DFT Investigations of the Magnetic Properties of Actinide Complexes




{kind=link}
{kind=link}
{kind=link}
{kind=link}
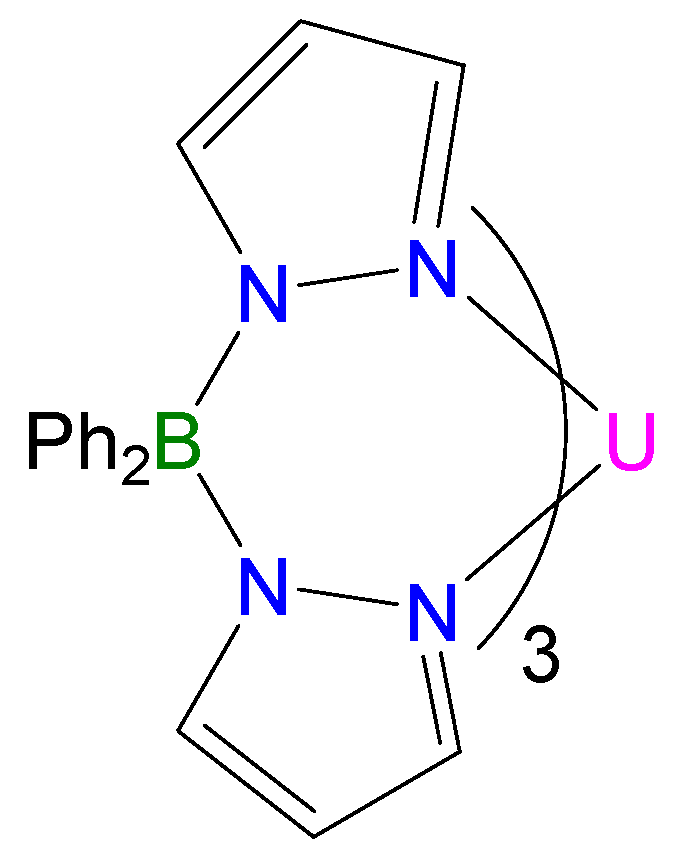{kind=link}
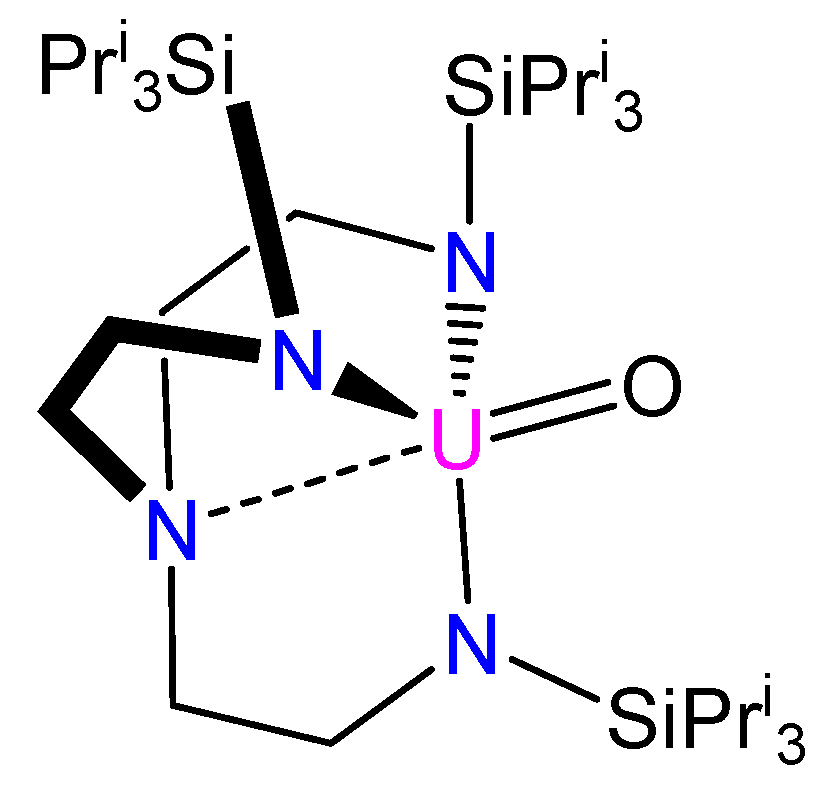{kind=link}
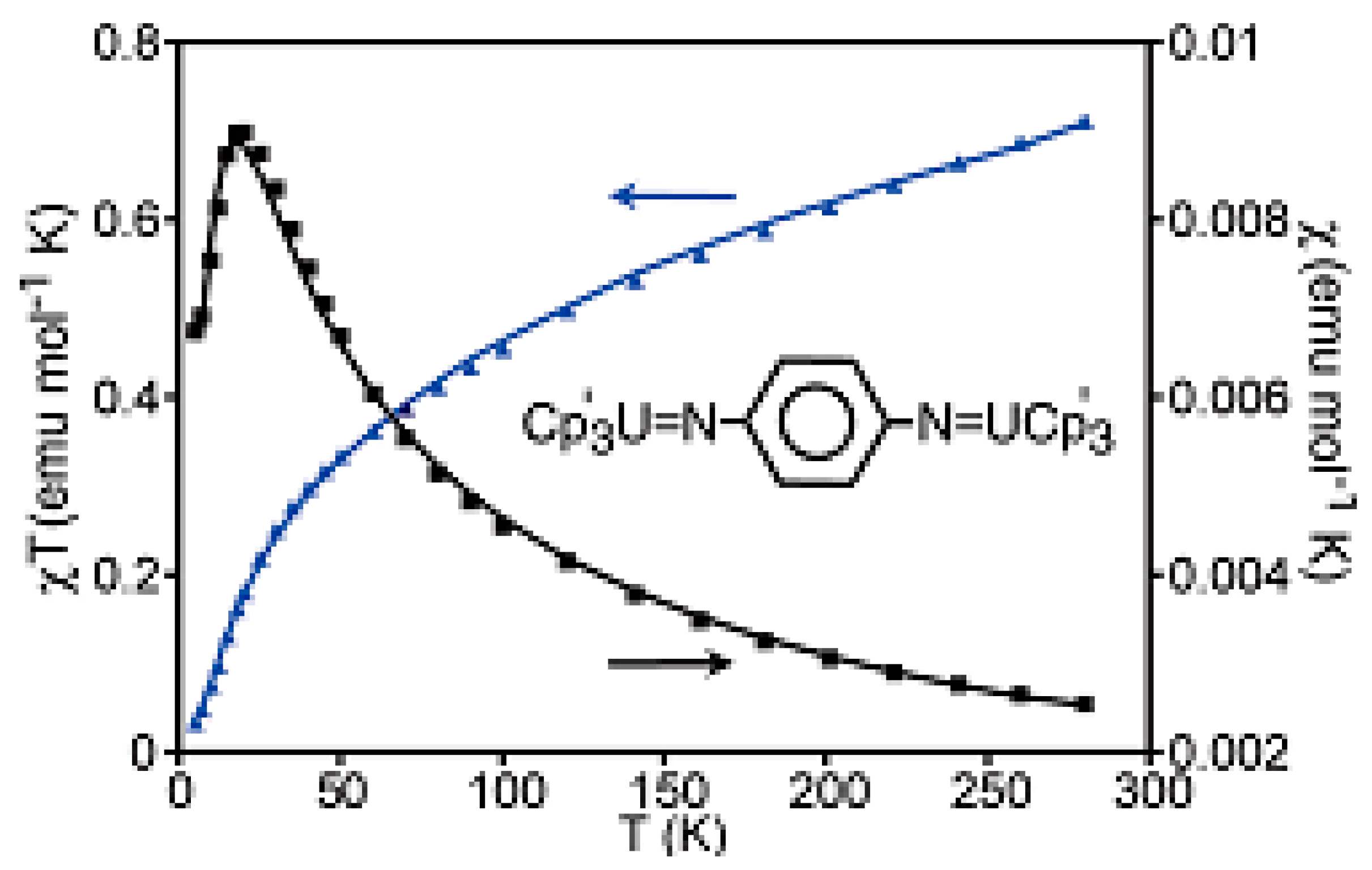{kind=link}
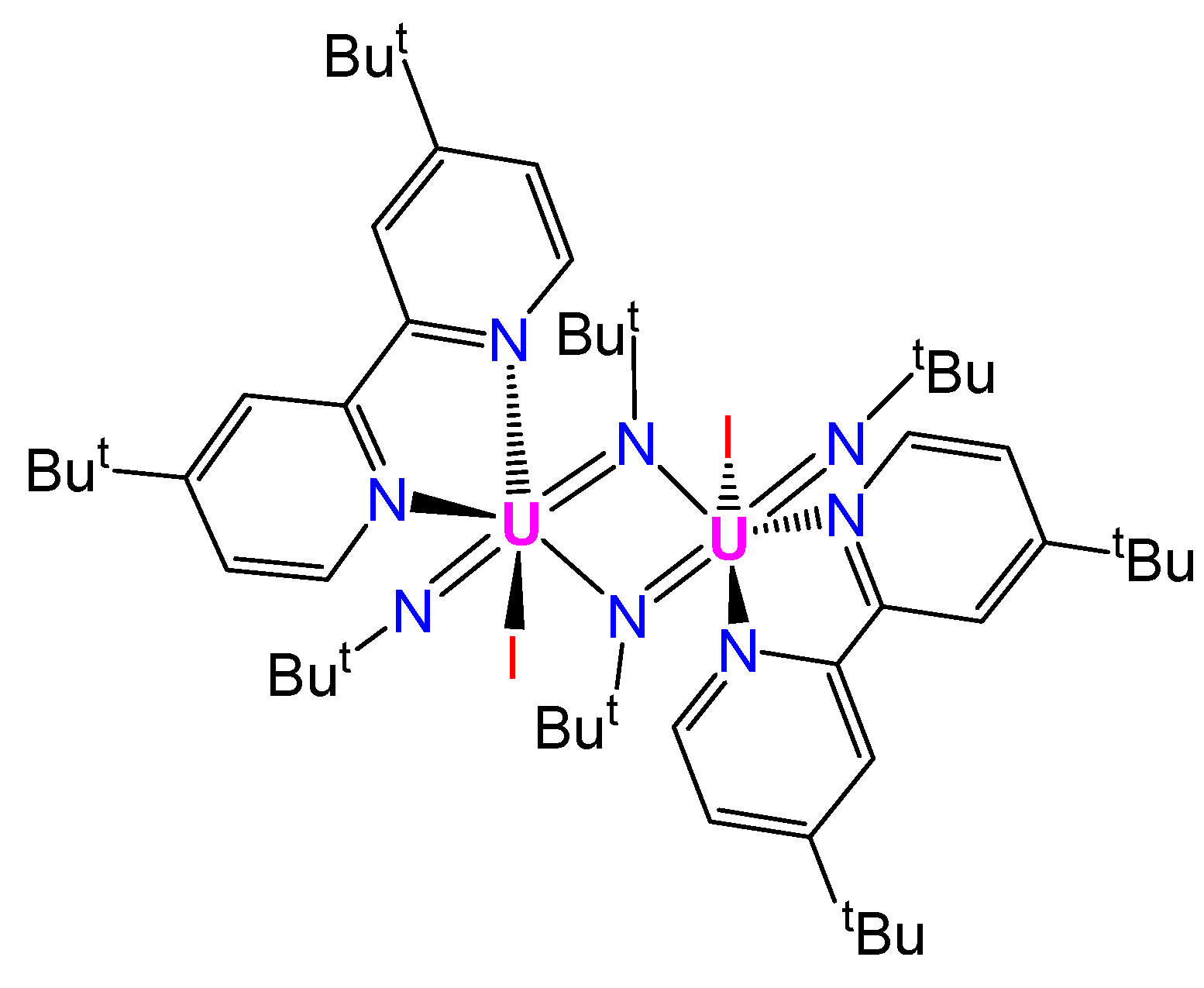{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
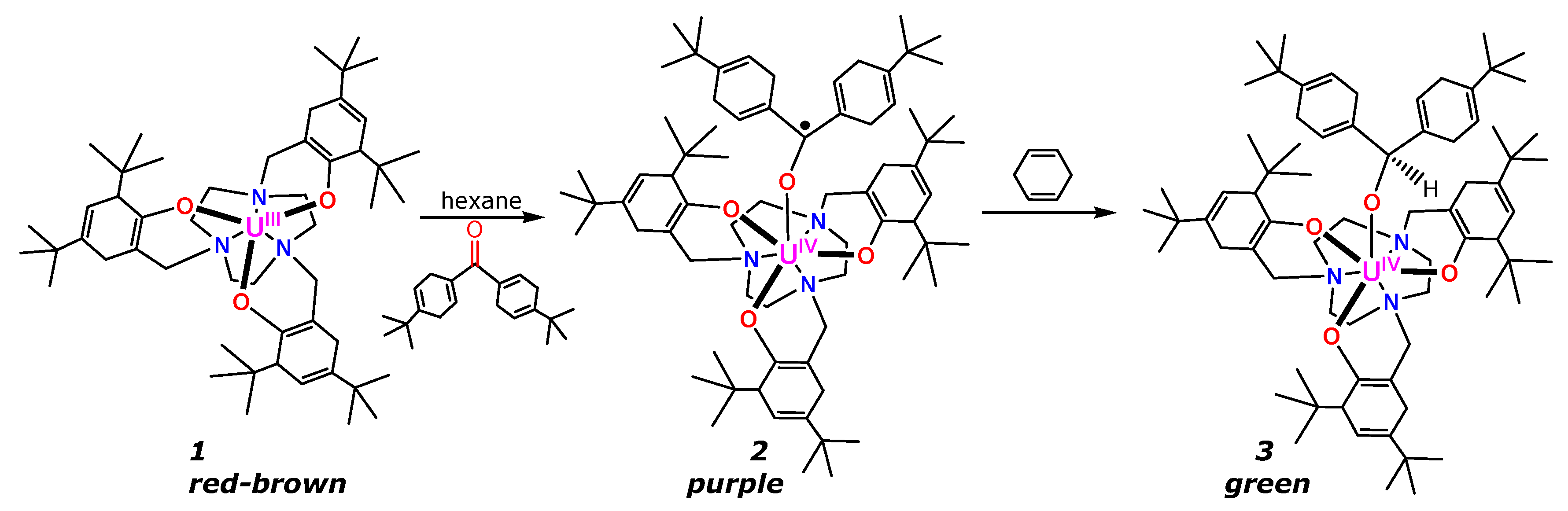{kind=link}
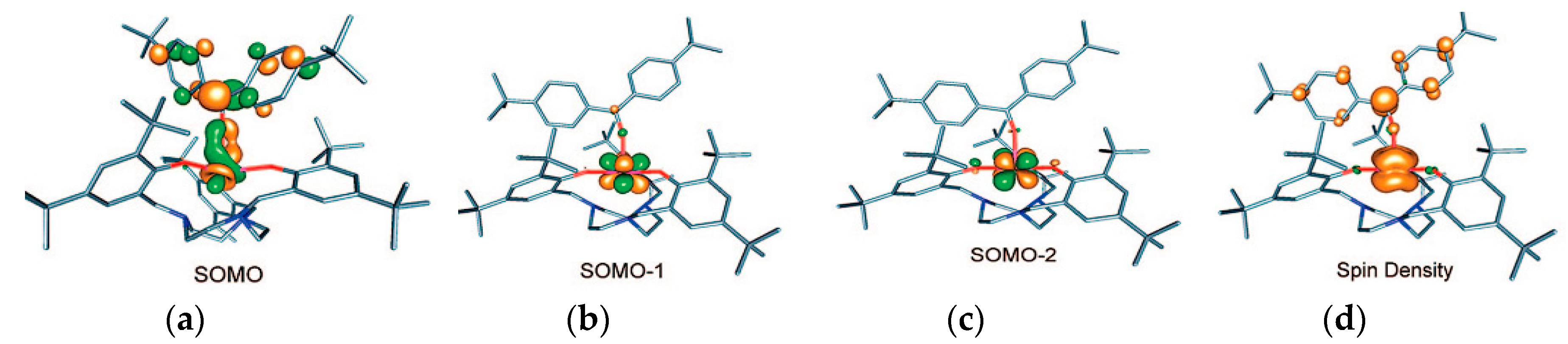{kind=link}
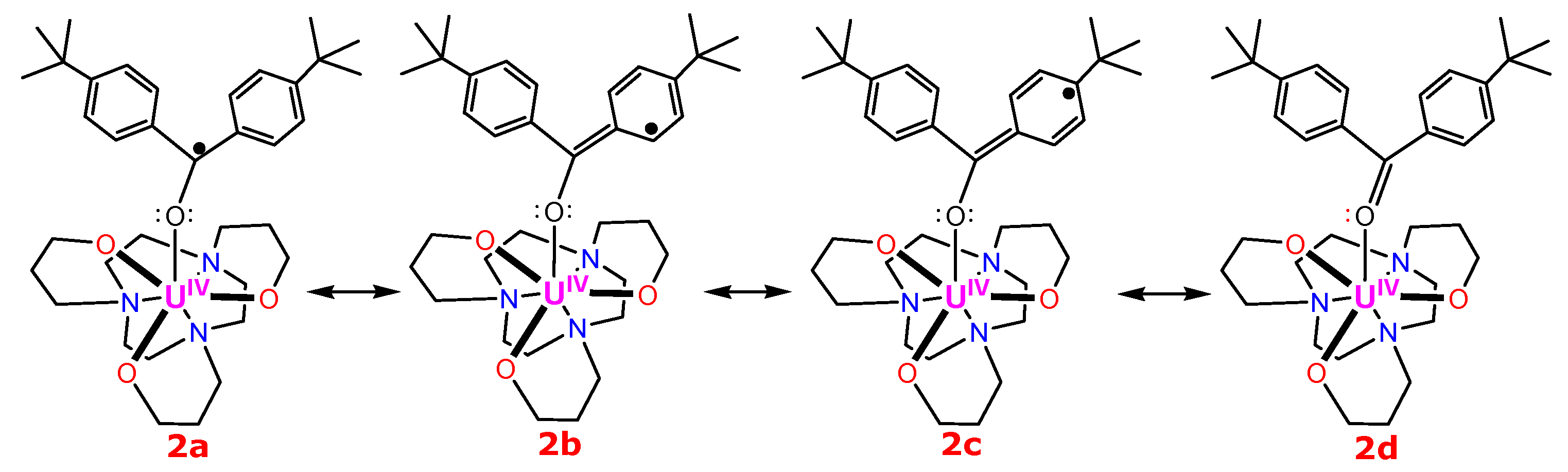{kind=link}
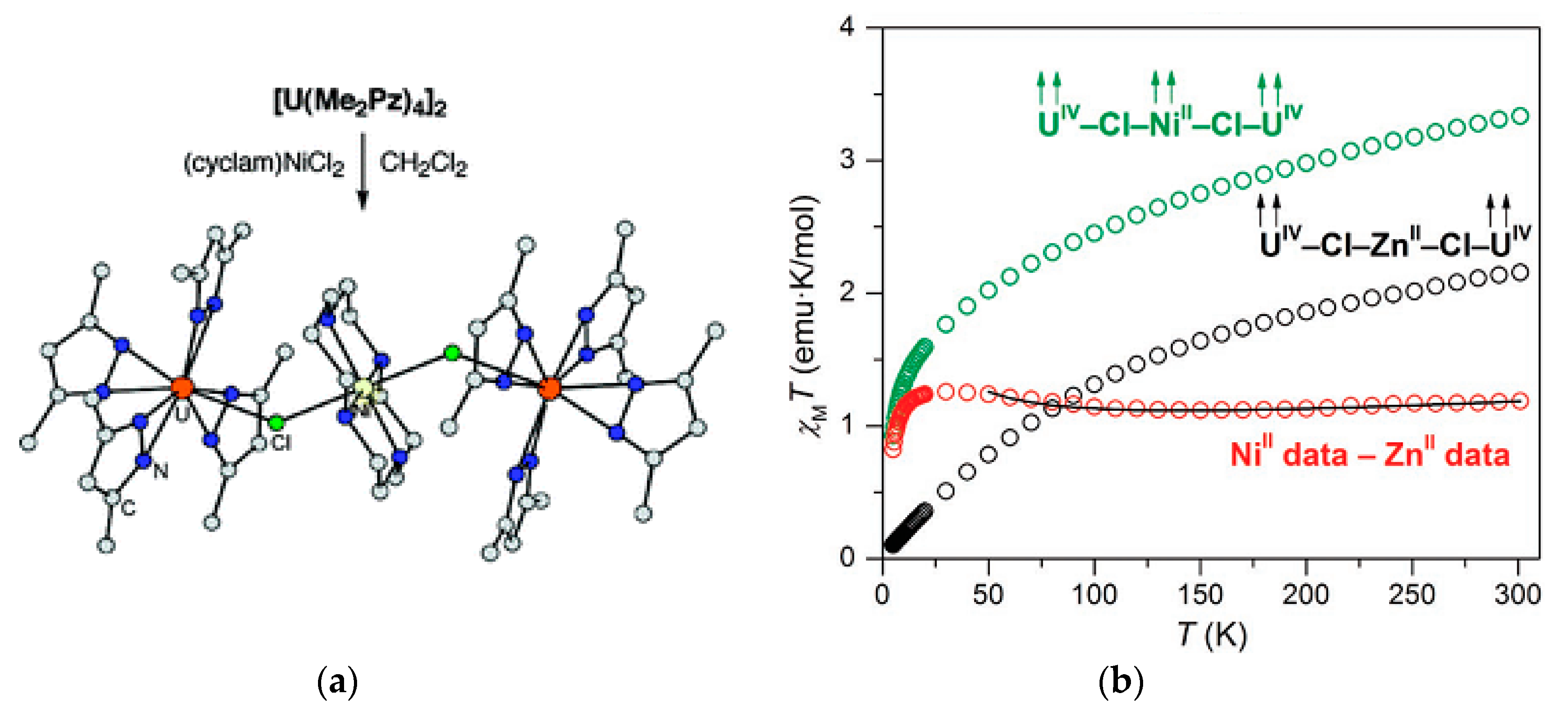{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
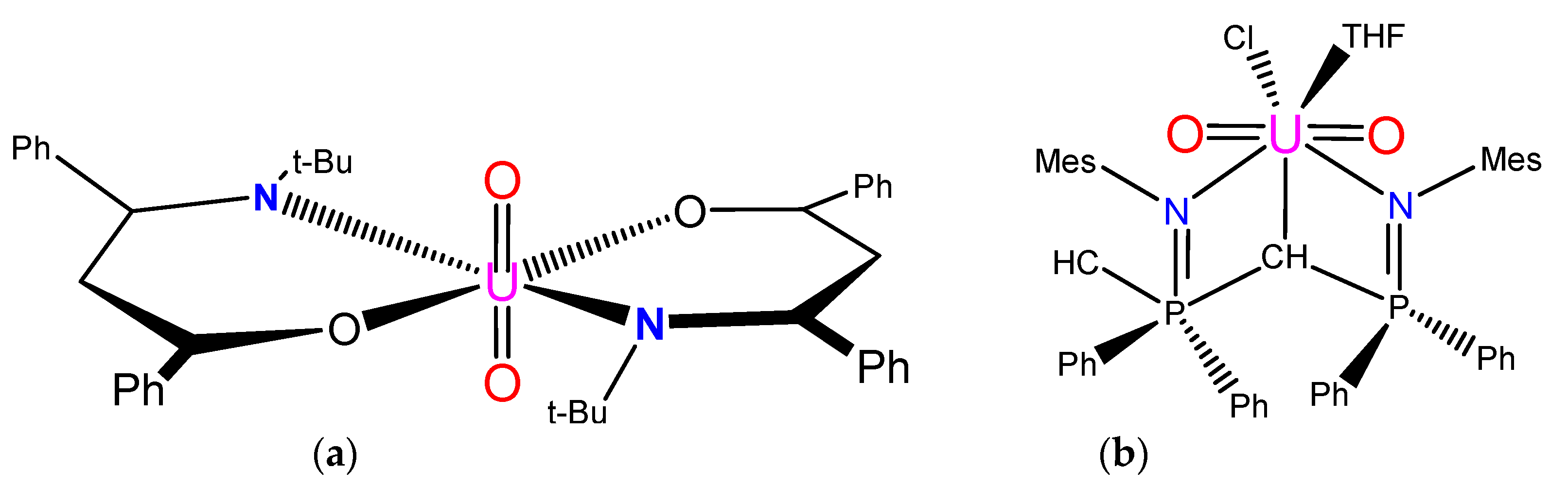{kind=link}
Abstract
1. Introduction
2. Survey of Molecules Potentially Exhibiting Magnetic Behavior
2.1. Magnetic Coupling Interactions
2.2. SMM Behavior
3. DFT Investigations of Actinide Complexes Magnetism
3.1. Theoretical Approaches for Computing Exchange Coupling Constants
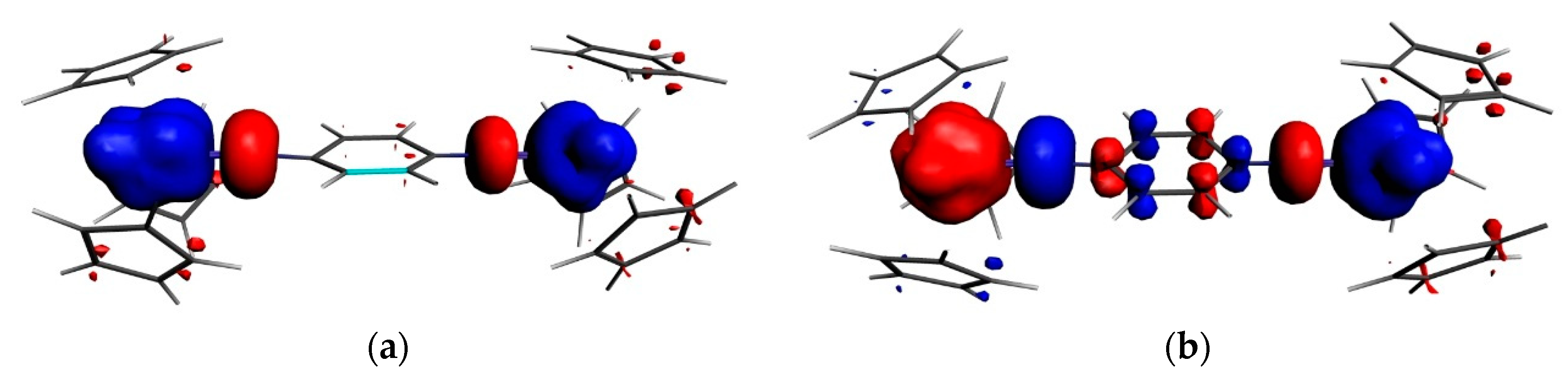
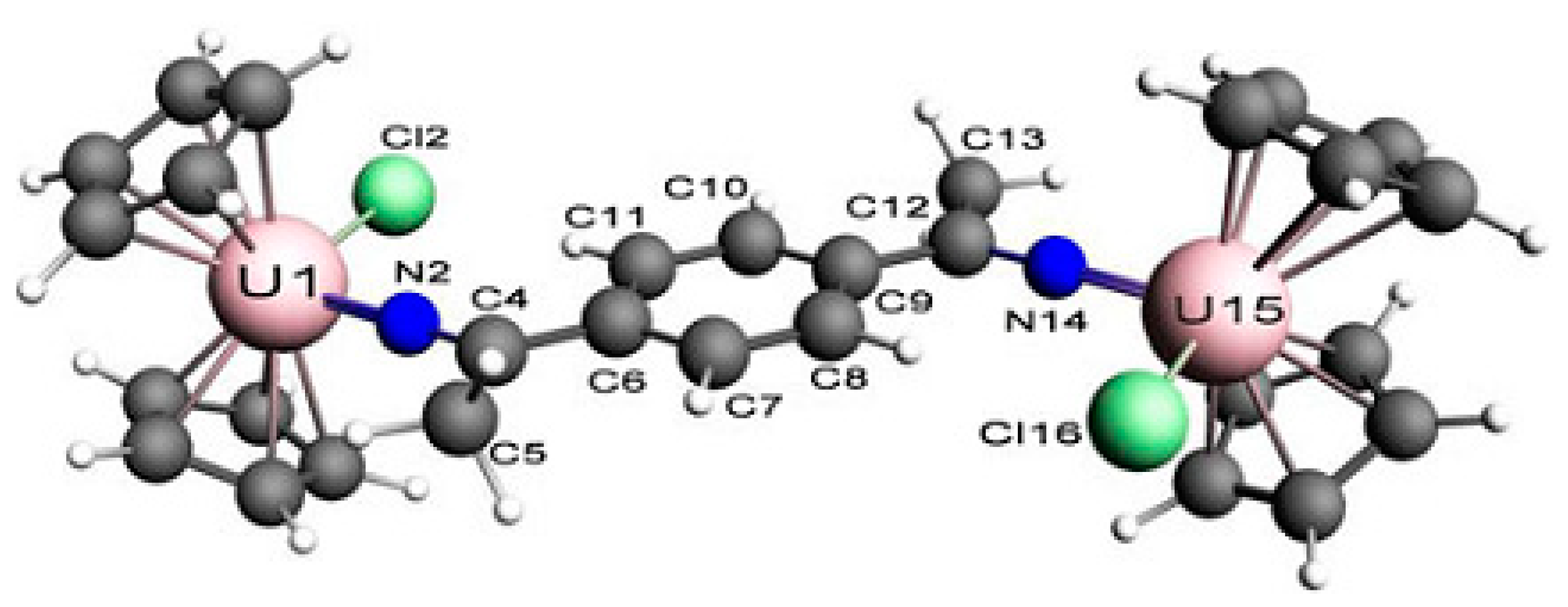
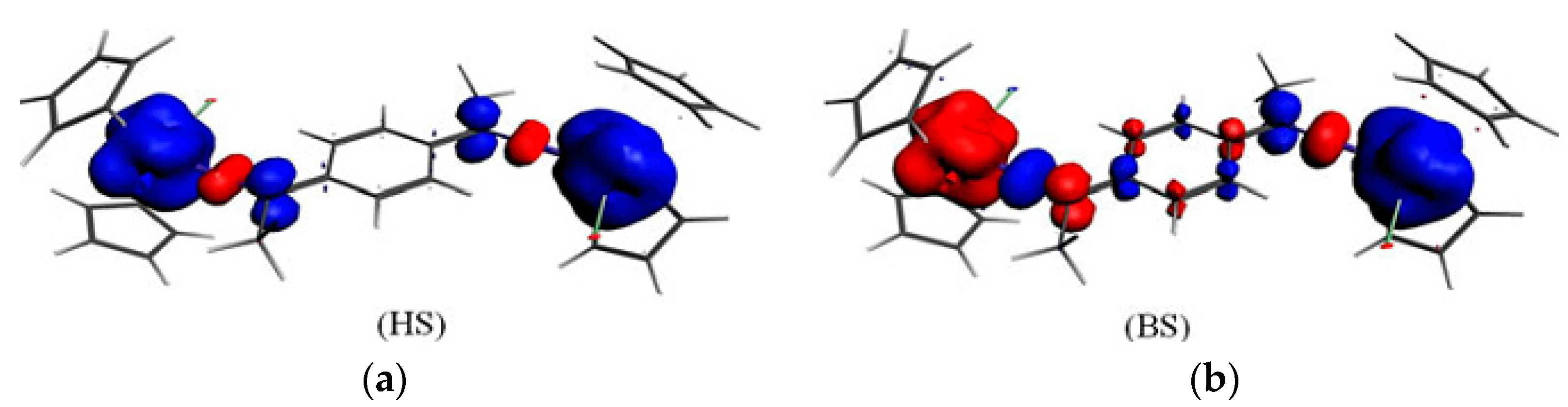
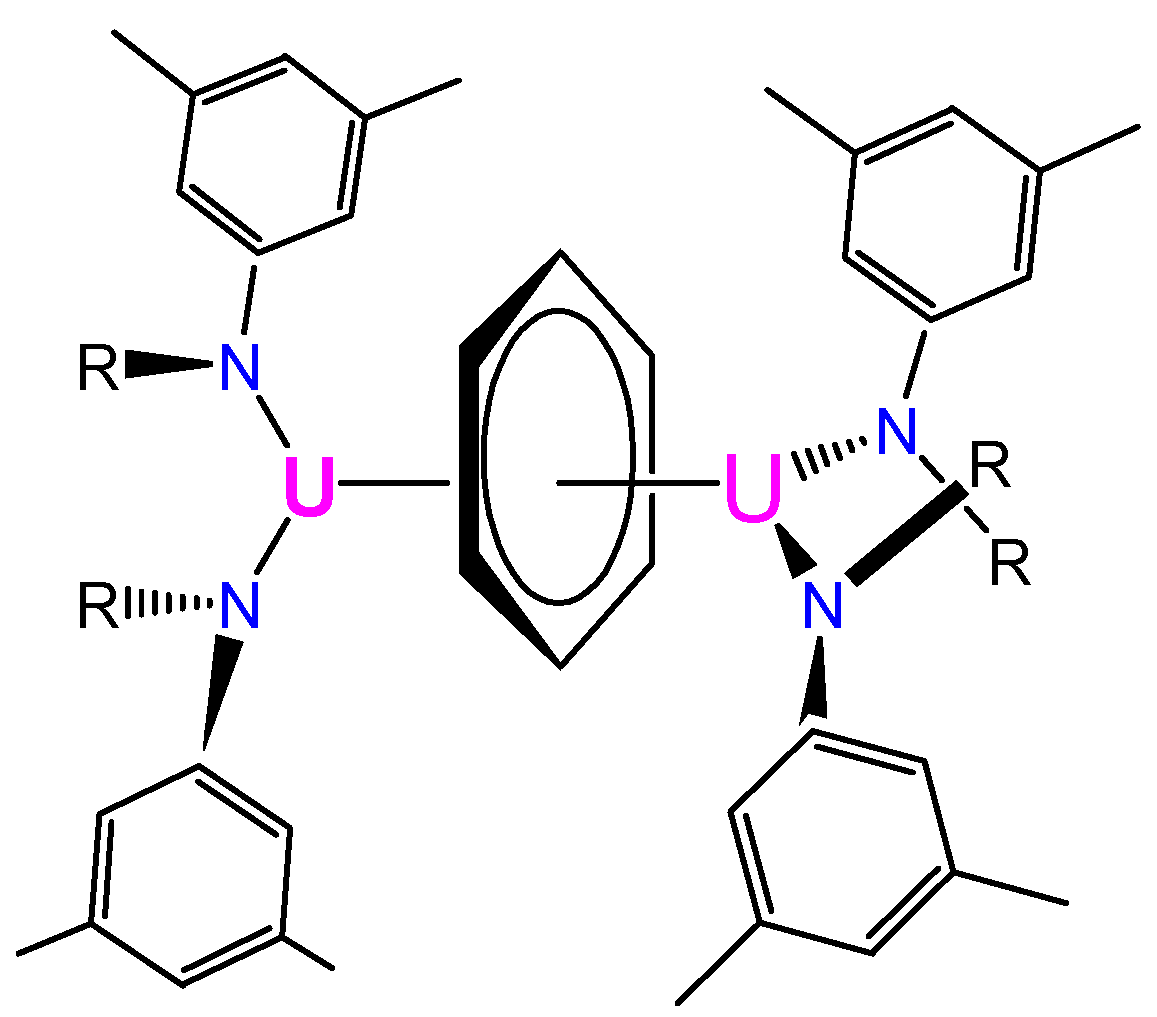
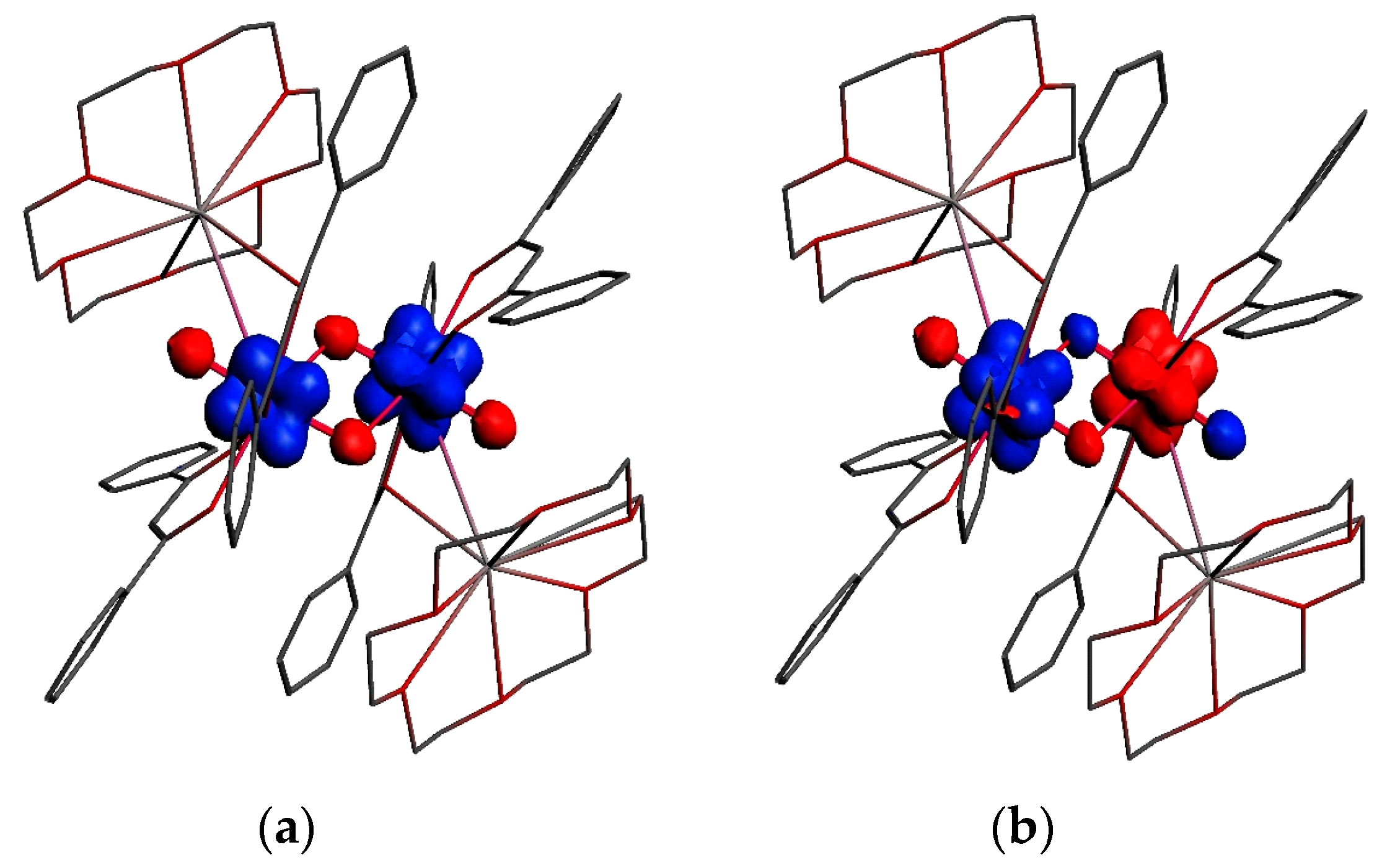
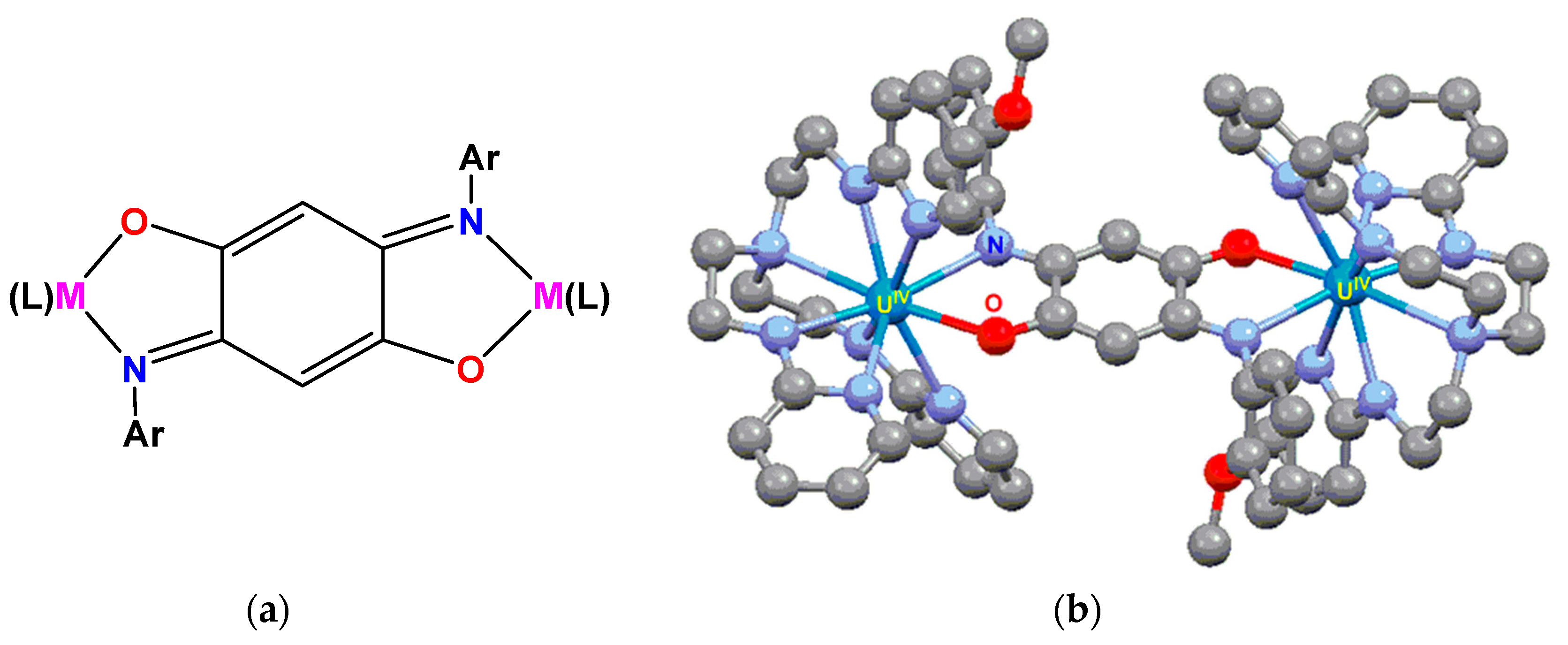
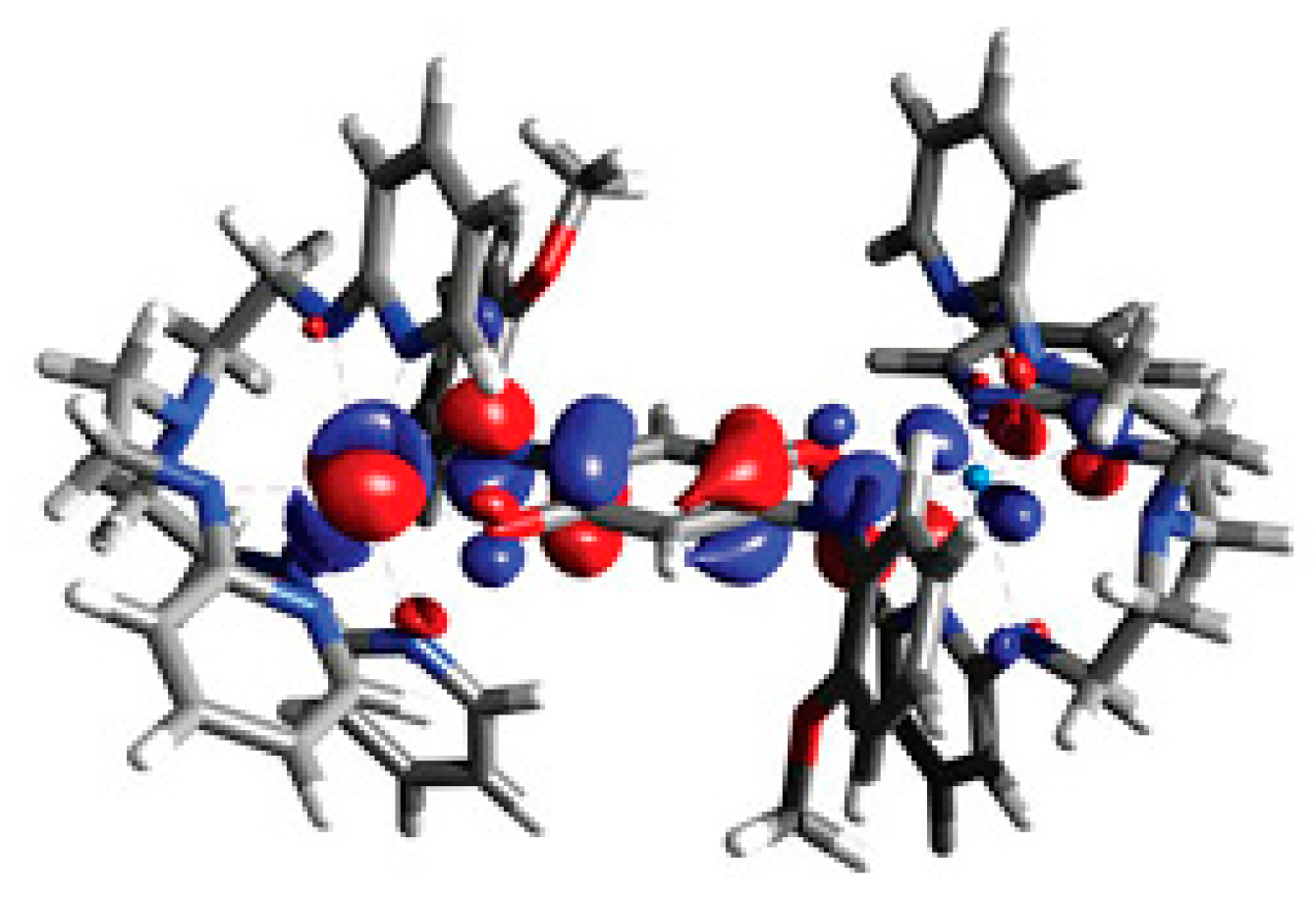
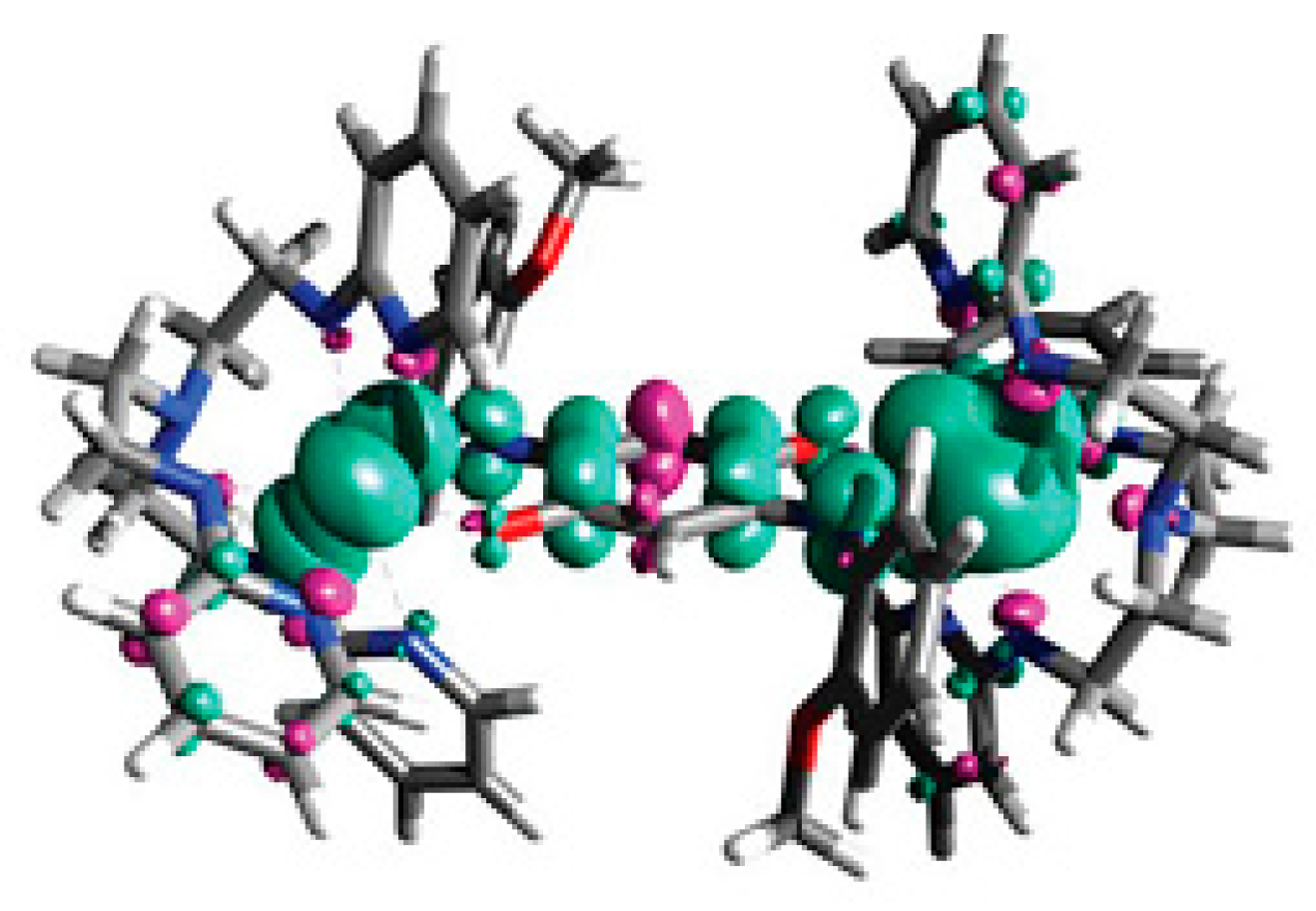
3.2. Magnetic Exchange Coupling in Actinide Bimetallic Systems
3.3. Mononuclear Actinide Complexes
3.4. Mixed Actinide/Transition Metal (5f-3d) and Actinide/Lanthanide (5f-4f) Complexes
3.5. Magnetic Susceptibility and EPR/NMR Spectra of Actinide Complexes
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rinehart, J.D.; Harris, T.D.; Kozimor, S.A.; Bartlett, B.M.; Long, J.R. Magnetic Exchange Coupling in Actinide-Containing Molecules. Inorg. Chem. 2009, 48, 3382–3395. [Google Scholar] [CrossRef] [PubMed]
- Lukens, W.W.; Walter, M.D. Quantifying Exchange Coupling in f-Ion Pairs Using the Diamagnetic Substitution Method. Inorg. Chem. 2010, 49, 4458–4465. [Google Scholar] [CrossRef] [PubMed]
- Liddle, S.T.; van Slageren, J. Lanthanides and Actinides. In Molecular Magnetism; Layfield, R.A., Murugesu, M., Eds.; Wiley-VCH: Weinheim, Germany, 2015; pp. 315–340. [Google Scholar]
- Feng, M.; Tong, M.-L. Single Ion Magnets from 3d to 5f: Developmentsand Strategies. Chem. Eur. J. 2018, 24, 7574–7594. [Google Scholar] [CrossRef] [PubMed]
- Boucekkine, A.; Belkhiri, L. f-Element Complexes. In Comprehensive Inorganic Chemistry II; Reedijk, J., Poeppelmeier, K., Eds.; Elsevier: Oxford, UK, 2013; Volume 9, pp. 277–319. [Google Scholar]
- Kindra, D.R.; Evans, W. Magnetic Susceptibility of Uranium Complexes. J. Chem. Rev. 2014, 114, 8865–8882. [Google Scholar] [CrossRef] [PubMed]
- Magnani, N. Spectroscopic and magnetic investigations of actinide-based nanomagnets. Int. J. Quantum Chem. 2014, 114, 755–759. [Google Scholar] [CrossRef]
- Kaltsoyannis, N. Transuranic Computational Chemistry. Chem. Eur. J. 2018, 24, 2815–2825. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.K.; Andersen, R.A.; Edelstein, N.M.J. [(MeC5H4)3U]2[μ.-1, 4-N2C6H4]: A bimetallic molecule with antiferromagnetic coupling between the uranium centers. J. Am. Chem. Soc. 1990, 112, 4588–4590. [Google Scholar] [CrossRef]
- Diaconescu, P.L.; Arnold, P.L.; Baker, T.A.; Mindiola, D.J.; Cummins, C.C. Arene-Bridged Diuranium Complexes: Inverted Sandwiches Supported by δ Backbonding. J. Am. Chem. Soc. 2000, 122, 6108–6109. [Google Scholar] [CrossRef]
- Diaconescu, P.L.; Cummins, C.C. Diuranium Inverted Sandwiches: Involving Naphthalene and Cyclooctatetraene. J. Am. Chem. Soc. 2002, 124, 7660–7661. [Google Scholar] [CrossRef]
- Odom, A.L.; Arnold, P.L.; Cummins, C.C. Heterodinuclear uranium/molybdenum dinitrogen complexes. J. Am. Chem. Soc. 1998, 120, 5836. [Google Scholar] [CrossRef]
- Fox, A.R.; Arnold, P.L.; Cummins, C.C. Uranium Nitrogen Multiple Bonding: Isostructural Anionic, Neutral, and Cationic Uranium Nitride Complexes Featuring a Linear U = N = U Core. J. Am. Chem. Soc. 2010, 132, 3250–3251. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.R.; Creutz, S.E.; Cummins, C.C. A bimetallic uranium μ-dicarbide complex: Synthesis, X-ray crystal structure, and bonding. Dalton Trans. 2010, 39, 6632–6634. [Google Scholar] [CrossRef] [PubMed]
- Diaconescu, P.L.; Cummins, C.C. μ-η6,η6-Arene-Bridged Diuranium Hexakisketimide Complexes Isolable in Two States of Charge. Inorg. Chem. 2012, 51, 2902–2916. [Google Scholar] [CrossRef] [PubMed]
- Vlaisavljevich, B.; Diaconescu, P.L.; Lukens, W.L.; Gagliardi, L.; Cummins, C.C. Investigations of the Electronic Structure of Arene-Bridged Diuranium Complexes. Organometallics 2013, 32, 1341–1352. [Google Scholar] [CrossRef]
- Korobkov, I.; Gambarotta, S.; Yap, G.P.A. Dinuclear Trivalent and Mixed-Valence Uranium [(-CH2-)5]4-calix[4]tetrapyrrole Complexes with Short Intermetallic Distances. Organometallics 2001, 20, 5440–5445. [Google Scholar] [CrossRef]
- Monreal, M.J.; Carver, C.T.; Diaconescu, P.L. Redox Processes in a Uranium Bis(1,1′-diamidoferrocene) Complex. Inorg. Chem. 2007, 46, 7226–7228. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.J.; Kozimor, S.A.; Ziller, J.W.; Kaltsoyannis, N. Structure, Reactivity, and Density Functional Theory Analysis of the Six-Electron Reductant, [(C5Me5)2U]2(μ-η6:η6-C6H6), Synthesized via a New Mode of (C5Me5)3M Reactivity. J. Am. Chem. Soc. 2004, 126, 14533–14547. [Google Scholar] [CrossRef]
- Evans, W.J.; Miller, K.A.; DiPasquale, A.G.; Rheingold, A.L.; Stewart, T.J.; Bau, R. A Crystallizable f-Element Tuck-In Complex: The Tuck-In Tuck-over Uranium Metallocene (C5Me5)U[µ-η5:η1:η1-C5Me3(CH2)2](µ-H)2U(C5Me5)2. Angew. Chem. Int. Ed. 2008, 47, 5075–5078. [Google Scholar] [CrossRef]
- Rinehart, J.D.; Bartlett, B.M.; Kozimor, S.A.; Long, J.R. Ferromagnetic exchange coupling in the linear, chloride-bridged cluster (cyclam)CoII[(μ-Cl)UIV(Me2Pz)4]2. Inorg. Chim. Acta 2008, 361, 3534–3538. [Google Scholar] [CrossRef]
- Spencer, L.P.; Yang, P.; Scott, B.L.; Batista, E.R.; Boncella, J.M. Oxidative Addition to U(V)-U(V) Dimers: Facile Routes to Uranium(VI) Bis(imido) Complexes. Inorg. Chem. 2009, 48, 11615–11623. [Google Scholar] [CrossRef]
- Nocton, G.; Horeglad, P.; Pécaut, J.; Mazzanti, M. Polynuclear cation-cation complexes of pentavalent uranyl: Relating stability and magnetic properties to structure. J. Am. Chem. Soc. 2008, 130, 16633–16645. [Google Scholar] [CrossRef] [PubMed]
- Mougel, V.; Horeglad, P.; Nocton, G.; Pécaut, J.; Mazzanti, M. Stable Pentavalent Uranyl Species and Selective Assembly of a Polymetallic Mixed-Valent Uranyl Complex by Cation–Cation Interactions. Angew. Chem. Int. Ed. 2009, 121, 8629–8632. [Google Scholar] [CrossRef]
- Nocton, G.; Horeglad, P.; Vetere, V.; Pécaut, J.; Dubois, L.; Maldivi, P.; Edelstein, N.M.; Mazzanti, M. Synthesis, Structure, and Bonding of Stable Complexes of Pentavalent Urany. J. Am. Chem. Soc. 2010, 132, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Mougel, V.; Horeglad, P.; Nocton, G.; Pecaut, J.; Mazzanti, M. Cation-cation complexes of pentavalent uranyl: From disproportionation intermediates to stable clusters. Chem. Eur. J. 2010, 16, 14365–14377. [Google Scholar] [CrossRef] [PubMed]
- Chatelain, L.; Mougel, V.; Pécautand, J.; Mazzanti, M. Magnetic communication cation–cation trimer of pentavalent uranyl. Chem. Sci. 2012, 3, 1075–1079. [Google Scholar] [CrossRef]
- Mougel, V.; Pecaut, J.; Mazzanti, M. New polynuclear U(IV)–U(V) complexes from U(IV) mediated uranyl(V) Disproportionation. Chem. Commun. 2012, 48, 868–870. [Google Scholar] [CrossRef] [PubMed]
- Mougel, V.; Chatelain, L.; Hermle, J.; Caciuffo, R.; Colineau, E.; Tuna, F.; Magnani, N.; De Geyer, A.; Pécaut, J.; Mazzanti, M. A uranium-based UO2(+)-Mn2+ single-chain magnet assembled trough cation-cation interactions. Angew. Chem. Int. Ed. 2014, 53, 819–823. [Google Scholar] [CrossRef]
- Chatelain, L.; Pécaut, J.; Tuna, F.; Mazzanti, M. Heterometallic Fe2II–UV and Ni2II–UV Exchange-Coupled Single-Molecule Magnets: Effect of the 3 d Ion on the Magnetic Properties. Chem. Eur. J. 2015, 21, 18038–18042. [Google Scholar] [CrossRef]
- Chatelain, L.; Tuna, F.; Pécaut, J.; Mazzanti, M. A zig-zag uranyl(v)-Mn(ii) single chain magnet with a high relaxation barrier. Chem. Commun. 2015, 51, 11309–11312. [Google Scholar] [CrossRef]
- Chatelain, L.; Tuna, F.; Pécaut, J.; Mazzanti, M. Synthesis and SMM behaviour of trinuclear versus dinuclear 3d–5f uranyl(v)–cobalt(ii) cation–cation complexes. Dalton Trans. 2017, 46, 5498–5502. [Google Scholar] [CrossRef]
- Magnani, N.; Colineau, E.; Eloirdi, R.; Griveau, J.-C.; Caciuffo, R.; Cornet, S.M.; May, I.; Sharrad, C.A.; Collison, D.; Winpenny, R.E.P. Superexchange Coupling and Slow Magnetic Relaxation in a Transuranium Polymetallic Complex. Phys. Rev. Lett. 2010, 104, 197202. [Google Scholar] [CrossRef] [PubMed]
- Tsoureas, N.; Kilpatrick, A.F.R.; Inmana, C.J.; Cloke, F.G.N. Steric control of redox events in organo-uranium chemistry: Synthesis and characterisation of U(V) oxo and nitrido complexes. Chem. Sci. 2016, 7, 4624–4632. [Google Scholar] [CrossRef] [PubMed]
- Wooles, A.; Mills, D.; Tuna, F.; Mcinnes, E.; Law, G.; Fuller, A.; Kremer, F.; Ridgway, M.; Lewis, W.; Gagliardi, L.; et al. Uranium(III)-Carbon Multiple Bonding Supported by Arene δ-Bonding in Mixed-Valence Hexauranium Nanometre-Scale Rings. Nat. Commun. 2018, 9, 2097. [Google Scholar] [CrossRef] [PubMed]
- Wooles, A.J.; Lewis, W.; Blake, A.J.; Liddle, S.T. β-Diketiminate Derivatives of Alkali Metals and Uranium. Organometallics 2013, 32, 5058–5070. [Google Scholar] [CrossRef]
- Gardner, B.M.; King, D.M.; Tuna, F.; Wooles, A.J.; Chilton, N.F.; Liddle, S.T. Assessing Crystal Field and Magnetic Interactions in Diuranium UIV–E–UIV cores (E = S, Se, Te). Chem. Sci. 2017, 8, 6207–6217. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Moro, F.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. A Formal High Oxidation State Inverse-Sandwich Diuranium Complex: A New Route to f-Block-Metal Bonds. Angew. Chem. Int. Ed. 2011, 50, 10388–10392. [Google Scholar] [CrossRef] [PubMed]
- Schelter, E.J.; Yang, P.; Scott, B.L.; Thompson, J.D.; Martin, R.L.; Hay, P.J.; Morris, D.E.; Kiplinger, J.L. Systematic Studies of Early Actinide Complexes: Uranium(IV) Fluoroketimides. Inorg. Chem. 2007, 46, 7477–7488. [Google Scholar] [CrossRef]
- Graves, C.R.; Yang, P.; Kozimor, S.A.; Vaughn, A.E.; Clark, D.L.; Conradson, S.D.; Schelter, E.J.; Scott, B.L.; Thompson, J.D.; Hay, P.J.; et al. Organometallic Uranium(V)-Imido Halide Complexes: From Synthesis to Electronic Structure and Bonding. J. Am. Chem. Soc. 2008, 130, 5272–5285. [Google Scholar] [CrossRef]
- Schelter, E.J.; Wu, R.; Scott, B.L.; Thompson, J.D.; Morris, D.E.; Kiplinger, J.L. Mixed Valency in a Uranium Multimetallic Complex. Angew. Chem. Int. Ed. 2008, 47, 2993–2996. [Google Scholar] [CrossRef]
- Minasian, S.G.; Krinsky, J.L.; Rinehart, J.D.; Copping, R.; Tyliszczak, T.; Janousch, M.; Shuh, D.K.; Arnold, J. A comparison of 4f vs 5f metal-metal bonds in (CpSiMe3)3M-ECp* (M = Nd, U.; E = Al, Ga; Cp* = C5Me5): Synthesis, thermodynamics, magnetism, and electronic structure. J. Am. Chem. Soc. 2009, 131, 13767–13783. [Google Scholar] [CrossRef]
- Salmon, L.; Thuéry, P.; Rivière, E.; Marrot, J.; Girerd, J.-J.; Ephritikhine, M. Syntheses, X-Ray Crystal Structures, and Magnetic Properties of Novel Linear MII2UIV Complexes (M = Co, Ni, Cu, Zn). Chem. Eur. J. 2002, 8, 773–783. [Google Scholar] [CrossRef]
- Salmon, L.; Thuéry, P.; Rivière, E.; Girerd, J.-J.; Ephritikhine, M. Versatility of the nature of the magnetic Cu(II)–U(IV) interaction. Syntheses, crystal structures and magnetic properties of Cu2U and CuU compounds. Dalton Trans. 2003, 2872–2880. [Google Scholar] [CrossRef]
- Salmon, L.; Thuéry, P.; Rivière, E.; Girerd, J.-J.; Ephritikhine, M. Synthesis, Structure, and Magnetic Behavior of a Series of Trinuclear Schiff Base Complexes of 5f (UIV,ThIV) and 3d (CuII, ZnII) Ions. Inorg. Chem. 2006, 45, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Love, J.B.; Patel, D. Pentavalent uranyl complexes. Coord. Chem. Rev. 2009, 253, 1973–1978. [Google Scholar] [CrossRef]
- Arnold, P.L.; Cowie, B.E.; Suvova, M.; Zegke, M.; Magnani, N.; Colineau, E.; Griveau, J.-C.; Caciuffo, R.; Love, J.B. Axially Symmetric U−O−Ln- and U−O−U-Containing Molecules from the Control of Uranyl Reduction with Simple f-Block Halides. Angew. Chem. Int. Ed. 2017, 129, 10915–10919. [Google Scholar] [CrossRef]
- Arnold, P.L.; Jones, G.M.; Pan, Q.-J.; Schreckenbach, G.; Love, J.B. Co-linear, double-uranyl coordination by an expanded Schiff-base polypyrrole macrocycle. Dalton Trans. 2012, 41, 6595–6597. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Jones, G.M.; Odoh, S.O.; Schreckenbach, G.; Magnani, N.; Love, J.B. Strongly coupled binuclear uranium–oxo complexes from uranyl oxo rearrangement and reductive silylation. Nat. Chem. 2012, 4, 221–227. [Google Scholar] [CrossRef]
- Jones, G.M.; Arnold, P.L.; Love, J.B. Oxo–Group-14-Element Bond Formation in Binuclear Uranium(V) Pacman Complexes. Chem. Eur. J. 2013, 19, 10287–10294. [Google Scholar] [CrossRef]
- Cowie, B.E.; Nichol, G.S.; Love, J.B.; Arnold, P.L. Double uranium oxo cations derived from uranyl by borane or silane reduction. Chem. Commun. 2018, 54, 3839–3842. [Google Scholar] [CrossRef]
- Larch, C.P.; Cloke, F.G.N.; Hitchcock, P.B. Activation and reduction of diethyl ether by low valent uranium: Formation of the trimetallic, mixed valence uranium oxo species [U(CpRR’)(μ-I)2]3(μ3-O) (CpRR’=C5Me5, C5Me4H, C5H4SiMe3). Chem. Commun. 2008, 82–84. [Google Scholar] [CrossRef]
- Rinehart, J.D.; Long, J.R. Slow Magnetic Relaxation in a Trigonal Prismatic Uranium(III) Complex. J. Am. Chem. Soc. 2009, 131, 12558–12559. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.; Kaizu, Y. Lanthanide Double-Decker Complexes Functioning as Magnets at the Single-Molecular Level. J. Am. Chem. Soc. 2003, 125, 8694. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.; Kaizu, Y. Mononuclear Lanthanide Complexes with a Long Magnetization Relaxation Time at High Temperatures: A New Category of Magnets at the Single-Molecular Level. J. Phys. Chem. B 2004, 108, 11265–11271. [Google Scholar] [CrossRef]
- Rinehart, J.D.; Meihaus, K.R.; Long, J.R. Observation of a Secondary Slow Relaxation Process for the Field-Induced Single-Molecule Magnet U(H2BPz2)3. J. Am. Chem. Soc. 2010, 132, 7572–7573. [Google Scholar] [CrossRef] [PubMed]
- Meihaus, K.R.; Rinehart, J.D.; Long, J.R. Dilution-Induced Slow Magnetic Relaxation and Anomalous Hysteresis in Trigonal Prismatic Dysprosium(III) and Uranium(III) Complexes. Inorg. Chem. 2011, 50, 8484–8489. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, J.D.; Long, J.R. Slow magnetic relaxation in homoleptic trispyrazolylborate complexes of neodymium(III) and uranium(III). Dalton Trans. 2012, 41, 13572. [Google Scholar] [CrossRef] [PubMed]
- Antunes, M.A.; Pereira, L.C.; Santos, I.C.; Mazzanti, M.; Marcalo, J.; Almeida, M. U(Tp(Me2))(2)(bipy) (+): A Cationic Uranium(III) Complex with Single-Molecule-Magnet Behavior. Inorg. Chem. 2011, 50, 9915–9917. [Google Scholar] [CrossRef]
- Mills, D.P.; Moro, F.; McMa ster, J.; van Slageren, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. A Delocalized Arene-Bridged Diuranium Single-Molecule Magnet. Nat. Chem. 2011, 3, 454–460. [Google Scholar] [CrossRef]
- Mougel, V.; Chatelain, L.; Pecaut, J.; Caciuffo, R.; Colineau, E.; Griveau, J.C.; Mazzanti, M. Uranium and Manganese Assembled in a Wheel-Shaped Nanoscale Single-Molecule Magnet with High Spin-Reversal Barrier. Nat. Chem. 2012, 4, 1011–1017. [Google Scholar] [CrossRef]
- King, D.M.; Tuna, F.; McMaster, J.; Lewis, W.; Blake, A.J.; McInnes, E.J.; Liddle, S.T. Single-Molecule Magnetism in a Single-Ion Triamidoamine Uranium (V) Terminal Mono-Oxo Complex. Angew. Chem. Int. Ed. 2013, 52, 4921–4924. [Google Scholar] [CrossRef]
- Spivak, M.; Vogiatzis, K.D.; Cramer, C.J.; de Graaf, C.; Gagliardi, L. Quantum Chemical Characterization of Single Molecule Magnets Based on Uranium. J. Phys. Chem. A 2017, 121, 1726–1733. [Google Scholar] [CrossRef] [PubMed]
- Gaggioli, C.A.; Gagliardi, L. Theoretical Investigation of Plutonium-Based Single-Molecule Magnets. Inorg. Chem. 2018, 57, 8098–8105. [Google Scholar] [CrossRef]
- Patel, D.; Tuna, F.; McInnes, E.J.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. A triamido-uranium(V) inverse-sandwich 10π-toluene tetraanion arene complex. Dalton Trans. 2013, 42, 5224–5227. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.C.; Camp, C.; Coutinho, J.T.; Chatelain, L.; Maldivi, P.; Almeida, M.; Mazzanti, M. Single-Molecule-Magnet Behavior in Mononuclear Homoleptic Tetrahedral Uranium(III) Complexes. Inorg. Chem. 2014, 53, 11809–11811. [Google Scholar] [CrossRef] [PubMed]
- Gendron, F.; Le Guennic, B.; Autschbach, J. Magnetic Properties and Electronic Structures of Ar3UIV–L Complexes with Ar = C5(CH3)4H− or C5H5− and L = CH3, NO, and C. Inorg. Chem. 2014, 53, 13174–13187. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, J.J.; Gorelsky, S.I.; Korobkov, I.; Murugesu, M. Slow Magnetic Relaxation in U(III) and Nd(III) Cyclooctatetraenyl Complexes. Organometallics 2015, 34, 1415–1418. [Google Scholar] [CrossRef]
- Chatelain, L.; Walsh, J.P.S.; Pécaut, J.; Tuna, F.; Mazzanti, M. Self-Assembly of a 3d–5f Trinuclear Single-Molecule Magnet from a Pentavalent Uranyl Complex. Angew. Chem. Int. Ed. 2014, 53, 13434–13438. [Google Scholar] [CrossRef]
- Rinehart, J.D.; Long, J.R. Exploiting Single-Ion Anisotropy in the Design of f-Element Single-Molecule Magnets. Chem. Sci. 2011, 2, 2078–2085. [Google Scholar] [CrossRef]
- Meihaus, K.R.; Long, J.R. Actinide-Based Single-Molecule Magnets. Dalton Trans. 2015, 44, 2517–2528. [Google Scholar] [CrossRef]
- Liddle, S.T.; van Slageren, J. Improving f-element single molecule magnets. Chem. Soc. Rev. 2015, 44, 6655–6668. [Google Scholar] [CrossRef]
- Pedersen, K.S.; Dreiser, J.; Weihe, H.; Sibille, R.; Johannesen, H.V.; Sorensen, M.A.; Nielsen, B.E.; Sigrist, M.; Mutka, H.; Rols, S.; et al. Design of Single-Molecule Magnets: Insufficiency of the Anisotropy Barrier as the Sole Criterion. Inorg. Chem. 2015, 54, 7600–7606. [Google Scholar] [CrossRef] [PubMed]
- McAdams, S.G.; Ariciu, A.-M.; Kostopoulos, A.K.; Walsh, J.P.S.; Tuna, F. Molecular single-ion magnets based on lanthanides and actinides: Design considerations and new advances in the context of quantum technologies. Coord. Chem. Rev. 2017, 346, 216–239. [Google Scholar] [CrossRef]
- Magnani, N.; Caciuffo, R. Future Directions for Transuranic Single Molecule Magnets. Inorganics 2018, 6, 26. [Google Scholar] [CrossRef]
- Jung, J.; Atanasov, M.; Neese, F. Ab Initio Ligand-Field Theory Analysis and Covalency Trends in Actinide and Lanthanide Free Ions and Octahedral Complexes. Inorg. Chem. 2017, 56, 8802–8816. [Google Scholar] [CrossRef] [PubMed]
- King, D.M.; Tuna, F.; McInnes, E.J.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. Isolation and Characterization of a Uranium(VI)–Nitride Triple Bond. Nat. Chem. 2013, 5, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Neidig, M.L.; Clark, D.L.; Martin, R.L. Covalency in f-element complexes. Coord. Chem. Rev. 2013, 257, 394–406. [Google Scholar] [CrossRef]
- Reta, D.; Ortu, F.; Randall, S.; Mills, D.P.; Chilton, N.F.; Winpenny, R.E.P.; Natrajan, L.; Edwards, B.; Kaltsoyannis, N. The performance of density functional theory for the description of ground and excited state properties of inorganic and organometallic uranium compounds. J. Organomet. Chem. 2018, 857, 58–74. [Google Scholar] [CrossRef]
- Söderlind, P.; Kotliar, G.; Haule, K.; Oppeneer, P.M.; Guillaumont, D. Computational Modeling of Actinide Materials and Complexes. MRS Bull. 2010, 35, 883–888. [Google Scholar] [CrossRef]
- Gryaznov, D.; Heifets, E.; Sedmidubsky, D. Density functional theory calculations on magnetic properties of actinide compounds. Phys. Chem. Chem. Phys. 2010, 12, 12273–12278. [Google Scholar] [CrossRef]
- Bencini, A. Some considerations on the proper use of computational tools in transition metal chemistry. Inorg. Chim. Acta 2008, 361, 3820–3831. [Google Scholar] [CrossRef]
- Neese, F. Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling. Coord. Chem. Rev. 2009, 253, 526–563. [Google Scholar] [CrossRef]
- Wan, D.; van Gunsteren, W.F.; Chai, Z. Recent advances in computational actinoid chemistry. Chem. Soc. Rev. 2012, 41, 5836–5865. [Google Scholar] [CrossRef]
- Schreckenbach, G.; Shamov, G.A. Theoretical actinide molecular science. Acc. Chem. Res. 2010, 43, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785. [Google Scholar] [CrossRef]
- Noodleman, L.J.; Davidson, E.R. Ligand spin polarization and antiferromagnetic coupling in transition metal dimers. Chem. Phys. 1986, 109, 131–143. [Google Scholar] [CrossRef]
- Noodleman, L.J.; Peng, C.Y.; Case, D.A.; Mouesca, J.M. Orbital interactions, electron delocalization and spin coupling in iron-sulfur clusters. Coord. Chem. Rev. 1995, 144, 199–244. [Google Scholar] [CrossRef]
- Døssing, A. Recent advances in the coordination chemistry of hydroxo-bridged complexes. Coord. Chem. Rev. 2014, 280, 38–53. [Google Scholar] [CrossRef]
- Selmi, W.; Abdelhak, J.; Marchivie, M.; Chastanet, G.; Zid, M.F. An investigation by DFT of the electronic structure and magnetic properties of a novel l-oxo-iron(III) complex with the 1,10-phenathroline ligand. Polyhedron 2017, 123, 441–452. [Google Scholar] [CrossRef]
- Ouilia, S.; Beghidja, C.; Beghidja, A.; Belkhiri, L.; Rabu, P. Synthesis, crystal structure, magnetic properties and DFT calculations of new dihydroxo-bridged binuclear chromium(III) based on monodentate mixed ligand. Inorg. Chim. Acta 2018, 476, 54–60. [Google Scholar] [CrossRef]
- Ruiz, E.; Cano, J.; Alvarez, S.; Alemany, P. Broken Symmetry Approach to Calculation of Exchange Coupling Constants for Homobinuclear and Heterobinuclear Transition Metal Complexes. J. Comput. Chem. 1999, 20, 1391–1400. [Google Scholar] [CrossRef]
- Ruiz, E.; Rodríguez-Fortea, A.; Cano, J.; Alvarez, S.; Alemany, P. About the calculation of exchange coupling constants in polynuclear transition metal complexes. J. Comput. Chem. 2003, 24, 982–989. [Google Scholar] [CrossRef] [PubMed]
- Ciofini, I.; Daul, C.A. DFT calculations of molecular magnetic properties of coordination compounds. Coord. Chem. Rev. 2003, 187, 238–239. [Google Scholar] [CrossRef]
- Kortus, J. Molecular magnets explored by density functional theory calculations. C. R. Chimie 2007, 10, 65–67. [Google Scholar] [CrossRef]
- Fouqueau, A.; Casida, M.E.; Daku, L.M.L.; Hauser, A.; Neese, F. Comparison of density functionals for energy and structural differences between the high- [5T2g:(t2g)4(eg)2] and low- [1A1g:(t2g)6(eg)0] spin states of iron(II) coordination compounds. II. More functionals and the hexaminoferrous cation, [Fe(NH3)6]2+. J. Chem. Phys. 2005, 122, 044110. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V.; Bencini, A.; Totti, F.; Ciofini, I. On the Calculation and Modeling of Magnetic Exchange Interactions in Weakly Bonded Systems: The Case of the Ferromagnetic Copper(II) í2-Azido Bridged Complexes. Inorg. Chem. 1999, 38, 1996–2004. [Google Scholar] [CrossRef] [PubMed]
- Korzeniak, T.; Desplanches, C.; Podgajny, R.; Giménez-Saiz, C.; Stadnicka, K.; Rams, M.; Sieklucka, B. Magnetostructural Correlations in CuII–NC–WV Linkage: The Case of [CuII(diimine)]2+–[WV(CN)8]3−0D Assemblies. Inorg. Chem. 2009, 48, 2865–2872. [Google Scholar] [CrossRef]
- Moreira, I.D.; Costa, R.; Filatov, M.; Illas, F. Restricted Ensemble-Referenced Kohn–Sham versus Broken Symmetry Approaches in Density Functional Theory: Magnetic Coupling in Cu Binuclear Complexes. J. Chem. Theory Comput. 2007, 3, 764–774. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. Density functional theory for transition metals and transition metal chemistry. Phys. Chem. Chem. Phys. 2009, 11, 10757–10816. [Google Scholar] [CrossRef]
- Onofrio, N.; Mouesca, J.M. Analysis of the Singlet Triplet Splitting Computed by the Density Functional TheoryBroken-Symmetry Method: Is It an Exchange Coupling Constant? Inorg. Chem. 2011, 50, 5577–5586. [Google Scholar] [CrossRef]
- Zhekova, H.; Seth, M.; Ziegler, T. Introduction of a New Theory for the Calculation of Magnetic Coupling Based on Spin–Flip Constricted Variational Density Functional Theory. Application to Trinuclear Copper Complexes which Model the Native Intermediate in Multicopper Oxidases. J. Chem. Theory Comput. 2011, 7, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Peralta, J.E.; Melo, J.I. Magnetic Exchange Couplings with Range-Separated Hybrid Density Functionals. J. Chem. Theory Comput. 2010, 6, 1894–1899. [Google Scholar] [CrossRef] [PubMed]
- Páez-Hernández, D.; Murillo-López, J.A.; Arratia-Pérez, R. Optical and Magnetic Properties of the Complex Bis(dicyclooctatetraenyl)diuranium. A Theoretical View. Organometallics 2012, 31, 6297–6304. [Google Scholar] [CrossRef]
- Spencer, L.P.; Schelter, E.J.; Yang, P.; Gdula, R.L.; Scott, B.L.; Thompson, J.D.; Kiplinger, J.L.; Batista, E.R.; Boncella, J.M. Cation-cation interactions, magnetic communication, and reactivity of the pentavalent uranium ion [U(NtBu)2]+. Angew. Chem. Int. Ed. 2009, 48, 3795–3798. [Google Scholar] [CrossRef] [PubMed]
- Newell, B.S.; Rapp, A.K.; Shores, M.P. Experimental Evidence for Magnetic Exchange in Di- and Trinuclear Uranium(IV) Ethynylbenzene Complexes. Inorg. Chem. 2010, 49, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.E.; Zhao, Y.; Truhlar, D.G. Density Functionals for Inorganometallic and Organometallic Chemistry. J. Phys. Chem. A 2005, 109, 11127–11143. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.N. On the Accuracy of Density Functional Theory in Transition Metal Chemistry. Annu. Rep. Prog. Chem. Sect. C Phys. Chem. 2006, 102, 203–226. [Google Scholar] [CrossRef]
- Cohen, J.; Mori-Sanchez, A.P.; Yang, W.T. Insights into Current Limitations of Density Functional Theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef]
- Monreal, M.J.; Diaconescu, P.L. A Weak Interaction between Iron and Uranium in Uranium Alkyl Complexes Supported by Ferrocene Diamide Ligands. Organometallics 2008, 27, 1702–1706. [Google Scholar] [CrossRef]
- Schelter, E.J.; Veauthier, J.M.; Thompson, J.D.; Scott, B.L.; John, K.D.; Morris, D.E.; Kiplinger, J.L. 4f-5f heterotrimetallic complexes exhibiting electrochemical and magnetic communication. J. Am. Chem. Soc. 2006, 128, 2198–2199. [Google Scholar] [CrossRef]
- Natrajan, L.; Burdet, F.; Pécaut, J. Mazzanti, M. Synthesis and structure of a stable pentavalent-uranyl coordination polymer. J. Am. Chem. Soc. 2006, 128, 7152–7153. [Google Scholar] [CrossRef] [PubMed]
- Lam, O.P.; Heinemann, F.W.; Meyer, K. Activation of elemental S, Se and Te with uranium(III): Bridging U–E–U (E = S, Se) and diamond-core complexes U–(E)2–U (E = O, S, Se, Te). Chem. Sci. 2011, 2, 1538–1547. [Google Scholar] [CrossRef]
- Schmidt, A.-C.; Heinemann, F.W.; Lukens, W.W., Jr.; Meyer, K. Molecular and Electronic Structure of Dinuclear Uranium Bis-μ-Oxo Complexes with Diamond Core Structural Motifs. J. Am. Chem. Soc. 2014, 136, 11980–11993. [Google Scholar] [CrossRef] [PubMed]
- Gardner, B.M.; Stewart, J.C.; Davis, A.L.; McMaster, J.; Lewis, W.; Blake, A.J.; Liddle, S.T. Homologation and functionalization of carbon monoxide by a recyclable uranium complex. Proc. Natl. Acad. Sci. USA 2012, 109, 9265–9270. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne, T.; Rivière, E.; Marrot, J.; Girerd, J.-J.; Ephritikhine, M. Synthesis, Crystal Structure, and Magnetic Behavior of Linear MUIV Complexes (M = Co, Ni, Cu, Zn). Angew. Chem., Int. Ed. 2000, 39, 1647. [Google Scholar] [CrossRef]
- Ephritikhine, M. The vitality of uranium molecular chemistry at the dawn of the XXIst century. Dalton Trans. 2006, 2501. [Google Scholar] [CrossRef] [PubMed]
- Arliguie, T.; Lance, M.; Nierlich, M.; Vigner, J.; Ephritikhine, M. Inverse cycloheptatrienyl sandwich complexes. Crystal structure of [U(BH4)2(OC4H8)5][(BH4)3U(µ-η7,η7-C7H7)U(BH4)3]. J. Chem. Soc. Chem. Commun. 1994, 847–848. [Google Scholar] [CrossRef]
- Arliguie, T.; Lance, M.; Nierlich, M.; Vigner, J.; Ephritikhine, M. Synthesis and crystal structure of [K(C12H24O6)][U(η-C7H7)2], the first cycloheptatrienyl sandwich compound. J. Chem. Soc. Chem. Commun. 1995, 183–184. [Google Scholar] [CrossRef]
- Camp, C.; Toniolo, D.; Andrez, J.; Pécaut, J.; Mazzanti, M. A versatile route to homo- and hetero-bimetallic 5f–5f and 3d–5f complexes supported by a redox active ligand framework. Dalton Trans. 2017, 46, 11145–11148. [Google Scholar] [CrossRef] [PubMed]
- Kozimor, S.A.; Bartlett, B.M.; Rinehart, J.D.; Long, J.R. Magnetic Exchange Coupling in Chloride-Bridged 5f-3d Heterometallic Complexes Generated via Insertion into a Uranium(IV) Dimethylpyrazolate Dimer. J. Am. Chem. Soc. 2007, 129, 10672–10674. [Google Scholar] [CrossRef]
- Arnold, P.L.; Hollis, E.; Nichol, G.S.; Love, J.B.; Griveau, J.C.; Caciuffo, R.; Magnani, N.; Maron, L.; Castro, L.; Yahia, A.; et al. Oxo-functionalization and reduction of the uranyl ion through lanthanide-element bond homolysis: Synthetic, structural, and bonding analysis of a series of singly reduced uranyl-rare earth 5f1-4fn complexes. J. Am. Chem. Soc. 2013, 135, 3841–3854. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Patel, D.; Wilson, C.; Love, J.B. Reduction and selective oxo group silylation of the uranyl dication. Nature 2008, 451, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Pecharman, A.F.; Hollis, E.; Yahia, A.; Maron, L.; Parsons, S.; Love, J.B. Uranyl oxo activation and functionalization by metal cation coordination. Nat. Chem. 2010, 2, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Hollis, E.; White, F.J.; Magnani, N.; Caciuffo, R.; Love, J.B. Single Electron Uranyl Reduction by a Rare Earth Cation. Angew. Chem. Int. Ed. 2011, 123, 917–920. [Google Scholar] [CrossRef]
- Liddle, S.T. The Renaissance of Non-Aqueous Uranium Chemistry. Angew. Chem. Int. Ed. 2015, 54, 8604–8641. [Google Scholar] [CrossRef] [PubMed]
- Schelter, E.J.; Veauthier, J.M.; Graves, C.R.; John, K.D.; Scott, B.L.; Thompson, J.D.; Pool-Davis-Tournear, J.A.; Morris, D.E.; Kiplinger, J.L. 1,4-dicyanobenzene as a scaffold for the preparation of bimetallic actinide complexes exhibiting metal-metal communication. Chem. Eur. J. 2008, 14, 7782–7790. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.A.P.; Tuna, F.; McInnes, E.J.L.; Liddle, S.T.; McMaster, J.; Vitorica-Yrezabal, I.J.; Mills, D.P. [UIII{N(SiMe2tBu)2}3]: A Structurally Authenticated Trigonal Planar Actinide Complex. Chem. Eur. J. 2014, 20, 14579–14583. [Google Scholar] [CrossRef]
- Meihaus, K.R.; Minasian, S.G.; Lukens, W.W., Jr.; Kozimor, S.A.; Shuh, D.K.; Tyliszczak, T.; Long, J.R. Influence of Pyrazolate vs. N-Heterocyclic Carbene Ligands on the Slow Magnetic Relaxation of Homoleptic Trischelate Lanthanide(III) and Uranium(III) Complexes. J. Am. Chem. Soc. 2014, 136, 6056–6068. [Google Scholar] [CrossRef]
- Coutinho, J.T.; Antunes, M.A.; Pereira, L.C.J.; Bolvin, H.; Marcalo, J.; Mazzanti, M.; Almeida, M. Single-ion magnet behaviour in [U(TpMe2)2I]. Dalton Trans. 2012, 41, 13568–13571. [Google Scholar] [CrossRef]
- Cirera, J.; Jiang, Y.; Qin, L.; Zheng, Y.-Z.; Li, G.; Wu, G.; Ruiz, E. Ferromagnetism in polynuclear systems based on non-linear [MnII2MnIII] building blocks. Inorg. Chem. Front. 2016, 3, 1272–1279. [Google Scholar] [CrossRef]
- Ishikawa, N.; Iino, T.; Kaizu, Y. Interaction between f-Electronic Systems in Dinuclear Lanthanide Complexes with Phthalocyanines. J. Am. Chem. Soc. 2002, 124, 11440–11447. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M.L.; Ballou, R.; Porcher, P.; Kahn, O.; Sutter, J.P. Analytical Determination of the {Ln–Aminoxyl Radical} Exchange Interaction Taking into Account Both the Ligand-Field Effect and the Spin–Orbit Coupling of the Lanthanide Ion (Ln=DyIII and HoIII). Chem. Eur. J. 2002, 8, 525–531. [Google Scholar] [CrossRef]
- Przychodzen, P.; Pelka, R.; Lewinski, K.; Supel, J.; Rams, M.; Tomala, K.; Sieklucka, B. Tuning of Magnetic Properties of Polynuclear Lanthanide(III)–Octacyanotungstate(V) Systems: Determination of Ligand-Field Parameters and Exchange Interaction. Inorg. Chem. 2007, 46, 8924–8938. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Sunatsuki, Y.; Ishida, H.; Kojima, M.; Akashi, H.; Re, N.; Matsumoto, N.; Pochaba, A.; Mrozinski, J. Synthesis, Structures, and Magnetic Properties of Face-Sharing Heterodinuclear Ni(II)–Ln(III) (Ln = Eu, Gd, Tb, Dy) Complexes. Inorg. Chem. 2008, 47, 5736–5745. [Google Scholar] [CrossRef] [PubMed]
- Sorace, L.; Sangregorio, C.; Figuerola, A.; Benelli, C.; Gatteschi, D. Magnetic Interactions and Magnetic Anisotropy in Exchange Coupled 4f–3d Systems: A Case Study of a Heterodinuclear Ce3+–Fe3+ Cyanide-Bridged Complex. Chem. Eur. J. 2009, 15, 1377–1388. [Google Scholar] [CrossRef] [PubMed]
- Tangoulis, V.; Estrader, M.; Figuerola, A.; Ribas, J.; Diaz, C. Anisotropic exchange interactions in hetero-one-dimensional Ln3+–M3+ systems (Ln3+ = Er, Yb; M3+ = Cr, FeLS): Magnetometry and Dual Mode X-band Electron Paramagnetic Resonance spectroscopic studies. Chem. Phys. 2007, 336, 74–82. [Google Scholar] [CrossRef]
- Santini, P.; Carretta, S.; Amoretti, G.; Caciuffo, R.; Magnani, N.; Lander, G.H. Multipolar interactions in f-electron systems: The paradigm of actinide dioxides. Rev. Mod. Phys. 2009, 81, 807–863. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Jensen, F.; Dorigo, A.; Houk, K.N. A spin correction procedure for unrestricted Hartree-Fock and Møller-Plesset wavefunctions for singlet diradicals and polyradicals. Chem. Phys. Lett. 1988, 149, 537. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Namimoto, H.; Fueno, T.; Nogami, T.; Shirota, Y. Possibilities of organic ferromagnets and ferrimagnets by the use of charge-transfer (CT) complexes with radical substituents. Ab initio MO studies. Chem. Phys. Lett. 1990, 166, 408. [Google Scholar] [CrossRef]
- Soda, T.; Kitagawa, Y.; Onishi, T.; Takano, Y.; Shigeta, Y.; Nagao, H.; Yoshioka, Y.; Yamaguchi, K. Ab initio computations of effective exchange integrals for H–H, H–He–H and Mn2O2 complex: Comparison of broken-symmetry approaches. Chem. Phys. Lett. 2000, 319, 223. [Google Scholar] [CrossRef]
- Caballol, R.; Castell, O.; Ilias, F.; Moreira, I.P.; Malrieu, J.P. Remarks on the Proper Use of the Broken Symmetry Approach to Magnetic Coupling. J. Phys. Chem. A 1997, 101, 7860–7866. [Google Scholar] [CrossRef]
- Rajaraman, G.; Totti, F.; Bencini, A.; Caneschi, A.; Sessoli, R.; Gatteschi, D. Density functional studies on the exchange interaction of a dinuclear Gd(III)–Cu(II) complex: Method assessment, magnetic coupling mechanism and magneto-structural correlations. Dalton Trans. 2009, 3153–3161. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sonnenberg, J.L.; Schlegel, H.B. Theoretical Studies of AnII2(C8H8)2 (An = Th, Pa, U, and Np) Complexes: The Search for Double-Stuffed Actinide Metallocenes. Inorg. Chem. 2010, 49, 6545–6551. [Google Scholar] [CrossRef] [PubMed]
- Meskaldji, S.; Belkhiri, A.; Belkhiri, L.; Boucekkine, A.; Ephritikhine, M. Magnetic exchange coupling in imido bimetallic uranium(V) complexes. A relativistic DFT study. C. R. Chimie 2012, 15, 184–191. [Google Scholar] [CrossRef]
- Meskaldji, S.; Zaiter, A.; Belkhiri, L.; Boucekkine, A. A relativistic DFT study of magnetic exchange coupling in ketimide bimetallic uranium(IV) complexes. Theor. Chem. Acc. 2012, 131, 1151. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys. 1996, 105, 9982. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787. [Google Scholar] [CrossRef]
- Gagliardi, L.; Roos, B.O. Quantum chemical calculations show that the uranium molecule U2 has a quintuple bond. Nature 2005, 433, 848–851. [Google Scholar] [CrossRef]
- Macchia, G.L.; Brynda, M.; Gagliardi, L. Quantum Chemical Calculations Predict the Diphenyl Diuranium Compound [PhUUPh] To Have a Stable 1Ag Ground State. Chem. Int. Ed. 2006, 45, 6210–6213. [Google Scholar] [CrossRef]
- Cavigliasso, G.; Kaltsoyannis, N. Metal–metal bonding in molecular actinide compounds: Electronic structure of [M2X8]2− (M = U, Np, Pu; X = Cl, Br, I) complexes and comparison with d-block analogues. Dalton Trans. 2006, 5476–5483. [Google Scholar] [CrossRef] [PubMed]
- Roos, B.O.; Borin, A.C.; Gagliardi, L. Reaching the Maximum Multiplicity of the Covalent Chemical Bond. Angew. Chem. Int. Ed. 2007, 46, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Teyar, B.; Belkhiri, L.; Costuas, K.; Boucekkine, A.; Meyer, K. Electronic Structure and Magnetic Properties of Dioxo-Bridged Diuranium Complexes with Diamond-Core Structural Motifs: A Relativistic DFT Study. Inorg. Chem. 2016, 55, 2870–2881. [Google Scholar] [CrossRef] [PubMed]
- Hohloch, S.; Pankhurst, J.R.; Jaekel, E.E.; Parker, B.F.; Lussier, D.J.; Garner, M.E.; Booth, C.H.; Love, J.B.; Arnold, J. Benzoquinonoid-bridged dinuclear actinide complexes. Dalton Trans. 2017, 46, 11615–11625. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comp. Chem. 2011, 32, 1456. [Google Scholar] [CrossRef]
- Lu, J.; Guo, M.; Tang, J. Chem. Recent Developments in Lanthanide Single-Molecule Magnets. Asian J. 2017, 12, 2772–2779. [Google Scholar] [CrossRef] [PubMed]
- Antunes, M.A.; Coutinho, J.T.; Santos, I.C.; Marçalo, J.; Almeida, M.; Baldoví, J.J.; Pereira, L.C.J.; Ariño, A.-G.; Coronado, E. A Mononuclear Uranium(IV) Single-Molecule Magnet with an Azobenzene Radical Ligand. Chem. Eur. J. 2015, 21, 17817–17826. [Google Scholar] [CrossRef]
- Solis-Céspedes, E.; Páez-Hernández, D. Modeling the electronic states and magnetic properties derived from the f1 configuration in lanthanocene and actinocene compounds. Dalton Trans. 2017, 46, 4834–4843. [Google Scholar] [CrossRef]
- AlDamen, M.A.; Clemente-Juan, J.M.; Coronado, E.; Marti-Gastaldo, C.; Gaita-Arino, A. Mononuclear Lanthanide Single-Molecule Magnets Based on Polyoxometalates. J. Am. Chem. Soc. 2008, 130, 8874–8875. [Google Scholar] [CrossRef]
- AlDamen, M.A.; Cardona-Serra, S.; Clemente-Juan, J.M.; Coronado, E.; Gaita-Arino, A.; Marti-Gastaldo, C.; Luis, F.; Montero, O. Mononuclear Lanthanide Single Molecule Magnets Based on the Polyoxometalates [Ln(W5O18)2]9− and [Ln(β2-SiW11O39)2]13−(LnIII = Tb, Dy, Ho, Er, Tm, and Yb). Inorg. Chem. 2009, 48, 3467–3479. [Google Scholar] [CrossRef]
- Lukens, W.W.; Speldrich, M.; Yang, P.; Duignan, T.J.; Autschbach, J.; Kögerler, P. The roles of 4f- and 5f-orbitals in bonding: A magnetochemical, crystal field, density functional theory, and multi-reference wavefunction study. Dalton Trans. 2016, 45, 11508–11521. [Google Scholar] [CrossRef] [PubMed]
- Lam, O.P.; Anthon, C.; Heinemann, F.W.; O’Connor, J.M.; Meyer, K. Structural and Spectroscopic Characterization of a large-Separated Uranium Benzophenone Ketyl Radical Complex. J. Am. Chem. Soc. 2008, 130, 6567–6576. [Google Scholar] [CrossRef] [PubMed]
- Castro-Rodriguez, I.; Nakai, H.; Zakharov, L.N.; Rheingold, A.L.; Meyer, K. A linear, O-coordinated eta1-CO2 bound to uranium. Science 2004, 305, 1757–1759. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Q.; Luo, C.L.; Wang, B.W.; Gao, S. Understanding the magnetic anisotropy in a family of N2(3-) radical-bridged lanthanide complexes: Density functional theory and ab initio calculations. J. Phys. Chem. A 2013, 117, 10873–10880. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Jeon, L.-R.; Long, J.R.; Harris, D. Radical ligand-containing single-molecule magnets. Coord. Chem. Rev. 2015, 289–290. [Google Scholar] [CrossRef]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. General Performance of Density Functionals. J. Phys. Chem. A 2007, 111, 10439–10452. [Google Scholar] [CrossRef] [PubMed]
- Greif, A.H.; Hrobàrik, P.; Autschbach, J.; Kaupp, M. Giant spin–orbit effects on 1H and 13C NMR shifts for uranium(VI) complexes revisited: Role of the exchange–correlation response kernel, bonding analyses, and new predictions. Phys. Chem. Chem. Phys. 2016, 18, 30462–30474. [Google Scholar] [CrossRef] [PubMed]
- Mounce, A.M.; Yasuoka, H.; Koutroulakis, G.; Lee, J.A.; Cho, H.; Gendron, F.; Zurek, E.; Scott, B.L.; Trujillo, J.A.; Slemmons, A.K.; et al. Nuclear Magnetic Resonance Measurements and Electronic Structure of Pu(IV) in [(Me)4N]2PuCl6. Inorg. Chem. 2016, 55, 8371–8380. [Google Scholar] [CrossRef]
- Polinski, M.J.; Garner, E.B.; Maurice, R.; Planas, N.; Stritzinger, J.T.; Parker, T.G.; Cross, J.N.; Green, T.D.; Alekseev, E.V.; Van Cleve, S.M.; et al. Unusual structure, bonding and properties in a californium borate. Nat. Chem. 2014, 6, 387–392. [Google Scholar] [CrossRef]
- Cary, S.K.; Vasiliu, M.; Baumbach, R.E.; Stritzinger, J.T.; Green, T.D.; Diefenbach, K.; Cross, J.N.; Knappenberger, K.L.; Liu, G.; Silver, M.A.; et al. Emergence of californium as the second transitional element in the actinide series. Nat. Commun. 2015, 6, 6827. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Vydrov, O.A.; Scuseria, G.E. Assessment of a long-range corrected hybrid functional. J. Chem. Phys. 2006, 125, 234109. [Google Scholar] [CrossRef] [PubMed]
- Vydrov, O.A.; Heyd, J.; Krukau, A.; Scuseria, G.E. Importance of short-range versus long-range Hartree-Fock exchange for the performance of hybrid density functionals. J. Chem. Phys. 2006, 125, 074106. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.M.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Autillo, M.; Guerin, L.; Bolvin, H.; Moisya, P.; Berthon, C. Magnetic susceptibility of actinide(III) cations: An experimental and theoretical study. Phys. Chem. Chem. Phys. 2016, 18, 6515–6525. [Google Scholar] [CrossRef] [PubMed]
- King, D.M.; Cleaves, P.A.; Wooles, A.J.; Gardner, B.M.; Chilton, N.F.; Tuna, F.; Lewis, W.; McInnes, E.J.L.; Liddle, S.T. Molecular and electronic structure of terminal and alkali metal-capped uranium(V) nitride complexes. Nat. Commun. 2016, 7, 13773. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.J.; Rungthanaphatsophon, P.; Behrle, A.C.; Vilanova, S.P.; Kelley, S.P.; Lukens, W.W.; Walensky, J.R. Structure and properties of [(4,6-tBu2C6H2O)2Se]2An(THF)2, An = U, Np, and their reaction with p-benzoquinone. Chem. Commun. 2018, 54, 10435–10438. [Google Scholar] [CrossRef]


































© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belkhiri, L.; Le Guennic, B.; Boucekkine, A. DFT Investigations of the Magnetic Properties of Actinide Complexes. Magnetochemistry 2019, 5, 15. https://doi.org/10.3390/magnetochemistry5010015
Belkhiri L, Le Guennic B, Boucekkine A. DFT Investigations of the Magnetic Properties of Actinide Complexes. Magnetochemistry. 2019; 5(1):15. https://doi.org/10.3390/magnetochemistry5010015
Chicago/Turabian StyleBelkhiri, Lotfi, Boris Le Guennic, and Abdou Boucekkine. 2019. "DFT Investigations of the Magnetic Properties of Actinide Complexes" Magnetochemistry 5, no. 1: 15. https://doi.org/10.3390/magnetochemistry5010015
APA StyleBelkhiri, L., Le Guennic, B., & Boucekkine, A. (2019). DFT Investigations of the Magnetic Properties of Actinide Complexes. Magnetochemistry, 5(1), 15. https://doi.org/10.3390/magnetochemistry5010015