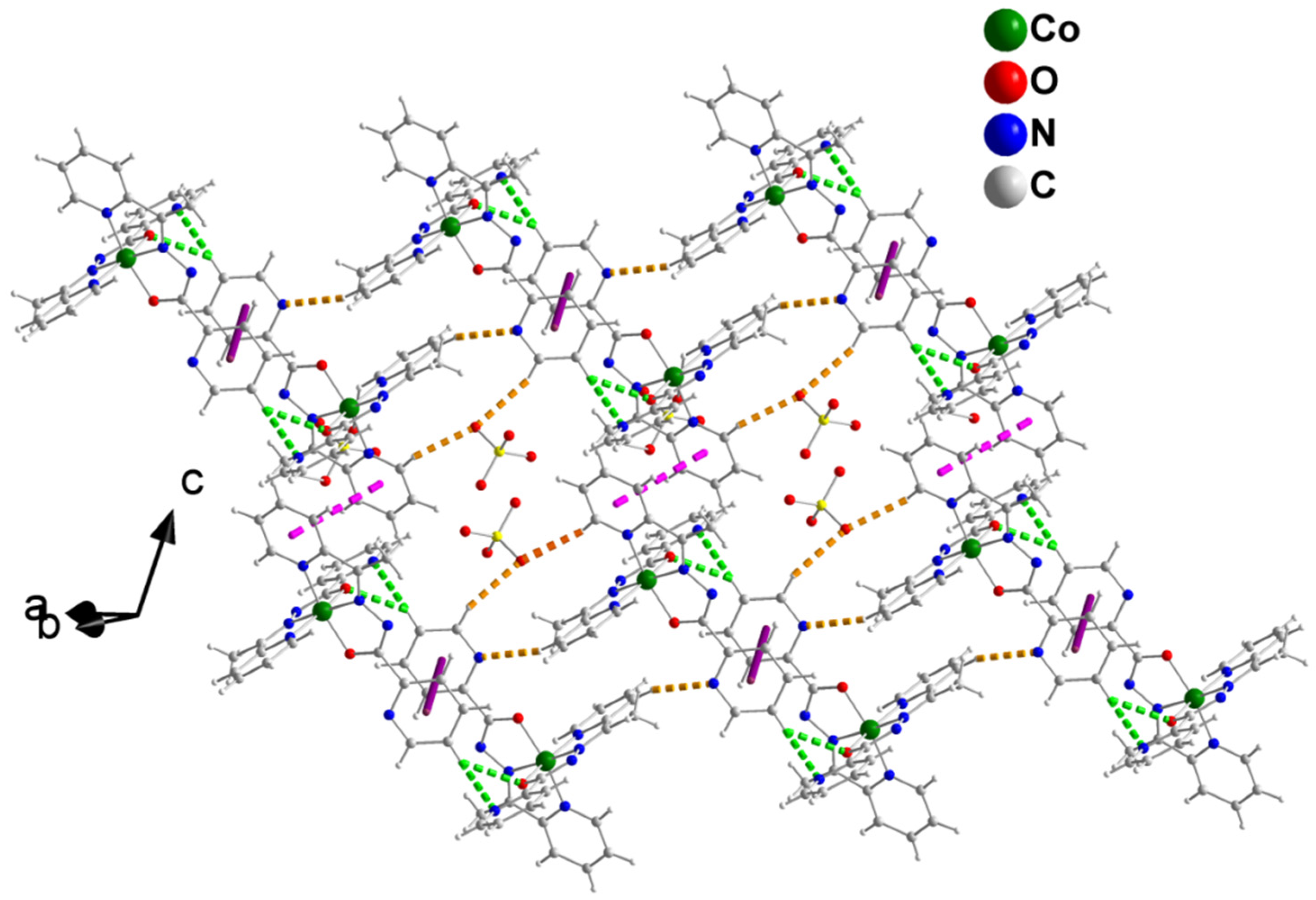
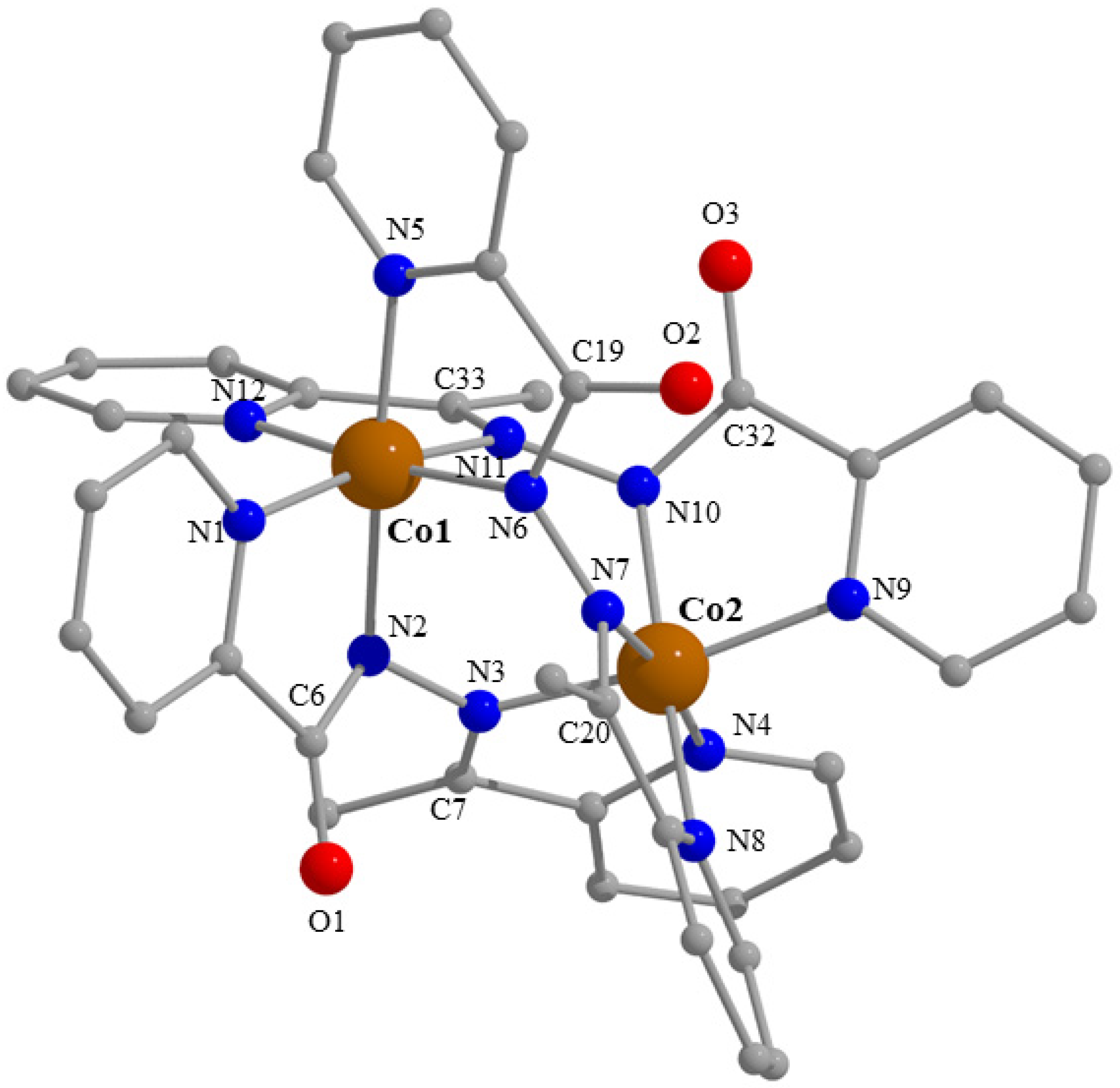
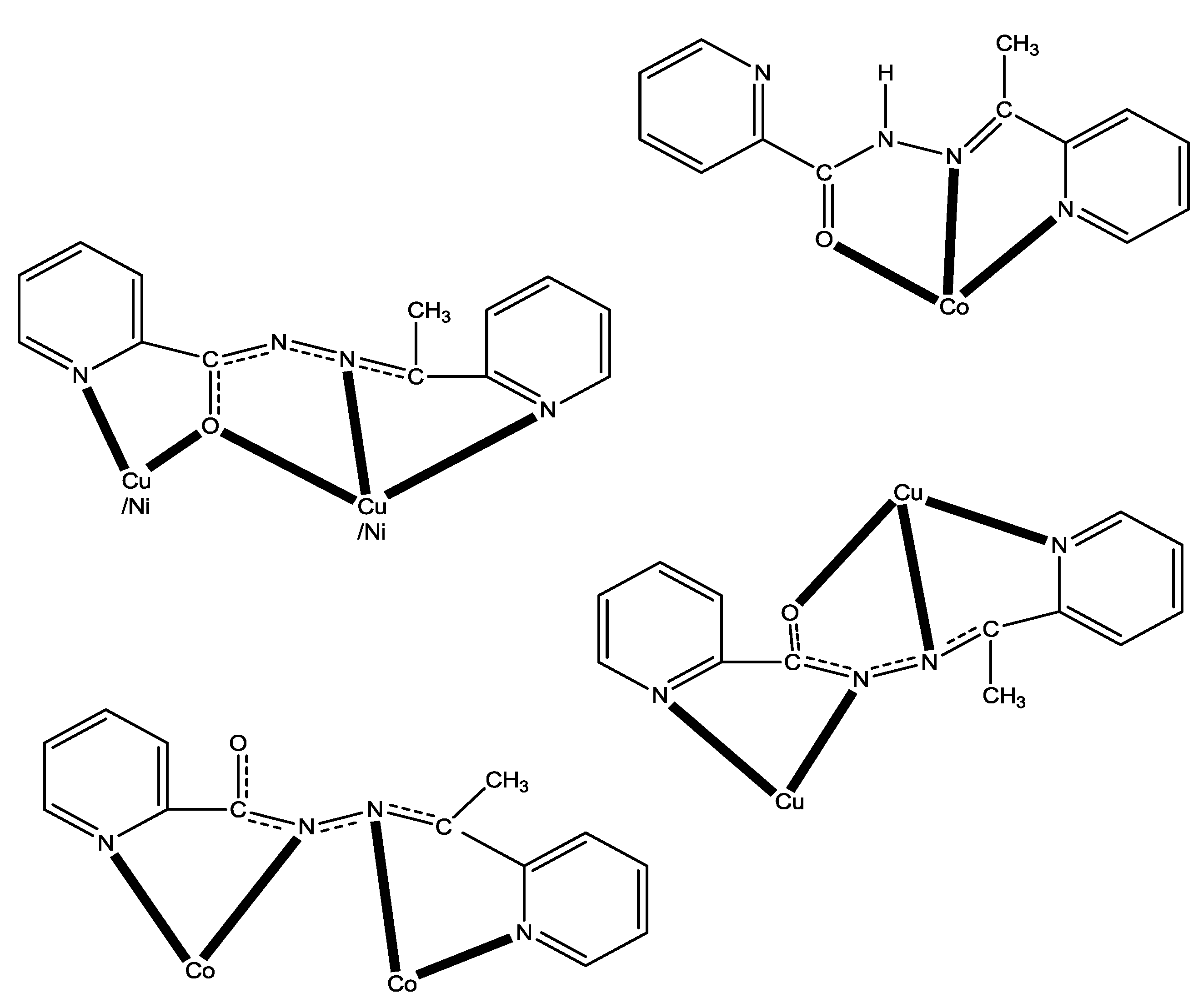
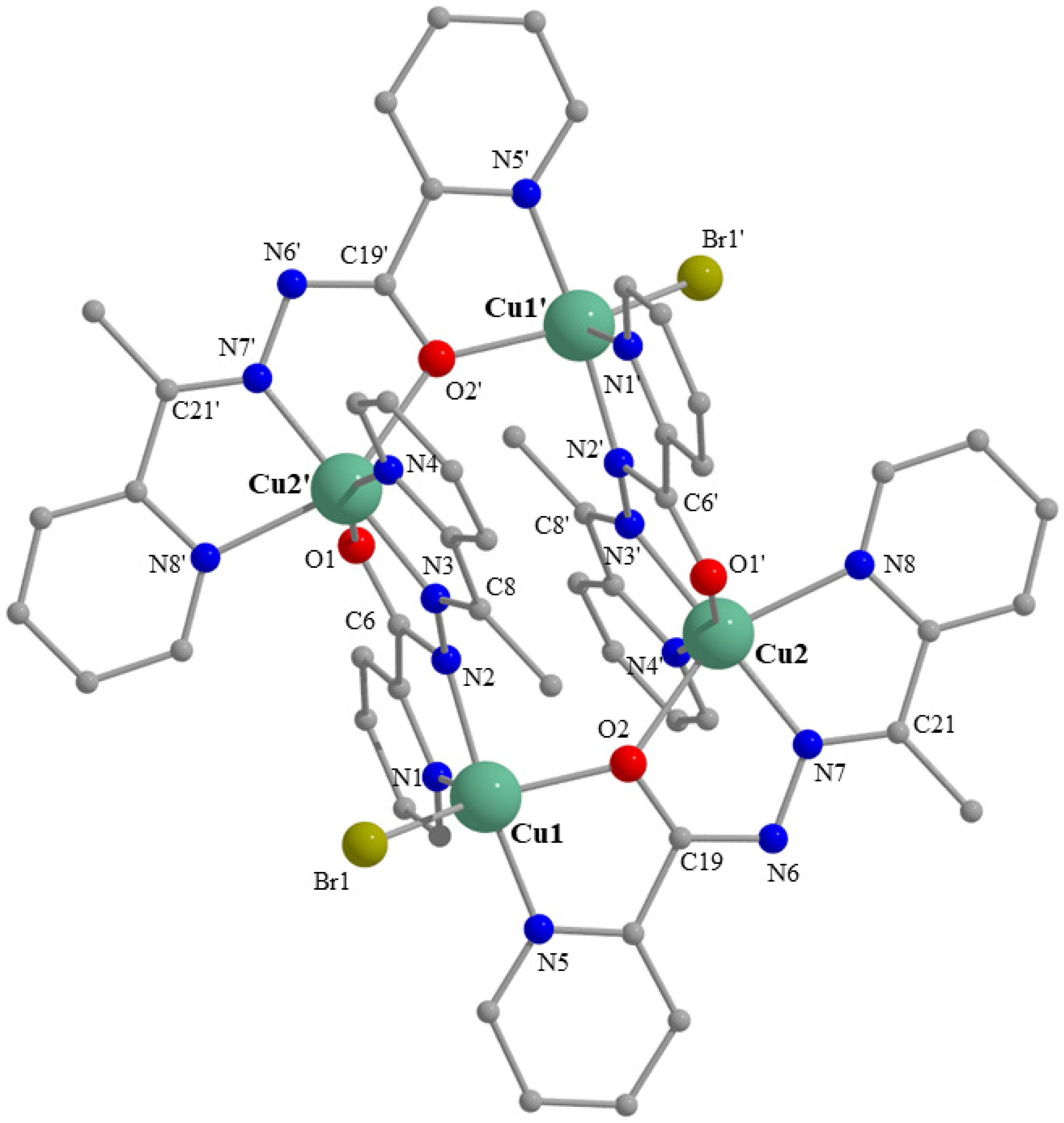
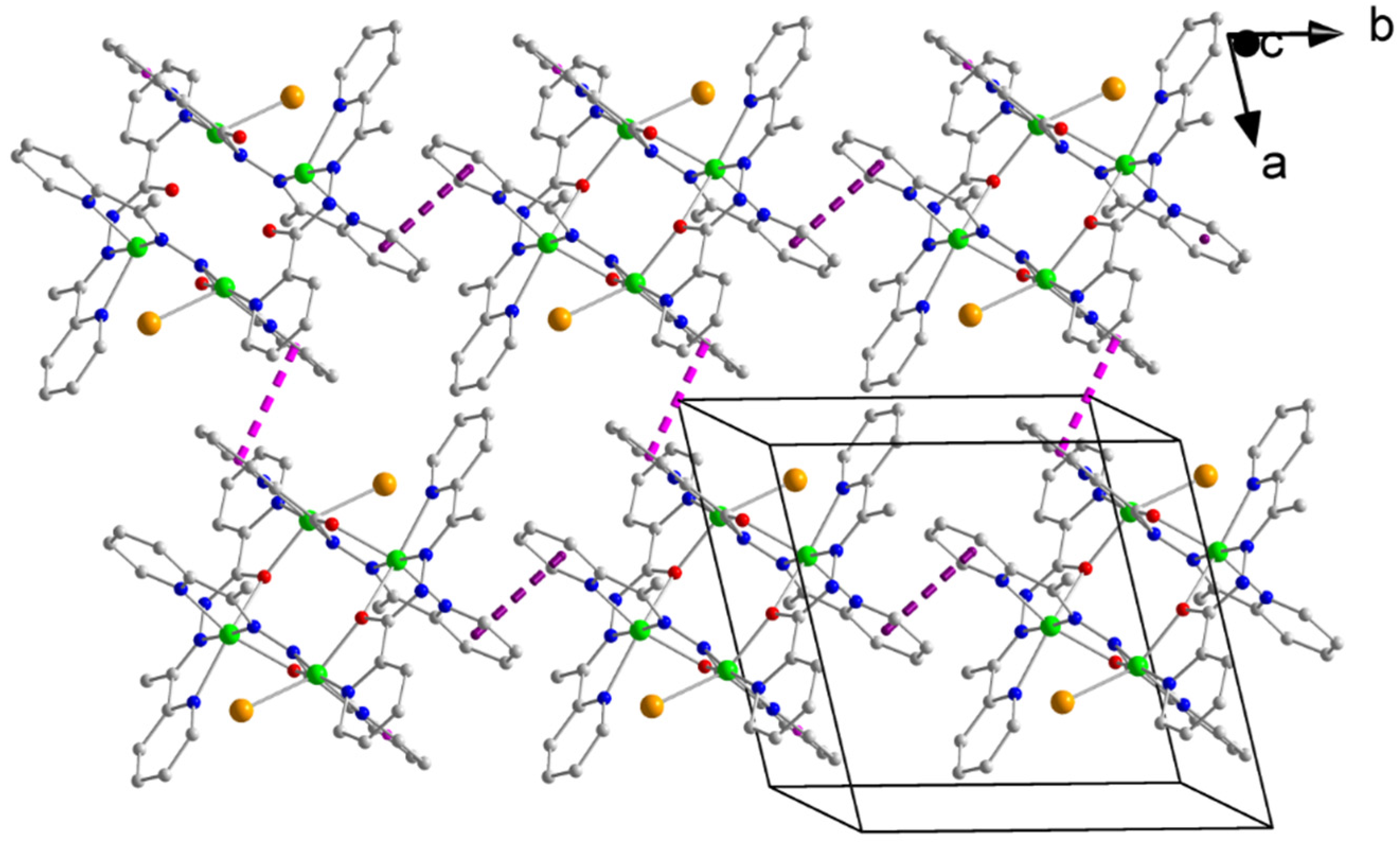
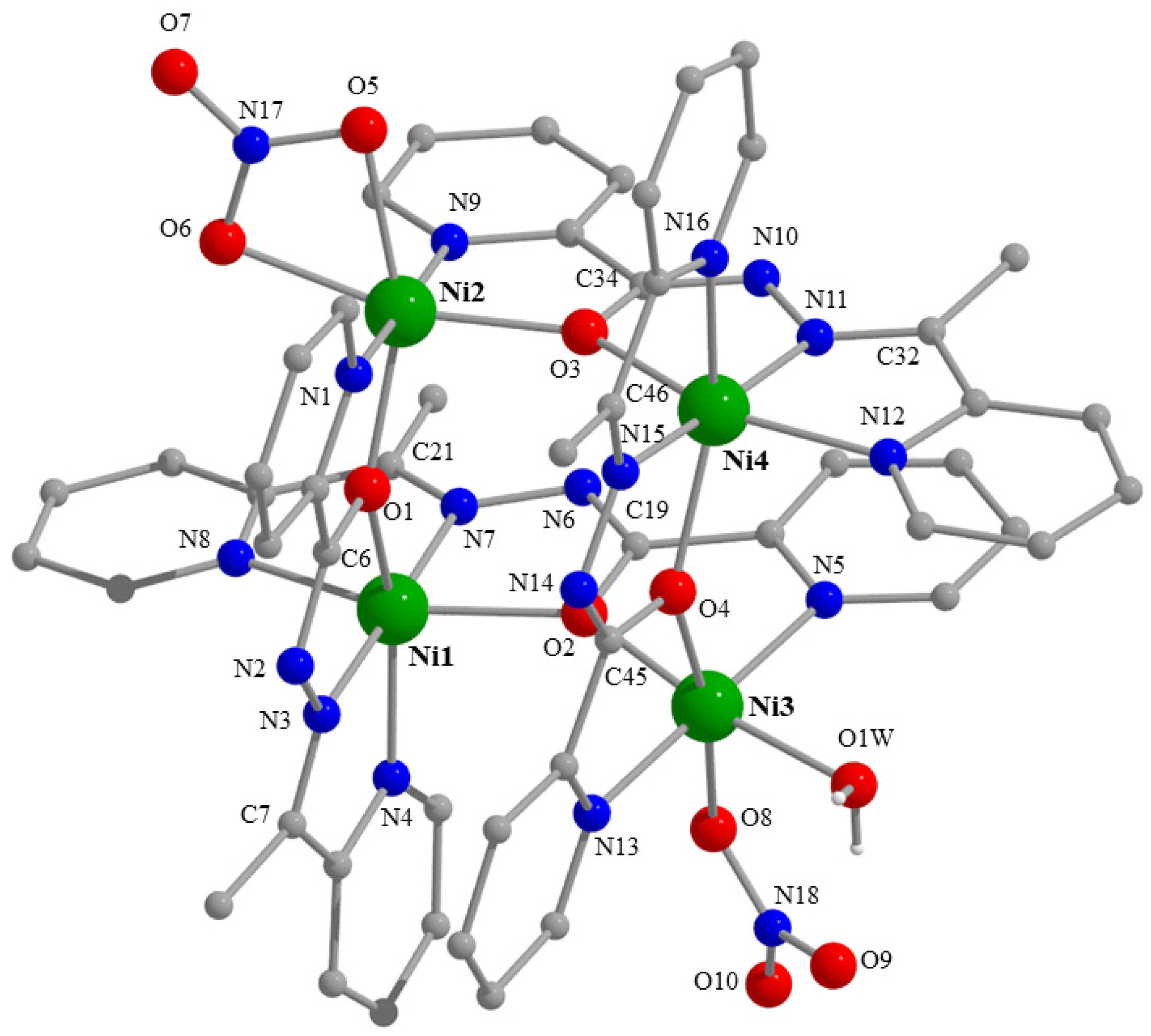
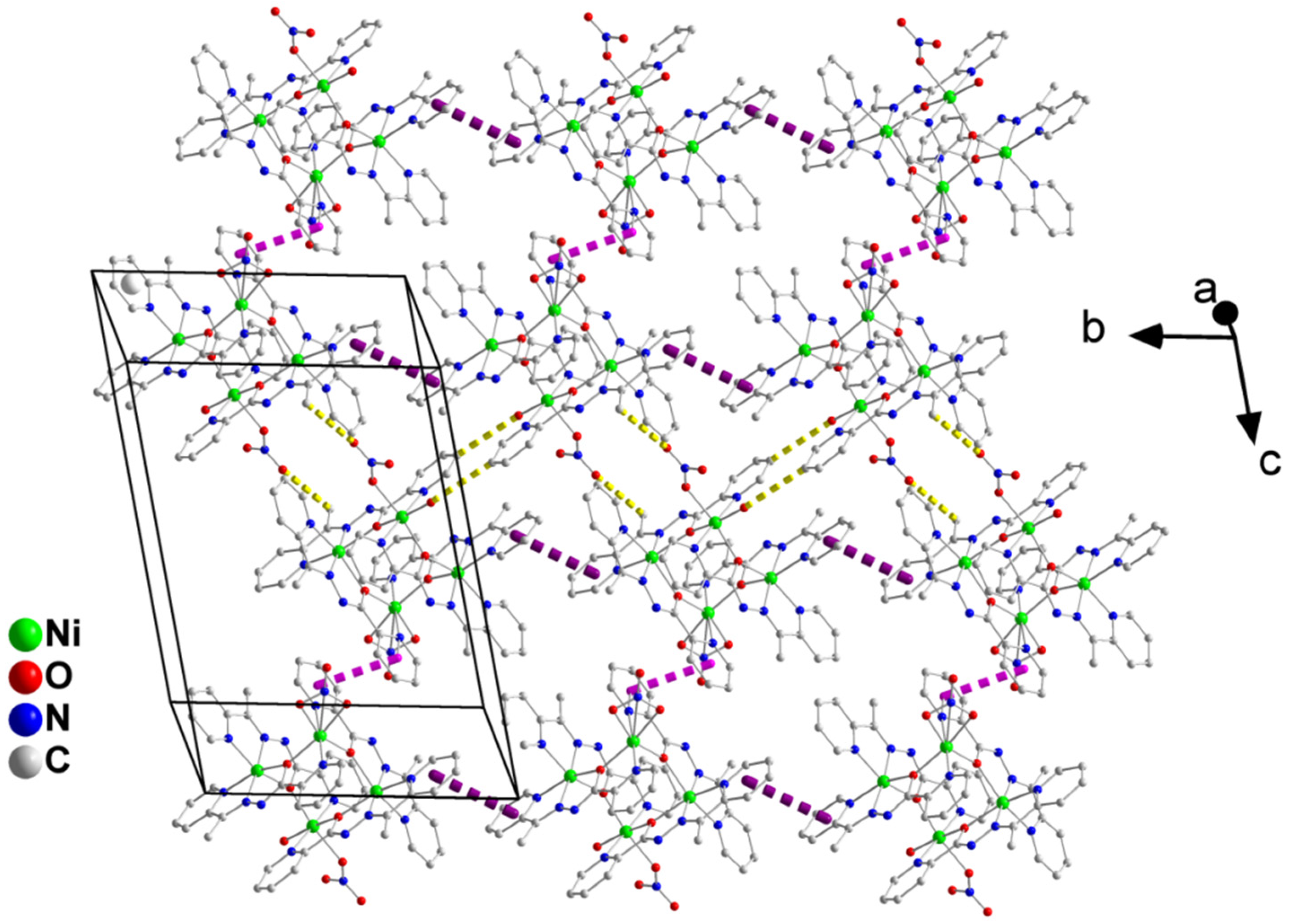
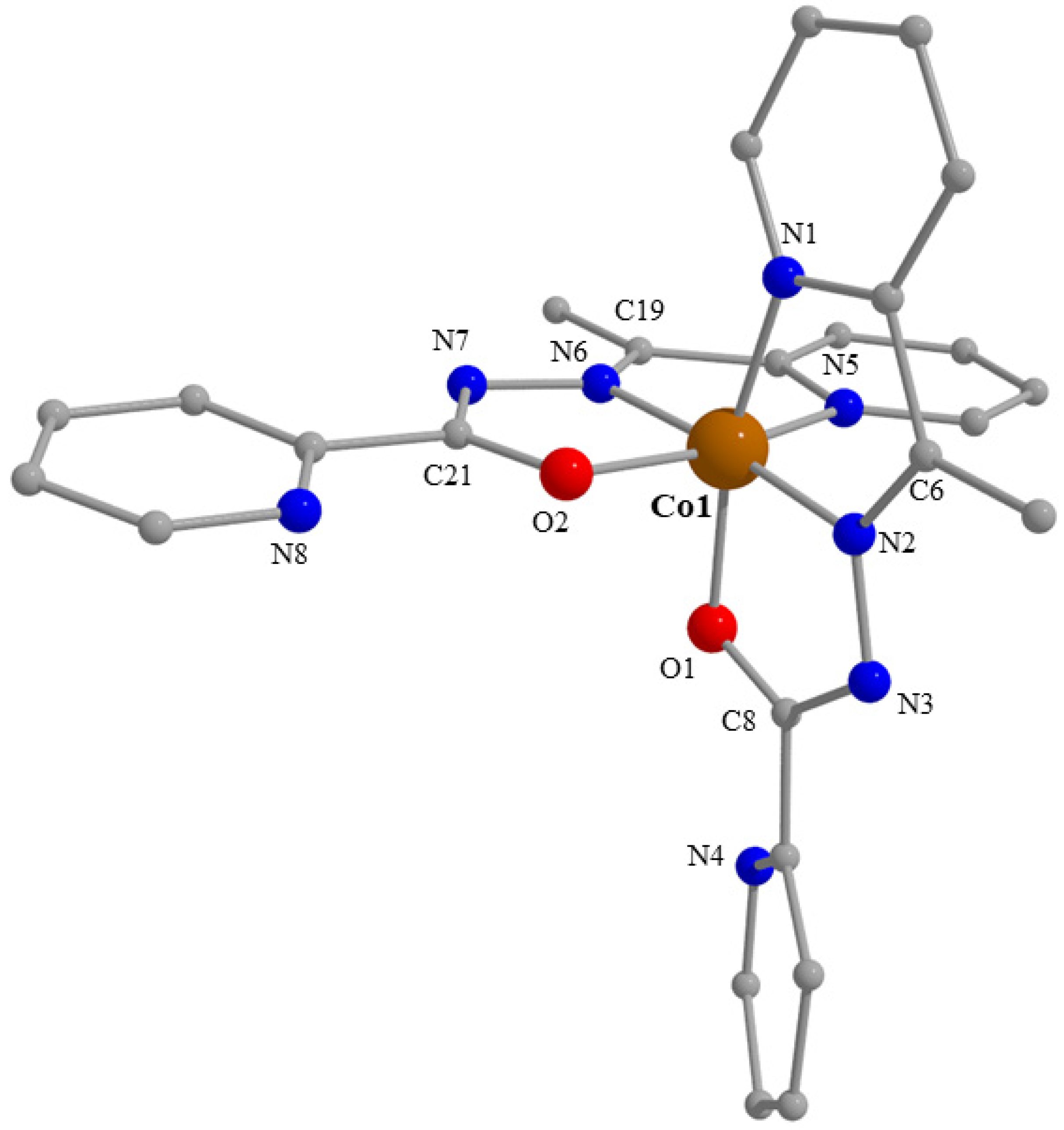
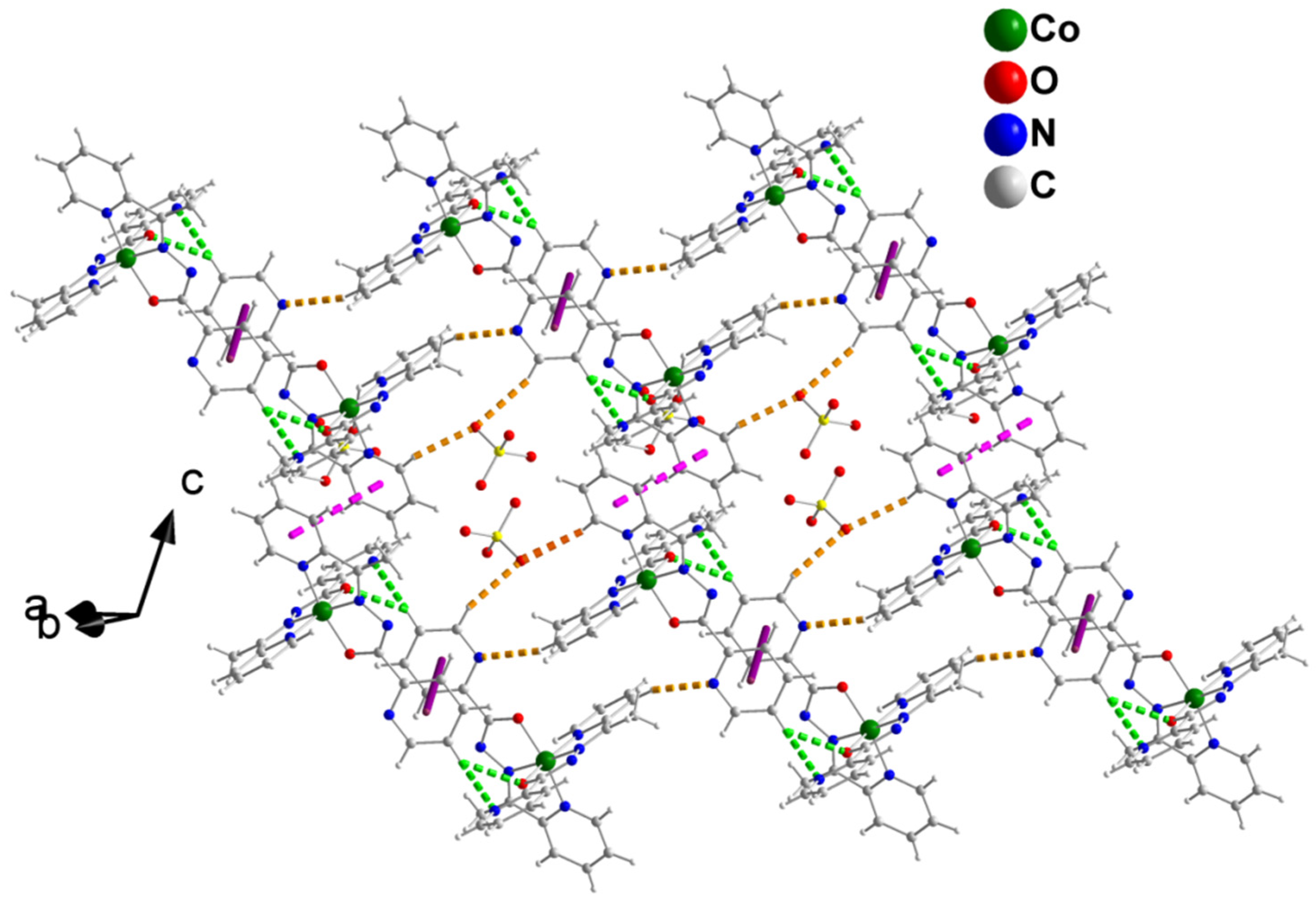
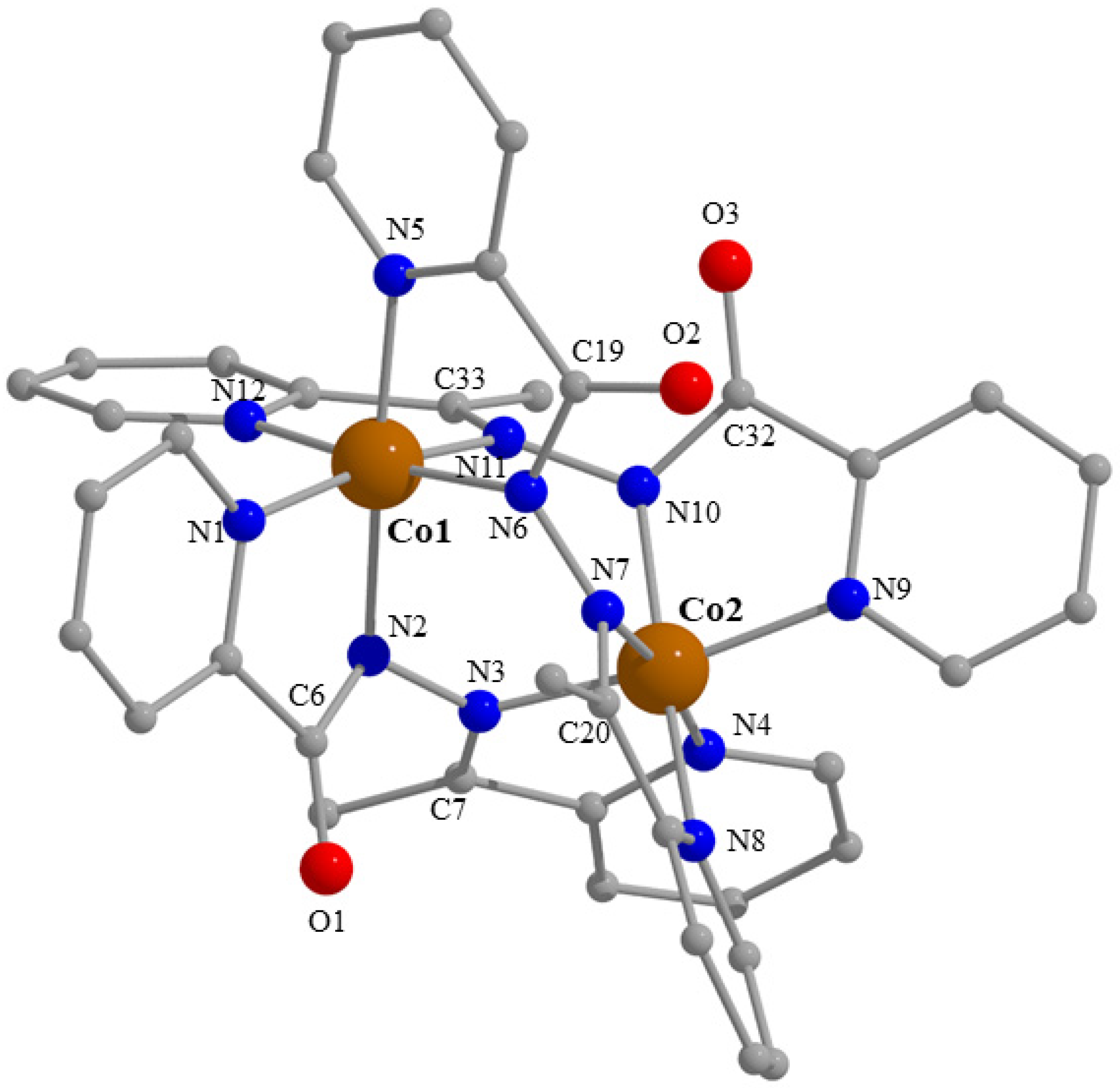
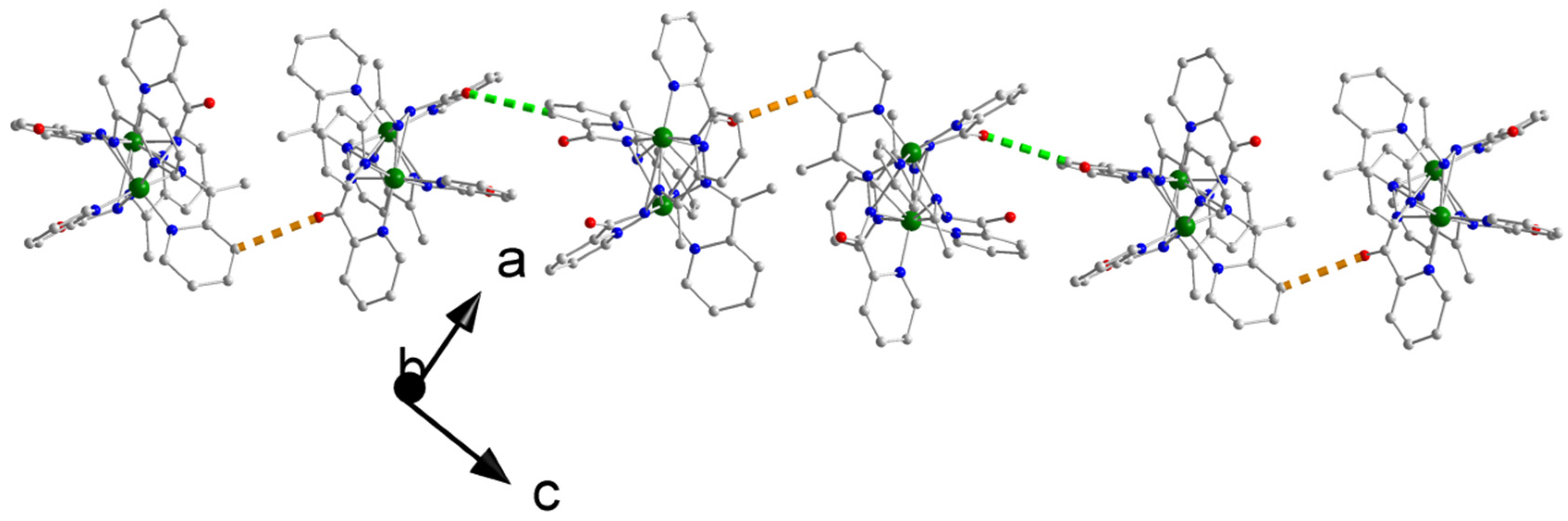
2.3. Description of Structures
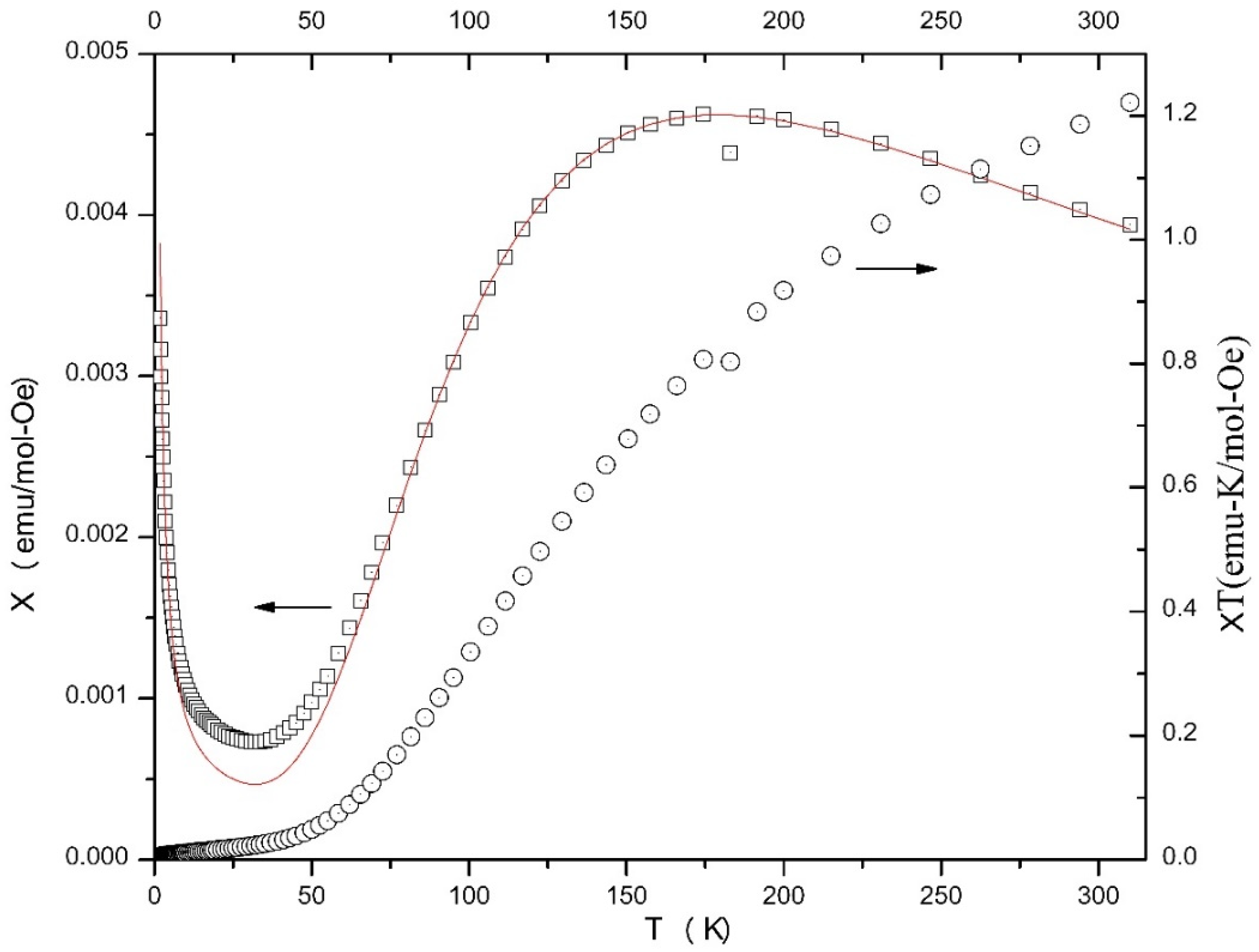
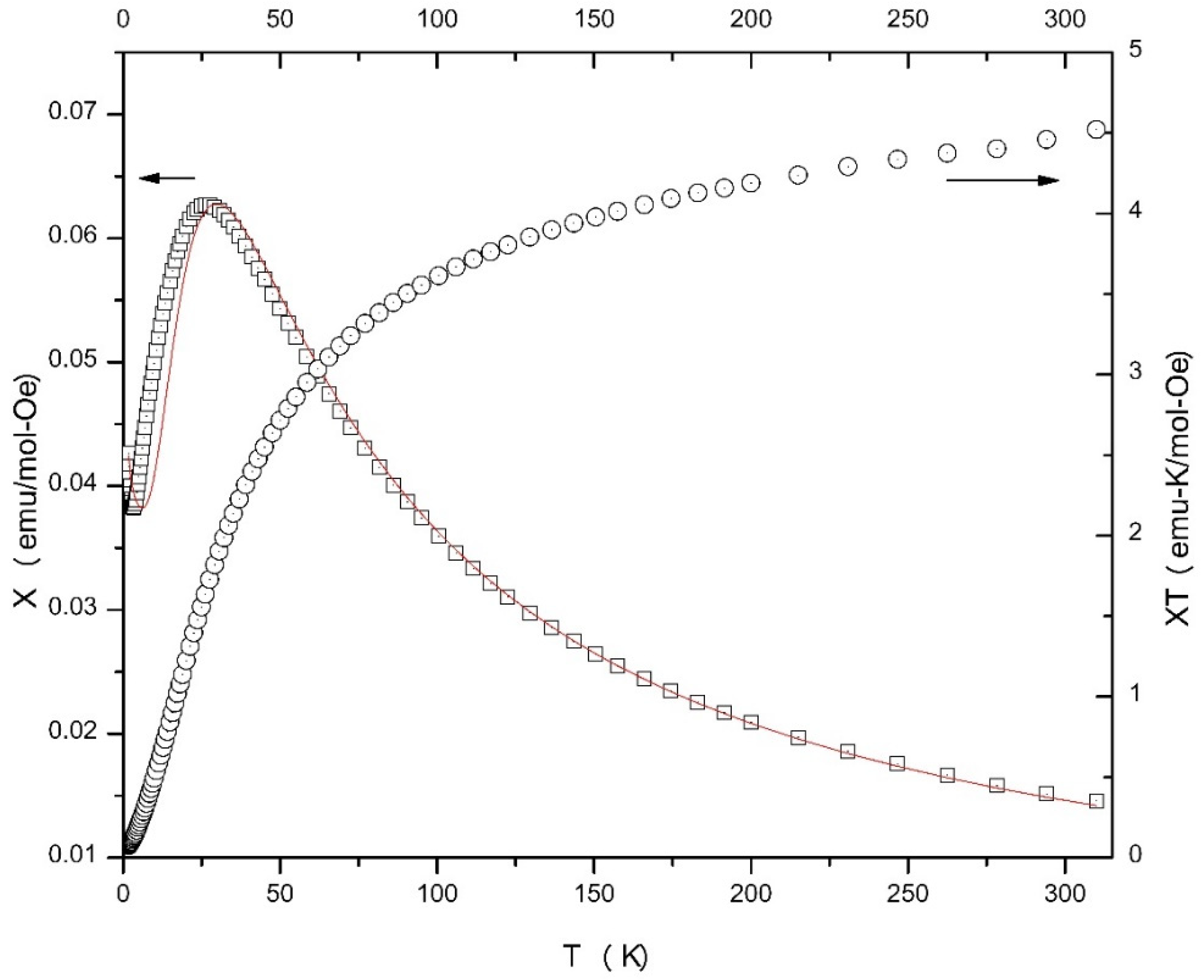
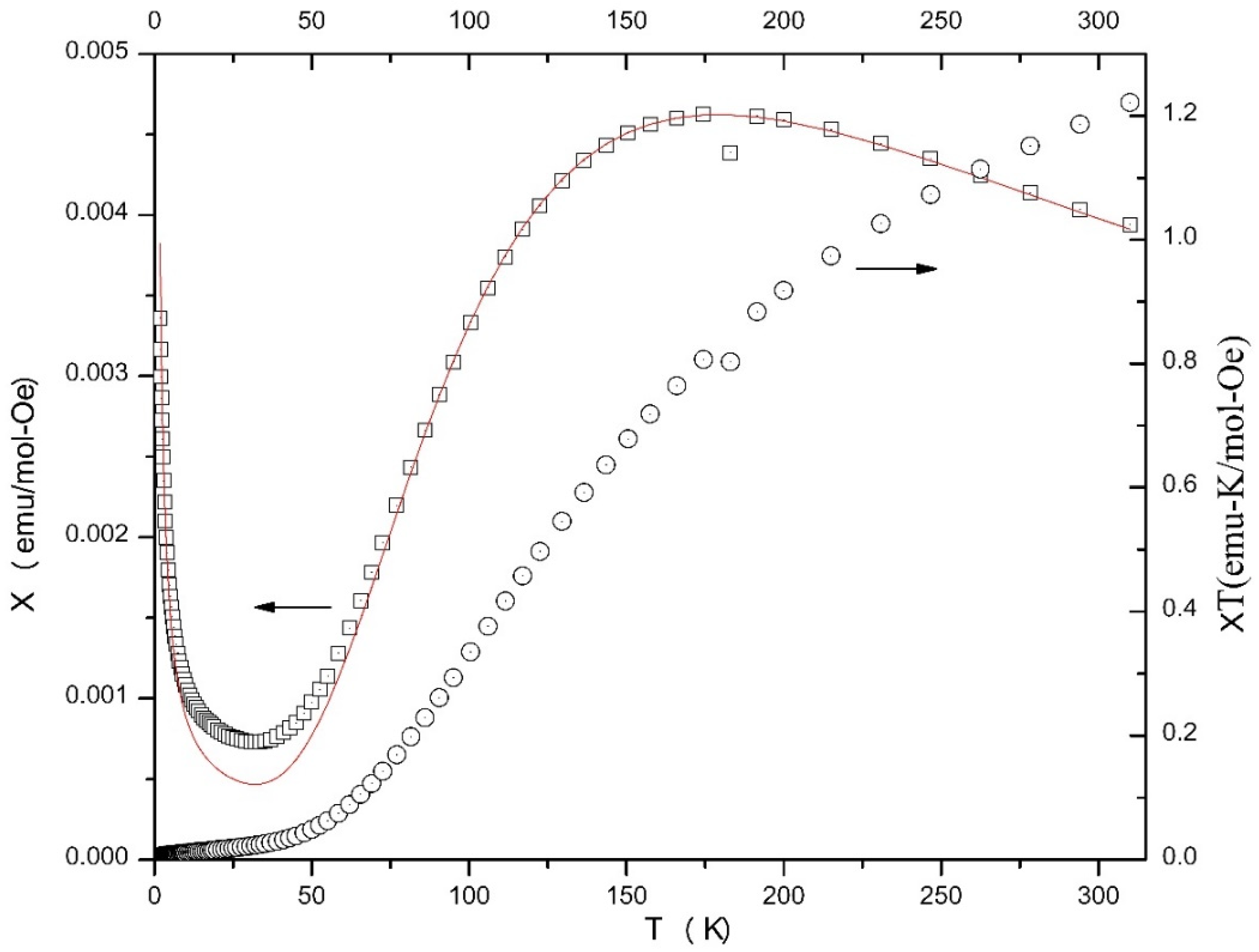
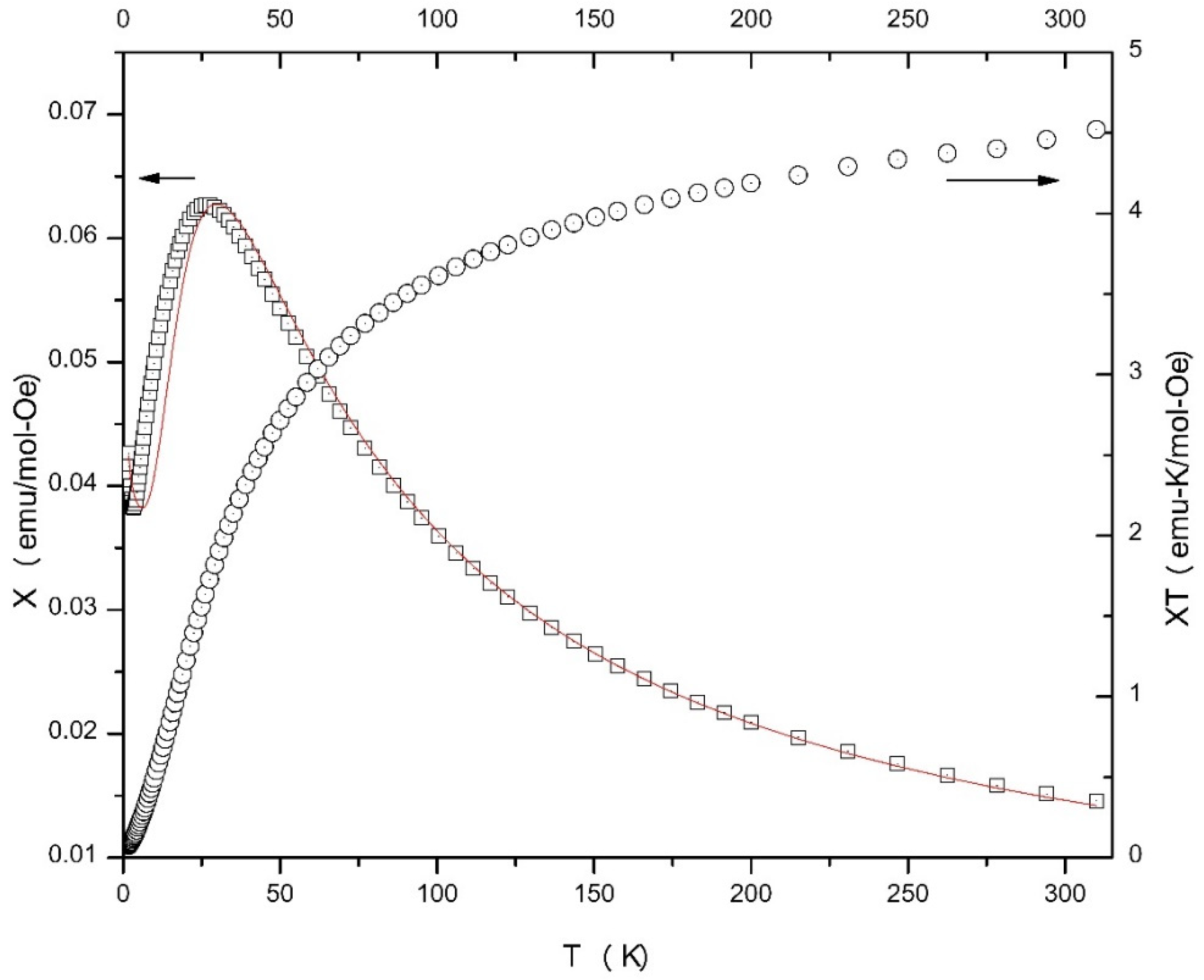
2.4. Magnetochemistry
3. Experimental Section
3.1. Materials, Physical and Spectroscopic Measurements
3.2. Synthesis of Complex [Cu4Br2(L)4]Br2·0.8H2O·MeOH (1·0.8H2O·MeOH)
3.3. Synthesis of Complex [Ni4(NO3)2(L)4(H2O](NO3)2·0.2H2O·3EtOH (2·0.2H2O·3EtOH)
3.4. Synthesis of Complex [Co2(L)3](ClO4)3·0.8H2O·1.3MeOH (3·0.8H2O·1.3MeOH)
3.5. Syntheses of Complex [Co(L)2](ClO4)(4)
3.6. Single-Crystal X-ray Crystallography
4. Concluding Comments and Perspectives
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 5th ed.; Pearson: Harlow, UK, 2018; pp. 237–239. [Google Scholar]
- Ribas Gispert, J. Coordination Chemistry; Wiley-VCH: Weinheim, Germany, 2008; pp. XXXIX, XL. [Google Scholar]
- Constable, E.C. Metals and Ligand Reactivity; VCH: Weinheim, Germany, 1996. [Google Scholar]
- Stamatatos, T.C.; Efthymiou, C.G.; Stoumpos, C.C.; Perlepes, S.P. Adventures in the Coordination Chemistry of Di-2-pyridyl Ketone and Related Ligands: From High-Spin Molecules and Single-Molecule Magnets to Coordination Polymers, and from Structural Aesthetics to an Exciting New Reactivity Chemistry of Coordinated Ligands. Eur. J. Inorg. Chem. 2009, 3361–3391. [Google Scholar] [CrossRef]
- Kahn, O. Molecular Magnetism; VCH Publishers: New York, NY, USA, 1993. [Google Scholar]
- Hearne, N.; Turnbull, M.M.; Landee, C.P.; van der Merwe, E.M.; Rademeyer, M. Halide-bi-bridged polymers of amide substituted-pyridines and –pyrazines: Polymorphism, structures, thermal stability and magnetism. CrystEngComm 2019, 21, 1910–1927. [Google Scholar] [CrossRef]
- Efthymiou, C.G.; Mylonas-Margaritis, I.; Raptopoulou, C.P.; Psycharis, V.; Escuer, A.; Papantriantafyllopoulou, C.; Perlepes, S.P. A Ni11 Coordination Cluster from the Use of the Di-2-Pyridyl Ketone/Acetate Ligand Combination: Synthetic, Structural and Magnetic Studies. Magnetochemistry 2016, 2, 30. [Google Scholar] [CrossRef]
- Escuer, A.; Esteban, J.; Perlepes, S.P.; Stamatatos, T.C. The bridging azido ligand as a central “player” in high-nuclearity 3d-metal cluster chemistry. Coord. Chem. Rev. 2014, 275, 87–129. [Google Scholar] [CrossRef]
- Lada, Z.G.; Katsoulakou, E.; Perlepes, S.P. Synthesis and Chemistry of Single-molecule Magnets. In Single-Molecule Magnets: Molecular Architectures and Building Blocks for Spintronics; Holynska, M., Ed.; Wiley-VCH: Weinheim, Germany, 2019; pp. 245–313. [Google Scholar]
- Milway, V.A.; Niel, V.; Abedin, T.S.M.; Xu, Z.; Thompson, L.K.; Grove, H.; Miller, D.O.; Parsons, S.R. Octanuclear and Nonanuclear Supramolecular Copper(II) Complexes with Linear “Tritopic” Ligands: Structural and Magnetic Studies. Inorg. Chem. 2004, 43, 1874–1884. [Google Scholar] [CrossRef]
- Zhao, L.; Xu, Z.; Grove, H.; Milway, V.A.; Dawe, L.N.; Abedin, T.S.M.; Thompson, L.K.; Kelly, T.L.; Harvey, R.G.; Miller, D.O.; et al. Supramolecular Mn(II) and Mn(II)/Mn(III) Grid Complexes with [Mn9(μ2-O)12] Core Structures. Structural, Magnetic, and Redox Properties and Surface Studies. Inorg. Chem. 2004, 43, 3812–3824. [Google Scholar] [CrossRef]
- Milway, V.A.; Abedin, S.M.T.; Thompson, L.K.; Miller, D.O. Ferromagnetic Cu8 pinwheel structures as building blocks for larger magnetic networks: Long-range structural and magnetic ordering. Inorg. Chim. Acta 2006, 359, 2700–2711. [Google Scholar] [CrossRef]
- Milway, V.A.; Abedin, S.M.T.; Niel, V.; Kelly, T.L.; Dawe, L.N.; Dey, S.K.; Thompson, D.W.; Miller, D.O.; Alam, M.S.; Müller, P.; et al. Supramolecular ‘flat’ Mn9 grid complexes—Towards functional molecular platforms. Dalton Trans. 2006, 2835–2851. [Google Scholar] [CrossRef]
- Grove, H.; Kelly, T.L.; Thompson, L.K.; Zhao, L.; Xu, Z.; Abedin, T.S.M.; Miller, D.O.; Goeta, A.E.; Wilson, C.; Howard, J.A.K. Copper (II) Complexes of a Series of Alkoxy Diazine Ligands: Mononuclear, Dinuclear and Tetranuclear Examples with Structural, Magnetic and DFT Studies. Inorg. Chem. 2004, 43, 4278–4288. [Google Scholar] [CrossRef]
- Dawe, L.N.; Abedin, T.S.M.; Kelly, T.K.; Thompson, L.K.; Miller, D.O.; Zhao, L.; Wilson, C.; Leech, M.A.; Howard, J.A.K. Self-assembled polymetallic square grids ([2×2] M4, [3×3] M9) and trigonal bipyramidal clusters (M5)-structural and magnetic properties. J. Mater. Chem. 2006, 16, 2645–2659. [Google Scholar] [CrossRef]
- Akkurt, M.; Khandar, A.A.; Tahir, M.N.; Yazdi, S.A.H.; Afkhami, F.A. Dibromido {N′-[1-pyridin-2-yl)ethylidene]picolinohydrazide-κ2N′,O} cadmium. Acta Crystallogr. E 2012, 68, m842. [Google Scholar]
- Xu, Z.-Q.; Mao, X.-J.; Zhang, X.; Cai, H.-X.; Bie, H.-Y.; Xu, J.; Jia, L. Syntheses, Crystal Structures and Antitumor Activities of Three Ln(III) Complexes with 2-Acetylpyridine Picolinohydrazone. Chin. J. Inorg. Chem. 2015, 31, 1–8. [Google Scholar]
- Kitamura, F.; Sawaguchi, K.; Mori, A.; Takagi, S.; Suzuki, T.; Kobayashi, A.; Kato, M.; Nakajima, K. Coordination Structure Conversion of Hydrazone-Palladium(II) Complexes in the Solid State and in Solution. Inorg. Chem. 2015, 54, 8436–8448. [Google Scholar] [CrossRef]
- Abedi, M.; Yesilel, O.Z.; Mahmoudi, G.; Bauzá, A.; Lofland, S.E.; Yerli, Y.; Kaminsky, W.; Garczarek, P.; Zareba, I.K.; Ienco, A.; et al. Tetranuclear Manganese(II) Complexes of Hydrazone and Carboxydrazone Ligands: Synthesis, Crystal Structures, Magnetic Properties, Hirshfeld Surface Analysis and DFT Calculations. Inorg. Chim. Acta 2016, 443, 101–109. [Google Scholar] [CrossRef]
- Mahmoudi, G.; Bauzá, A.; Gurbanov, A.V.; Zubkov, F.I.; Maniukiewicz, W.; Rodriguez-Dieguez, A.; López-Torres, E.; Frontera, A. The role of unconventional stacking interactions in the supramolecular assemblies of Hg(II) coordination compounds. CrystEngComm 2016, 18, 9056–9066. [Google Scholar] [CrossRef]
- Mahmoudi, G.; Stilinovic, V.; Bauzá, A.; Frontera, A.; Bartyzel, A.; Ruiz-Pérez, C.; Kirillov, A.M. Inorganic- organic hybrid materials based on PbBr2 and pyridine-hydrazone blocks—Structural and theoretical study. RSC Adv. 2016, 6, 60385–60393. [Google Scholar] [CrossRef]
- Maniaki, D.; Pilichos, E.; Perlepes, S.P. Coordination Clusters of 3d-metals That Behave as Single-Molecule Magnets (SMMs): Synthetic Routes and Strategies. Front. Chem. 2018, 6, 461. [Google Scholar] [CrossRef]
- Pilichos, E.; Mylonas-Margaritis, I.; Kontos, A.P.; Psycharis, V.; Klouras, N.; Raptopoulou, C.P.; Perlepes, S.P. Coordination and metal ion-mediated transformation of a polydentate ligand containing oxime, hydrazone and picolinoyl functionalities. Inorg. Chem. Commun. 2018, 94, 48–52. [Google Scholar] [CrossRef]
- Lada, Z.G.; Sanakis, Y.; Raptopoulou, C.P.; Psycharis, V.; Perlepes, S.P.; Mitrikas, G. Probing the electronic structure of a copper(II) complex by CW- and pure—EPR spectroscopy. Dalton Trans. 2017, 46, 8458–8475. [Google Scholar] [CrossRef]
- Anastasiadis, N.C.; Granadeiro, C.M.; Klouras, N.; Cunha-Silva, L.; Raptopoulou, C.P.; Psycharis, V.; Bekiari, V.; Balula, S.S.; Escuer, A.; Perlepes, S.P. Dinuclear Lanthanide(III) Complexes by Metal-Ion-Assisted Hydration of Di-2-pyridyl Ketone Azine. Inorg. Chem. 2013, 52, 4145–4147. [Google Scholar] [CrossRef]
- Duros, V.; Sartzi, H.; Teat, S.J.; Sanakis, Y.; Roubeau, O.; Perlepes, S.P. Tris{2,4-bis(2-pyridyl)-1,3,5-triazapentanedionato}manganese(III), a complex derived from a unique metal ion-assisted transformation of pyridine-2-amidoxime. Inorg. Chem. Commun. 2014, 50, 117–121. [Google Scholar] [CrossRef]
- Kitos, A.A.; Efthymiou, C.G.; Manos, M.J.; Tasiopoulos, A.J.; Nastopoulos, V.; Escuer, A.; Perlepes, S.P. Interesting copper(II)–assisted transformations of 2-acetylpyridine and 2-benzoylpyridine. Dalton Trans. 2016, 45, 1063–1077. [Google Scholar] [CrossRef]
- Nakamoto, K. Infrared and Raman Spectra of Inorganic and Coordination Compounds, 4th ed.; Wiley: New York, NY, USA, 1986; pp. 121–125, 130–139, 254–257. [Google Scholar]
- Mylonas-Margaritis, I.; Maniaki, D.; Mayans, J.; Ciammaruchi, L.; Bekiari, V.; Raptopoulou, C.P.; Psycharis, V.; Christodoulou, S.; Escuer, A.; Perlepes, S.P. Mononuclear Lanthanide(III)- Salicylideneaniline Complexes: Synthetic, Structural, Spectroscopic, and Magnetic Studies. Magnetochemistry 2018, 4, 45. [Google Scholar] [CrossRef]
- Mylonas-Margaritis, I.; Mayans, J.; Sakellakou, S.-M.; Raptopoulou, C.P.; Psycharis, V.; Escuer, A.; Perlepes, S.P. Using the Singly Deprotonated Triethanolamine to Prepare Dinuclear Lanthanide(III) Complexes: Synthesis, Structural Characterization and Magnetic Studies. Magnetochemistry 2017, 3, 5. [Google Scholar] [CrossRef]
- Tangoulis, V.; Raptopoulou, C.P.; Terzis, A.; Paschalidou, S.; Perlepes, S.P.; Bakalbassis, E.G. Octanuclearity in Copper(II) Chemistry: Preparation, Characterization, and Magnetochemistry of [Cu8(dpk·OH)8(O2CCH3)4](ClO4)4·9H2O (dpk·H2O = The hydrated, gem-Diol Form of Di-2-pyridyl Ketone). Inorg. Chem. 1997, 36, 3996–4006. [Google Scholar] [CrossRef]
- Lever, A.B.P. Inorganic Electronic Spectroscopy, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 1984; pp. 355, 356, 473–478, 507–520. [Google Scholar]
- Cotton, F.A.; Wilkinson, G.; Murillo, C.A.; Bochmann, M. Advanced Inorganic Chemistry, 6th ed.; Wiley: New York, NY, USA, 1999; pp. 824, 825, 838, 839. [Google Scholar]
- Alexopoulou, K.I.; Zagoraiou, E.; Zafiropoulos, T.F.; Raptopoulou, C.P.; Psycharis, V.; Terzis, A.; Perlepes, S.P. Mononuclear anionic octahedral cobalt(III) complexes based on N-salicylidene-o-aminophenol and its derivatives: Synthetic, structural and spectroscopic studies. Spectrochim. Acta 2015, A136, 122–130. [Google Scholar] [CrossRef]
- Coxall, R.A.; Harris, S.G.; Henderson, D.K.; Parsons, S.; Tasker, P.A.; Winpenny, R.E.P. Inter-ligand reactions: In situ formation of new polydentate ligands. J. Chem. Soc. Dalton Trans. 2000, 2349–2356. [Google Scholar] [CrossRef]
- Addison, A.W.; Rao, T.N.; Reedijk, J.; Rijn, J.V.; Verschoor, G.C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands: The crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. J. Chem. Soc. Dalton Trans. 1984, 1349–1356. [Google Scholar] [CrossRef]
- Papatriantafyllopoulou, C.; Stamatatos, T.C.; Wernsdorfer, W.; Teat, S.J.; Tasiopoulos, A.J.; Escuer, A.; Perlepes, S.P. Combining Azide, Carboxylate, and 2-Pyridyloximate Ligands in Transition-Metal Chemistry: Ferromagnetic NiII5 Clusters with a Bowtie Skeleton. Inorg. Chem. 2010, 49, 10486–10496. [Google Scholar] [CrossRef]
- Mylonas-Margaritis, I.; Mayans, J.; Perlepe, P.S.; Raptopoulou, C.P.; Psycharis, V.; Alexopoulou, K.I.; Escuer, A.; Perlepes, S.P. Nickel(II) Coordination Clusters Based on N-salicylidene-4-chloro-o-aminophenol: Synthetic and Structural Studies. Curr. Inorg. Chem. 2017, 7. [Google Scholar] [CrossRef]
- Polyzou, C.D.; Lada, Z.G.; Terzis, A.; Raptopoulou, C.P.; Psycharis, V.; Perlepes, S.P. The fac diastereoisomer of tris(2-pyridinealdoximato)cobalt(III) and a cationic cobalt(III) complex containing both the neutral and anionic forms of the ligand: Synthetic, structural and spectroscopic studies. Polyhedron 2014, 79, 29–36. [Google Scholar] [CrossRef]
- Polyzou, C.D.; Koumousi, E.S.; Lada, Z.G.; Raptopoulou, C.P.; Psycharis, V.; Rouzières, M.; Tsipis, A.C.; Mathonière, C.; Clérac, R.; Perlepes, S.P. “Switching on” the single- molecule magnet properties within a series of dinuclear cobalt(III)–dysprosium(III) 2-pyridyloximate complexes. Dalton Trans. 2017, 46, 14812–14825. [Google Scholar] [CrossRef]
- Howson, S.E.; Scott, P. Approaches to the synthesis of optically pure helicates. Dalton Trans. 2011, 40, 10268–10277. [Google Scholar] [CrossRef]
- Piguet, C.; Bernardinelli, G.; Bocquet, B.; Quattropani, A.; Williams, A.F. Self-Assembly of Double and Triple Helices Controlled by Metal Ion Stereochemical Preferences. J. Am. Chem. Soc. 1992, 114, 7440–7451. [Google Scholar] [CrossRef]
- Tuna, F.; Lees, M.R.; Clarkson, G.T.; Hannon, M.J. Readily Prepared Metallo-Supramolecular Triple Helicates Designed to Exhibit Spin-Crossover Behaviour. Chem. Eur. J. 2004, 10, 5737–5750. [Google Scholar] [CrossRef]
- Pelleteret, D.; Clèrac, R.; Mathonière, C.; Harté, E.; Schmitt, W.; Kruger, P.E. Asymmetric spin crossover behaviour and evidence of light-induced excited spin state trapping in a dinuclear iron(II) helicate. Chem. Commun. 2009, 221–223. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular Chemistry; VCH: Weinheim, Germany, 1995; p. 150. [Google Scholar]
- Steed, J.W.; Atwood, J.L. Supramolecular Chemistry; Wiley: New York, NY, USA, 2000; pp. 540–557. [Google Scholar]
- MAGMUN4.1/OW01.exe is Available as A Combined Package Free of Charge from the Authors. MAGMUN has been Developed by Dr. Zhiqiang Xu, in Collaboration with Prof. L.K. Thompson, and Dr. Oliver Waldmann. Available online: https://www.ucs.mun.ca./~lthomp/magmun (accessed on 18 June 2019).
- Xu, Z.; Thompson, L.K.; Miller, D.O. Dicopper(II) Complexes Bridged by Single N-N Bonds. Magnetic Exchange Dependence on the Rotation Angle between the Magnetic Planes. Inorg. Chem. 1997, 36, 3985–3995. [Google Scholar] [CrossRef]
- Thompson, L.K.; Xu, Z.; Goeta, A.E.; Howard, J.A.K.; Clase, H.J.; Miller, D.O. Structural and Magnetic Properties of Dicopper(II) Complexes of Polydentate Diazine Ligands. Inorg. Chem. 1998, 37, 3217–3229. [Google Scholar] [CrossRef]
- Klaaijsen, F.W.; Blöte, H.W.J.; Dokoupil, Z. Large Single-Ion Anisotropy and Ferromagnetic Intrachain Interaction in four Ni-Compounds of Formula NiX2L2 with X= Cl, Br; L= N2C3H4, NC5H5. Solid State Commun. 1974, 14, 607–612. [Google Scholar] [CrossRef]
- Warner, T.D.; Karnezos, M.; Friedberg, S.A.; Jafarey, S.; Wang, Y.-L. High-temperature series analysis of spin-one uniaxial ferromagnets: NiMF6·6H2O compounds. Phys. Rev. B 1981, 24, 2817–2824. [Google Scholar] [CrossRef]
- Iio, K.; Nagata, K. Observation of Tetragonal Optical Spectra of Divalent Nickel-Ion in Ba2NiF6. J. Phys. Soc. Jpn. 1976, 41, 1550–1555. [Google Scholar] [CrossRef]
- Nguyen, T.N.; Giaquinta, D.M.; zur Loye, H.-C. Synthesis of the New One-Dimentional Compound Sr3NiPtO6: Structure and Magnetic Properties. Chem. Mater. 1994, 6, 1642–1646. [Google Scholar] [CrossRef]
- Estes, W.E.; Weller, R.R.; Hatfield, W.E. Preparation and Magnetic Characterization of the Linear-Chain Series MIIL2Cl2 with M= Mn2+, Cu2+, and Ni2+ and L= 4-Phenylpyridine. Inorg. Chem. 1980, 19, 26–31. [Google Scholar] [CrossRef]
- Claridge, J.B.; Layland, R.C.; Henley, W.H.; zur Loye, H.-C. Crystal Growth and Magnetic Measurements on Aligned Single Crystals of the Oxides Sr3NiPtO6 and Sr3CuPtO6. Chem. Mater. 1999, 11, 1376–1380. [Google Scholar] [CrossRef]
- Stamatatos, T.C.; Escuer, A.; Abboud, K.A.; Raptopoulou, C.P.; Perlepes, S.P.; Christou, G. Unusual Structural Types in Nickel Cluster Chemistry from the Use of Pyridyl Oximes: Ni5, Ni12Na2, and Ni14 Clusters. Inorg. Chem. 2008, 47, 11825–11838. [Google Scholar] [CrossRef]
- Alexopoulou, K.I.; Terzis, A.; Raptopoulou, C.P.; Psycharis, V.; Escuer, A.; Perlepes, S.P. NiII20 “Bowls” from the use of Tridentate Schiff Bases. Inorg. Chem. 2015, 54, 5615–5617. [Google Scholar] [CrossRef]
- Carlin, R.L. Magnetochemistry; Springer: Berlin, Germany, 1986. [Google Scholar]
- CrystalClear; ver. 1.40; Rigaku/MSC: The Woodlands, TX, USA, 2005.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C 2015, 71, 3–8. [Google Scholar]
- DIAMOND, Crystal and Molecular Structure Visualization; ver. 3.1; Crystal Impact: Bonn, Germany, 2018.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interatomic Distances (Å) | Bond Angles (°) | ||
|---|---|---|---|
| Cu1-N1 | 2.143(5) | N2-Cu1-N5 | 172.7(2) |
| Cu1-N2 | 1.989(5) | O2-Cu1-Br1 | 147.7(1) |
| Cu1-O2 | 2.073(4) | N1-Cu1-Br1 | 117.0(1) |
| Cu1-N5 | 1.994(5) | N1-Cu1-N2 | 80.3(2) |
| Cu1-Br1 | 2.450(1) | N1-Cu1-N5 | 97.0(2) |
| Cu2-N7 | 1.980(5) | N1-Cu1-O2 | 95.3(2) |
| Cu2-N8 | 2.193(5) | N2-Cu1-O2 | 93.4(2) |
| Cu2-O2 | 2.331(4) | N5-Cu1-Br1 | 95.1(1) |
| Cu2-N3′ | 1.975(5) | N3′-Cu2-N7 | 175.5(2) |
| Cu2-N4′ | 2.054(5) | N8-Cu2-O2 | 148.2(2) |
| Cu2-O1′ | 2.117(4) | N4′-Cu2-O1′ | 155.4(2) |
| C6-O1 | 1.257(7) | N3′-Cu2-N4′ | 79.1(2) |
| C6-N2 | 1.346(8) | N7-Cu2-N8 | 77.4(2) |
| N2-N3 | 1.384(7) | O2-C19-N6 | 126.8(5) |
| N3-C8 | 1.290(8) | C19-N6-N7 | 110.3(5) |
| C19-O2 | 1.287(7) | N6-N7-C21 | 118.1(5) |
| C19-N6 | 1.331(7) | O1-C6-N2 | 125.0(5) |
| N6-N7 | 1.401(6) | C6-N2-N3 | 110.2(5) |
| N7-C21 | 1.281(7) | N2-N3-C8 | 123.7(5) |
| Interatomic Distances (Å) | Bond Angles (°) | ||
|---|---|---|---|
| Ni1⋯Ni2 | 3.922(1) | O1-Ni1-N4 | 154.1(1) |
| Ni1⋯N3 | 3.938(1) | O2-Ni1-N8 | 153.9(1) |
| Ni1⋯N4 | 5.526(1) | N3-Ni1-N7 | 177.9(1) |
| Ni2⋯Ni3 | 5.559(1) | O1-Ni1-O2 | 91.4(1) |
| Ni2⋯Ni4 | 3.921(1) | O2-Ni1-N3 | 105.3(1) |
| Ni3⋯Ni4 | 3.895(1) | N3-Ni1-N4 | 78.0(1) |
| Ni1-O1 | 2.147(2) | O1-Ni2-O5 | 158.2(1) |
| Ni1-O2 | 2.144(2) | O3-Ni2-O6 | 165.1(1) |
| Ni1-N3 | 1.988(2) | N1-Ni2-N9 | 177.5(1) |
| Ni1-N4 | 2.083(2) | O1-Ni2-O3 | 95.0(1) |
| Ni1-N7 | 1.984(2) | O5-Ni2-O6 | 60.7(1) |
| Ni1-N8 | 2.107(2) | O6-Ni2-N9 | 93.3(1) |
| Ni2-O1 | 2.049(2) | O2-Ni3-O1W | 169.9(1) |
| Ni2-O3 | 2.054(2) | O4-Ni3-O8 | 171.1(1) |
| Ni2-O5 | 2.161(2) | N5-Ni3-N13 | 179.1(1) |
| Ni2-O6 | 2.113(2) | O2-Ni3-O4 | 90.5(1) |
| Ni2-N1 | 2.041(2) | O1W-Ni3-O8 | 93.9(1) |
| Ni2-N9 | 2.036(2) | O8-Ni3-N5 | 86.8(1) |
| Ni3-O2 | 2.068(2) | O3-Ni4-N12 | 154.5(1) |
| Ni3-O4 | 2.052(2) | O4-Ni4-N16 | 154.7(1) |
| Ni3-O8 | 2.121(2) | N11-Ni4-N15 | 174.5(1) |
| Ni3-O1W | 2.080(2) | O3-Ni4-O4 | 94.4(1) |
| Ni3-N5 | 2.048(2) | N11-Ni4-N12 | 78.5(1) |
| Ni3-N13 | 2.055(2) | N15-Ni4-N16 | 78.4(1) |
| Ni4-O3 | 2.144(2) | O8-N18-O9 | 120.7(3) |
| Ni4-O4 | 2.126(2) | O8-N18-O10 | 118.8(3) |
| Ni4-N11 | 1.987(2) | O9-N18-O10 | 120.5(3) |
| Ni4-N12 | 2.093(2) | O5-N17-O6 | 116.2(3) |
| Ni4-N15 | 1.979(2) | O5-N17-O7 | 122.2(4) |
| Ni4-N16 | 2.091(2) | O6-N17-O7 | 121.6(4) |
| Bond Lengths (Å) a | Bond Angles (°) a | ||
|---|---|---|---|
| Co1-O1 | 1.899(2) | O1-Co1-N1 | 165.2(1) |
| Co1-N1 | 1.927(2) | O2-Co1-N5 | 165.4(1) |
| Co1-N2 | 1.855(2) | N2-Co1-N6 | 174.7(1) |
| Co1-O2 | 1.892(2) | O1-Co1-N2 | 82.5(1) |
| Co1-N5 | 1.915(2) | O1-Co1-O2 | 91.3(1) |
| Co1-N6 | 1.857(2) | O2-Co1-N6 | 82.6(1) |
| C8-O1 | 1.290(3) | N2-Co1-N5 | 100.7(1) |
| C8-N3 | 1.329(3) | O1-C8-N3 | 125.1(2) |
| N3-N2 | 1.391(3) | C8-N3-N2 | 106.3(2) |
| N2-C6 | 1.298(3) | N3-N2-C6 | 123.7(2) |
| C21-O2 | 1.306(3) | O2-C21-N7 | 124.2(2) |
| C21-N7 | 1.317(3) | C21-N7-N6 | 107.3(2) |
| N7-N6 | 1.383(3) | N7-N6-C19 | 123.9(2) |
| N6-C19 | 1.299(3) | O3-Cl1-O4 | 109.7(1) |
| Cl1-O3 | 1.440(2) | O3-Cl1-O6 | 110.7(1) |
| Cl1-O4 | 1.435(2) | O4-Cl1-O5 | 108.8(1) |
| Cl1-O5 | 1.442(2) | O5-Cl1-O6 | 108.8(1) |
| Cl1-O6 | 1.428(2) | ||
| Interatomic Distances (Å) | Bond Angles (°) | ||
|---|---|---|---|
| Co1⋯Co2 | 3.467(2) | N1-Co1-N11 | 170.6(3) |
| Co1-N1 | 1.953(6) | N2-Co1-N5 | 172.5(3) |
| Co1-N2 | 1.906(6) | N6-Co1-N12 | 172.0(3) |
| Co1-N5 | 1.951(6) | N1-Co1-N2 | 81.3(3) |
| Co1-N6 | 1.899(6) | N2-Co1-N6 | 92.0(3) |
| Co1-N11 | 1.910(6) | N5-Co1-N6 | 81.6(3) |
| Co1-N12 | 1.931(6) | N11-Co1-N12 | 82.1(3) |
| Co2-N3 | 1.900(6) | N3-Co2-N9 | 170.3(3) |
| Co2-N4 | 1.935(6) | N4-Co2-N7 | 171.1(2) |
| Co2-N7 | 1.901(6) | N8-Co2-N10 | 171.4(2) |
| Co2-N8 | 1.946(6) | N3-Co2-N4 | 81.7(2) |
| Co2-N9 | 1.942(7) | N4-Co2-N8 | 93.1(2) |
| Co2-N10 | 1.909(6) | N7-Co2-N8 | 81.6(3) |
| C6-O1 | 1.242(9) | N8-Co2-N9 | 94.1(3) |
| C6-N2 | 1.363(9) | O1-C6-N2 | 126.9(7) |
| N2-N3 | 1.407(7) | C6-N2-N3 | 114.6(6) |
| N3-C7 | 1.310(8) | N2-N3-C7 | 121.6(6) |
| C19-O2 | 1.319(9) | O2-C19-N6 | 126.1(8) |
| C19-N6 | 1.311(8) | C19-N6-N7 | 119.4(6) |
| N6-N7 | 1.384(8) | N6-N7-C20 | 120.5(6) |
| N7-C20 | 1.331(9) | O3-C32-N10 | 124.9(9) |
| C32-O3 | 1.253(9) | C32-N10-N11 | 117.9(6) |
| C32-N10 | 1.348(8) | N10-N11-C33 | 121.3(6) |
| N10-N11 | 1.400(8) | ||
| N11-C33 | 1.292(8) | ||
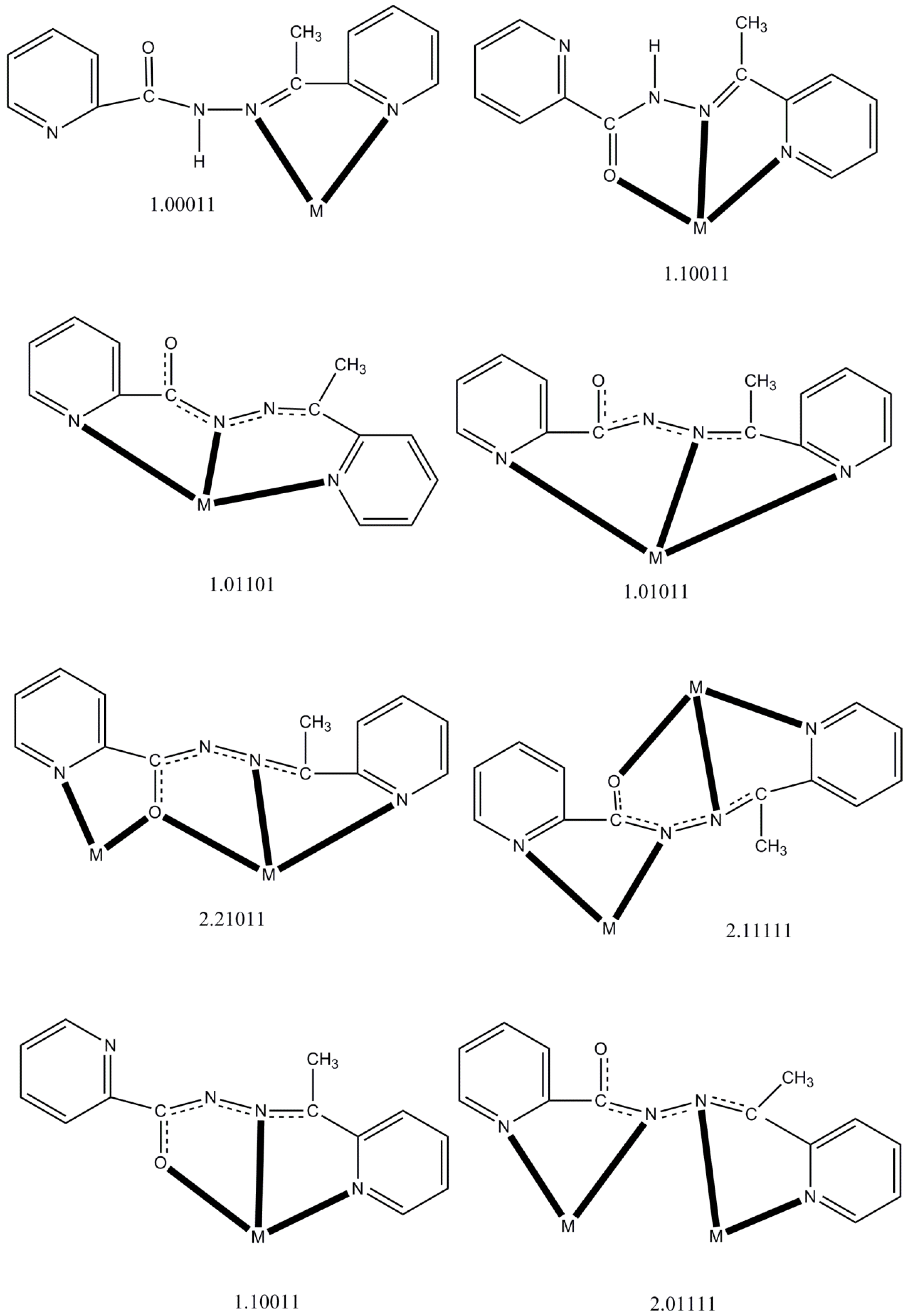
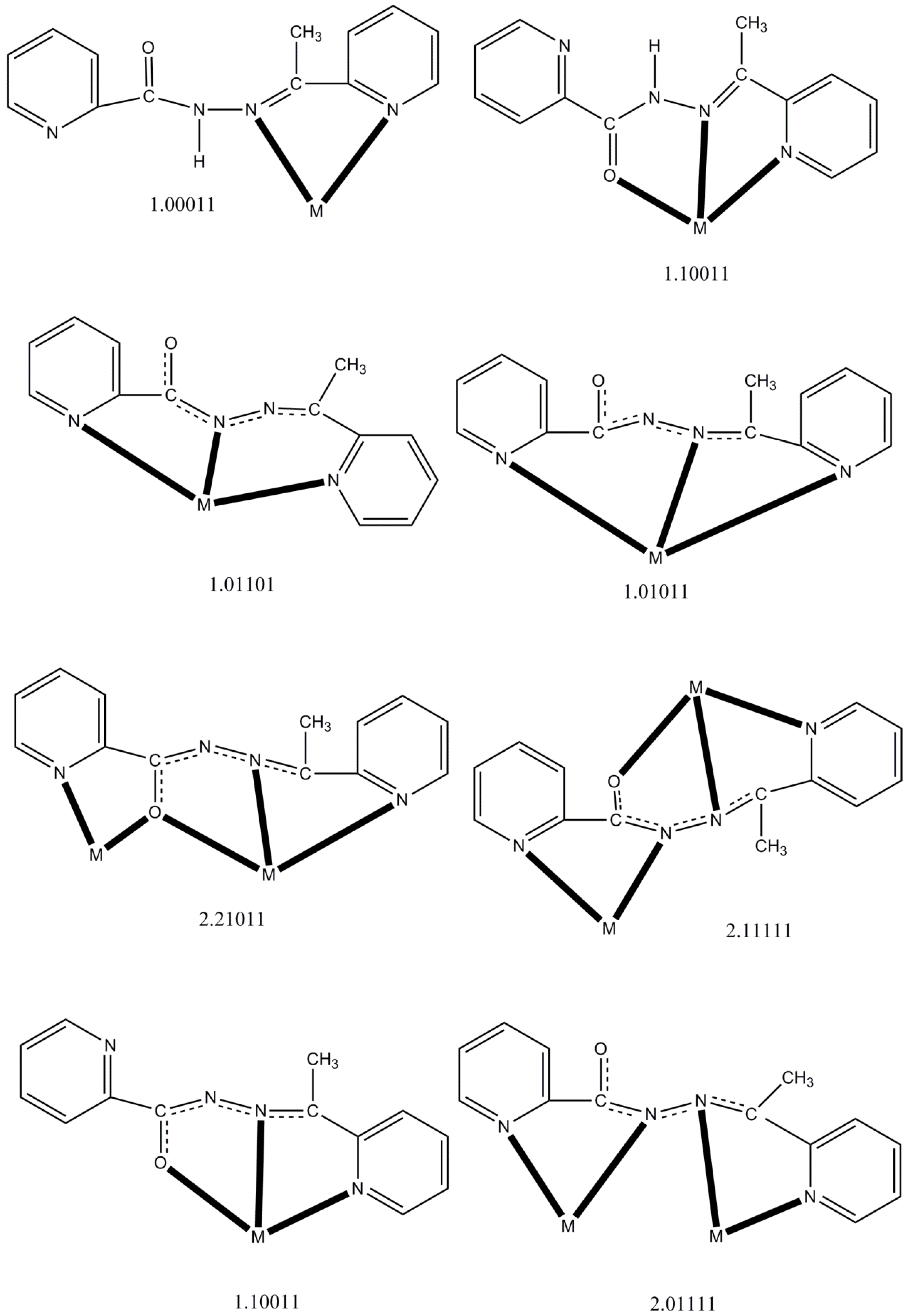
| Complex a | Coordination Mode b,c | Nuclearity/Dimensionality | Coordination Geometry | Ref. |
|---|---|---|---|---|
| [CdBr2(LH)] | η1:η1:η1(1.10011) | Mononuclear | Trigonal bipyramidal | [16] |
| [Ln(NO3)3(LH)(MeOH)2] (Ln = La, Ce) | η1:η1:η1(1.10011) | Mononuclear | Capped pentagonal antiprismatic | [17] |
| [Nd(NO3)3(LH)(H2O)] | η1:η1:η1(1.10011) | Mononuclear | Bicapped square antiprismatic | [17] |
| [PdCl2(LH)] | η1:η1(1.01100) | Mononuclear | Square planar | [18] |
| [HgX2(LH)] (X = Cl, Br) | η1:η1:η1(1.10011) | Mononuclear | Square pyramidal | [20] |
| [HgI2(LH)(H2O)] | η1:η1:η1(1.10011) | Mononuclear | Octahedral | [20] |
| {[Pb3Br6(LH)2]}n | η1:η1:η1(1.10011) | 1D metal-organic ribbon | 7-coordinate d, Octahedral | [21] |
| [PdCl(L)] | η1:η1:η1(1.01011) | Mononuclear | Square planar | [18] |
| [PdCl(L)] | η1:η1:η1(1.01101) | Mononuclear | Square planar | [18] |
| [Cu4(L)4(H2O)2](NO3)4 | η1:η2:η1:η1:μ2(2.21011), η1:η1:η1:η1:η1:μ2(2.11111) | Rectangular [2 × 2] grid | Square pyramidal, Octahedral | [14] |
| [Mn4(CF3SO3)(L)4(H2O)3](CF3SO3)3 | η1:η2:η1:η1:μ2(2.21011) | Square [2 × 2] grid | Octahedral | [15] |
| [Mn5(L)6](ClO4)4 | η1:η2:η1:η1:μ2(2.21011) | Trigonal bipyramidal {Mn5(μ2-OR)6}4+core | Octahedral | [15] |
| [Mn4(N3)4(L)4] | η1:η2:η1:η1:μ2(2.21011) | Square [2 × 2] grid | Octahedral | [19] |
| [Cu4Br2(L)4]Br2 (1) | η1:η2:η1:η1:μ2(2.21011), η1:η1:η1:η1:η1:μ2(2.11111) | Rectangular [2 × 2] grid | Square pyramidal, Octahedral | this work |
| [Ni4(NO3)2(L)4(H2O)](NO3)2 (2) | η1:η2:η1:η1:μ2(2.21011) | Square [2 × 2] grid | Octahedral | this work |
| [Co2(L)3](ClO4)3 (3) | η1:η1:η1:η1:μ2(2.01111) | Dinuclear helicate | Octahedral | this work |
| [Co(L)2](ClO4) (4) | η1:η1:η1(1.10011) | Mononuclear | Octahedral | this work |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilichos, E.; Spanakis, E.; Maniaki, E.-K.; Raptopoulou, C.P.; Psycharis, V.; Turnbull, M.M.; Perlepes, S.P. Diversity of Coordination Modes in a Flexible Ditopic Ligand Containing 2-Pyridyl, Carbonyl and Hydrazone Functionalities: Mononuclear and Dinuclear Cobalt(III) Complexes, and Tetranuclear Copper(II) and Nickel(II) Clusters. Magnetochemistry 2019, 5, 39. https://doi.org/10.3390/magnetochemistry5030039
Pilichos E, Spanakis E, Maniaki E-K, Raptopoulou CP, Psycharis V, Turnbull MM, Perlepes SP. Diversity of Coordination Modes in a Flexible Ditopic Ligand Containing 2-Pyridyl, Carbonyl and Hydrazone Functionalities: Mononuclear and Dinuclear Cobalt(III) Complexes, and Tetranuclear Copper(II) and Nickel(II) Clusters. Magnetochemistry. 2019; 5(3):39. https://doi.org/10.3390/magnetochemistry5030039
Chicago/Turabian StylePilichos, Evangelos, Evangelos Spanakis, Evangelia-Konstantina Maniaki, Catherine P. Raptopoulou, Vassilis Psycharis, Mark M. Turnbull, and Spyros P. Perlepes. 2019. "Diversity of Coordination Modes in a Flexible Ditopic Ligand Containing 2-Pyridyl, Carbonyl and Hydrazone Functionalities: Mononuclear and Dinuclear Cobalt(III) Complexes, and Tetranuclear Copper(II) and Nickel(II) Clusters" Magnetochemistry 5, no. 3: 39. https://doi.org/10.3390/magnetochemistry5030039
APA StylePilichos, E., Spanakis, E., Maniaki, E.-K., Raptopoulou, C. P., Psycharis, V., Turnbull, M. M., & Perlepes, S. P. (2019). Diversity of Coordination Modes in a Flexible Ditopic Ligand Containing 2-Pyridyl, Carbonyl and Hydrazone Functionalities: Mononuclear and Dinuclear Cobalt(III) Complexes, and Tetranuclear Copper(II) and Nickel(II) Clusters. Magnetochemistry, 5(3), 39. https://doi.org/10.3390/magnetochemistry5030039