Structure Confirmation and Evaluation of a Nonsteroidal Inhibitor of 17β-Hydroxysteroid Dehydrogenase Type 10


Abstract

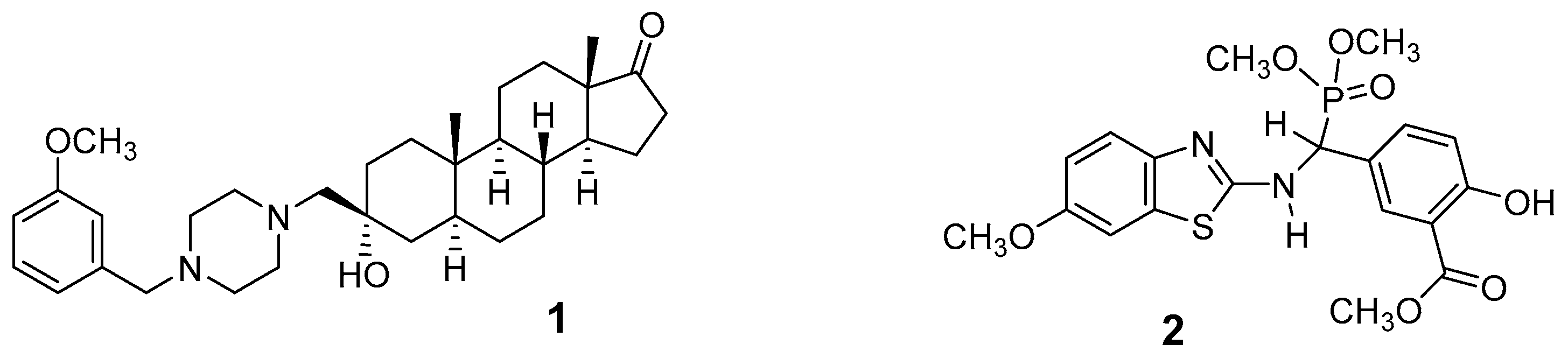
1. Introduction
2. Results and Discussion
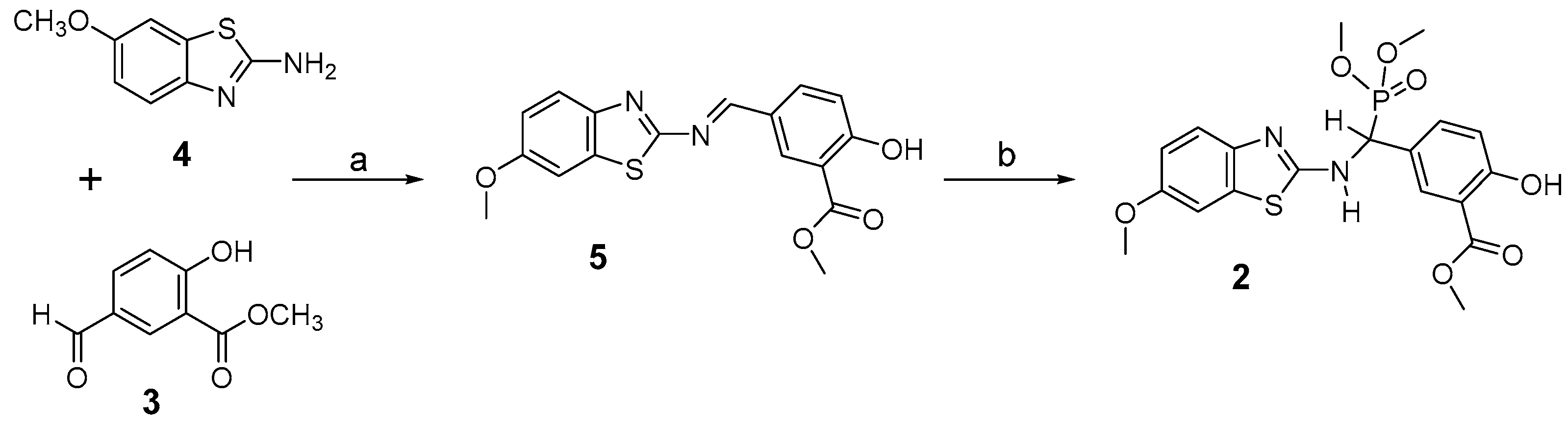
2.1. Chemical Synthesis of Nonsteroidal Inhibitor 2
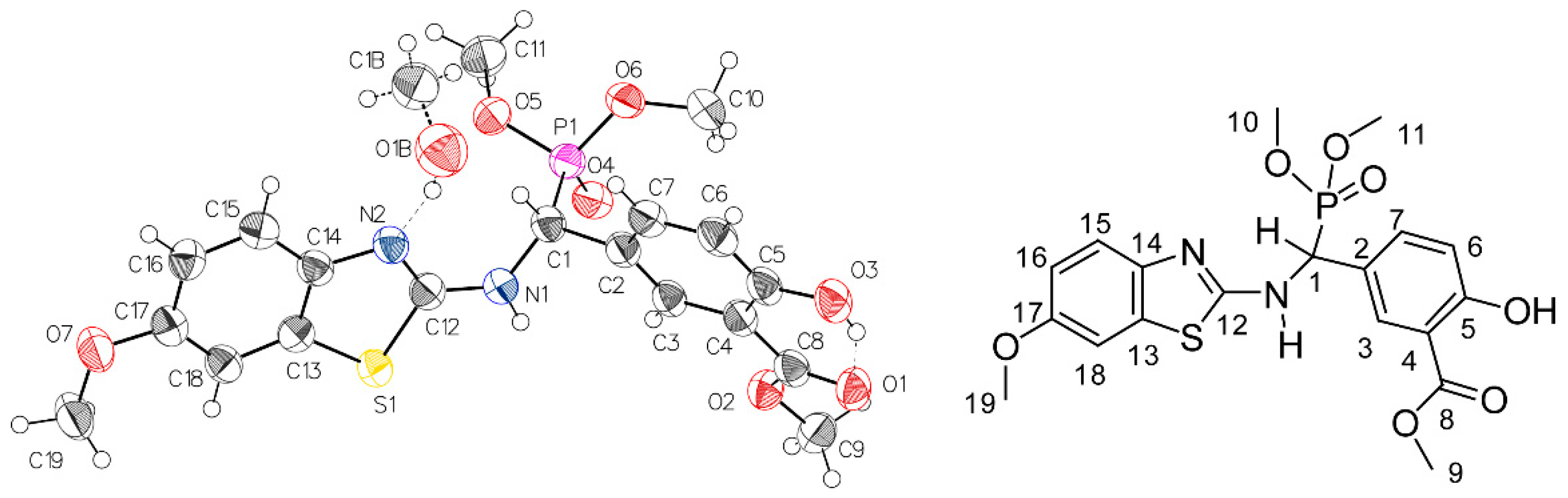
2.2. Preliminary Characterization of Compound 2
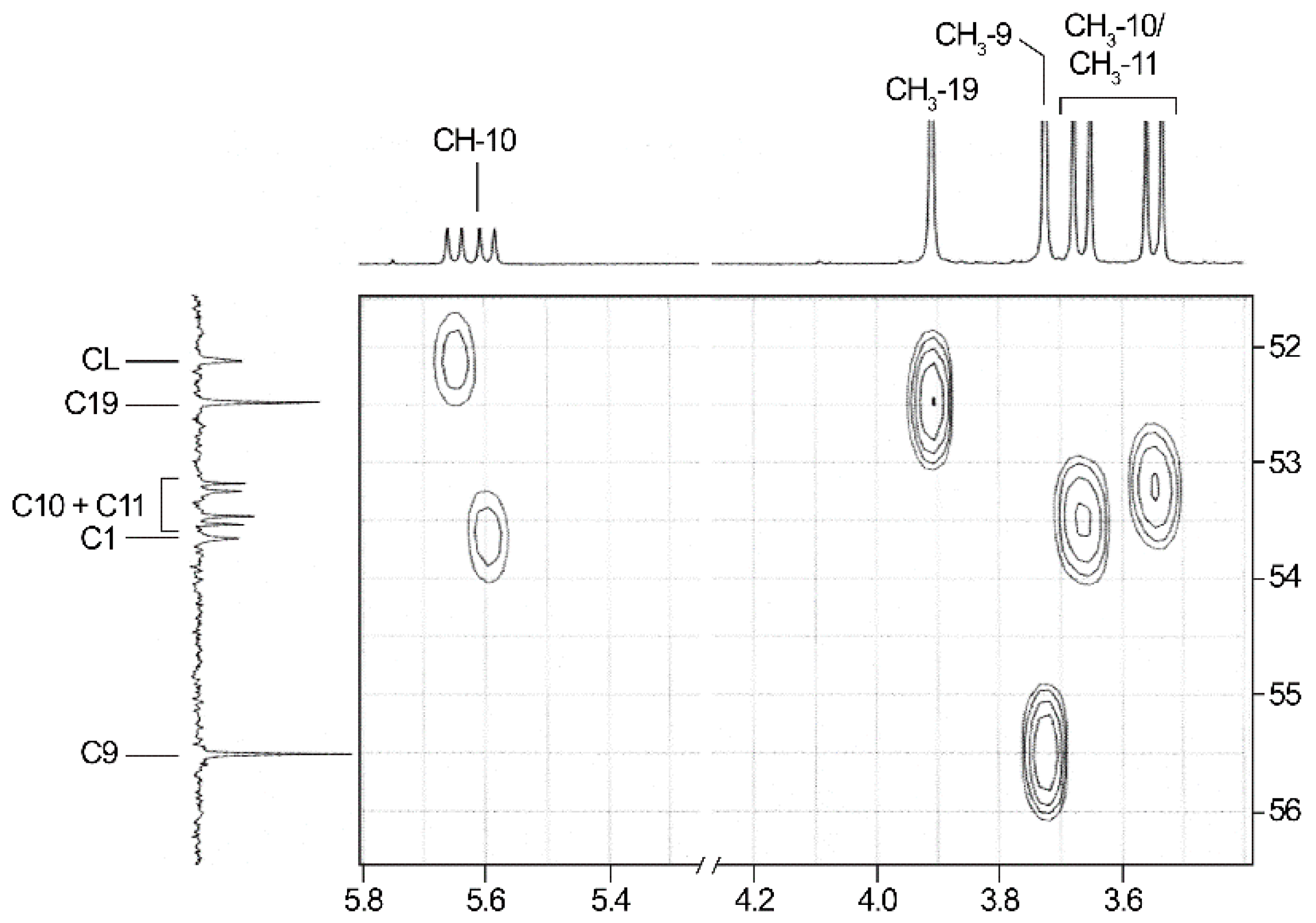
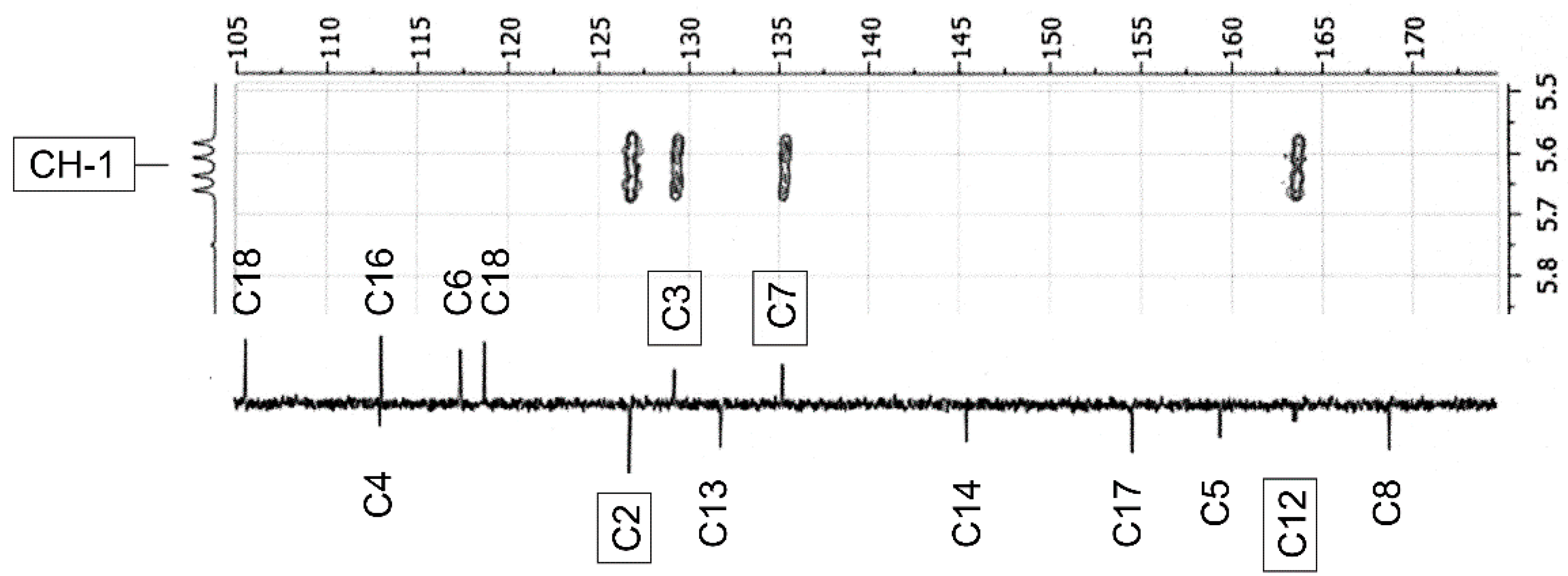
2.3. NMR-Analyses of Compound 2
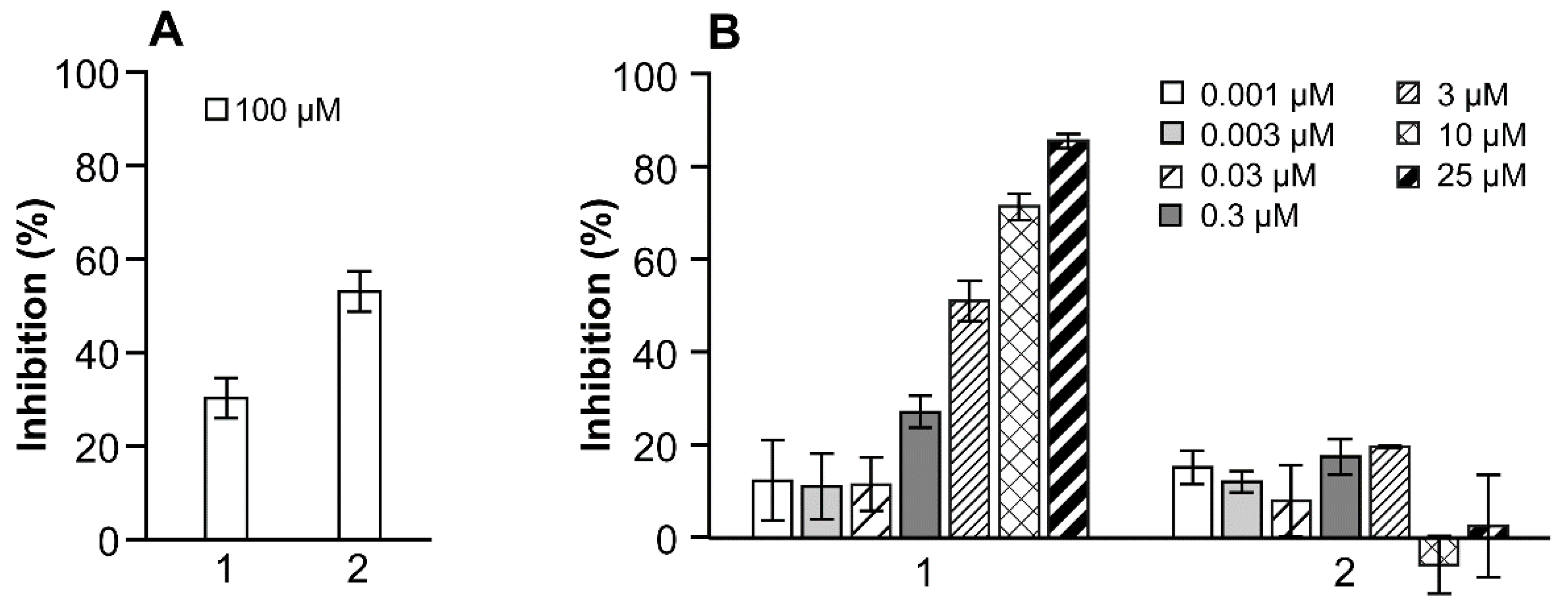
2.4. Enzyme Inhibition
3. Materials and Methods
3.1. Chemical Synthesis of 2
3.1.1. General
3.1.2. Synthesis of Imine 5
3.1.3. Synthesis of Inhibitor 2
3.2. Biological Evaluation of 1 and 2
3.2.1. Inhibition of E2 to E1 Transformation by 17β-HSD10 Protein
3.2.2. Inhibition of E2 to E1 Transformation by HEK-293 Cells Overexpressing 17β-HSD10
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goedert, M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349, 1255555. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; He, X.Y.; Isaacs, C.; Dobkin, C.; Miller, D.; Philipp, M. Roles of 17β-hydroxysteroid dehydrogenase type 10 in neurodegenerative disorders. J. Steroid Biochem. Mol. Biol. 2014, 143, 460–472. [Google Scholar] [CrossRef] [PubMed]
- He, X.Y.; Isaacs, C.; Yang, S.Y. Roles of mitochondrial 17β-hydroxysteroid dehydrogenase type 10 in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 62, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.X.; Yan, S.S. Role of mitochondrial amyloid-β in Alzheimer’s disease. J. Alzheimer’s Dis. 2010, 20, S569–S578. [Google Scholar] [CrossRef] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Aspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; et al. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.S.; Lee, K.W.; Kim, T.K.; Baek, I.S.; Im, J.Y.; Han, P.L. Behavioral stress causes mitochondrial dysfunction via ABAD up-regulation and aggravates plaque pathology in the brain of a mouse model of Alzheimer disease. Free Radic. Biol. Med. 2011, 50, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- He, X.Y.; Yang, S.Y. Roles of type 10 17beta-hydroxysteroid dehydrogenase in intracrinology and metabolism of isoleucine and fatty acids. Endocr. Metab. Immune Disord. Drug Targets 2006, 6, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Gouras, G.K.; Greenfield, J.P.; Vincent, B.; Naslund, J.; Mazzarelli, L.; Fried, G.; Jovanovic, J.N.; Seeger, M.; Relkin, N.R.; et al. Estrogen reduces neuronal generation of Alzheimer beta-amyloid peptides. Nat. Med. 1998, 4, 447–451. [Google Scholar] [CrossRef] [PubMed]
- Krištofiková, Z.; Bocková, M.; Hegnerová, K.; Bartoš, J.; Říčný, J.; Řípová, D.; Homola, J. Enhanced levels of mitochondrial enzyme 17β-hydroxysteroid dehydrogenase type 10 in patients with Alzheimer disease and multiple sclerosis. Mol. Biosyst. 2009, 5, 1174–1179. [Google Scholar]
- He, X.Y.; Wegiel, J.; Yang, S.Y. Intracellular oxidation of allopregnanolone by human brain type 10 17beta-hydroxysteroid dehydrogenase. Brain Res. 2005, 1040, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Maltais, R.; Ayan, D.; Trottier, A.; Barbeau, X.; Lagüe, P.; Bouchard, J.E.; Poirier, D. Discovery of a non-estrogenic inhibitor of 17β-hydroxysteroid dehydrogenase type 1 from 3-substituted-16β-(m-carbamoylbenzyl)-estradiol derivatives. J. Med. Chem. 2014, 57, 204–222. [Google Scholar] [CrossRef] [PubMed]
- Bydal, P.; Auger, S.; Poirier, D. Inhibition of type 2 17β-hydroxysteroid dehydrogenase by estradiol derivatives bearing a lactone on the D-ring: Structure-activity relationship. Steroids 2004, 69, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Fournier, M.A.; Maltais, R.; Kenmogne, L.C.; Poirier, D. In vitro and in vivo evaluation of a 3β-androsterone derivative as inhibitor of 17β-hydroxysteroid dehydrogenase type 3. J. Steroid Biochem. Mol. Biol. 2014, 141, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Bydal, P.; Luu-The, V.; Labrie, F.; Poirier, D. Steroidal lactones as inhibitors of 17β-hydroxysteroid dehydrogenase type 5: Chemical synthesis, enzyme inhibitory activity, and assessment of estrogenic and androgenic activities. Eur. J. Med. Chem. 2009, 44, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Bellavance, É.; Luu-The, V.; Poirier, D. Potent and selective inhibitors of 17β-hydroxysteroid dehydrogenase type 7, an enzyme that catalyzes the reduction of the key hormones estrone and dihydrotestosterone. J. Med. Chem. 2009, 52, 7488–7502. [Google Scholar] [CrossRef] [PubMed]
- Ayan, D.; Maltais, R.; Poirier, D. Identification of a 17β-hydroxysteroid dehydrogenase type 10 steroidal inhibitor: A tool to investigate the role of type 10 in Alzheimer’s disease and prostate cancer. ChemMedChem 2012, 7, 1181–1184. [Google Scholar] [CrossRef] [PubMed]
- Kissinger, C.R.; Rejto, P.A.; Pelletier, L.A.; Thomson, J.A.; Showalter, R.E.; Abreo, M.A.; Agree, C.S.; Margosiak, S.; Meng, J.J.; Aust, R.M.; et al. Crystal structure of human ABAD/HSD10 with a bound inhibitor: Implications for design of Alzheimer’s disease therapeutics. J. Mol. Biol. 2004, 342, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Valasani, K.R.; Hu, G.; Chaney, M.O.; Yan, S.S. Structure-based design and synthesis of benzothiazole phosphonate analogues with inhibitors of human ABAD-Ab for treatment of Alzheimer’s diseases. Chem. Biol. Drug. Des. 2013, 81, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Benek, O.; Hroch, L.; Aitken, L.; Dolezal, R.; Guest, P.; Benkova, M.; Soukup, O.; Musil, K.; Kuka, K.; Smith, T.K.; et al. 6-Benzothiazolyl ureas, thioureas and guanidines are potent inhibitors of ABAD/17β-HSD10 and potential drugs for Alzheimer’s disease treatment: Design, synthesis and in vitro evaluation. Med. Chem. 2017, 13, 345–358. [Google Scholar] [CrossRef]
- Poirier, D. Advances in development oh inhibitors of 17β-hydroxysteroid dehydrogenases. Anti-Cancer Agents Med. Chem. 2009, 9, 642–660. [Google Scholar] [CrossRef]
- Yan, S.; Valasani, K.R. Phosphonate Derivatives and Methods of Use Thereof in the Treatment of Alzheimer’s Disease. U.S. Patent Application 2015/0065463 A1, 5 March 2015. [Google Scholar]
- Chakraborti, A.K.; Bhagat, S.; Rudrawar, S. Magnesium perchlorate as an efficient catalyst for the synthesis of imines and phenylhydrazones. Tetrahedron Lett. 2004, 45, 7641–7644. [Google Scholar] [CrossRef]
- Yang, S.Y.; He, X.Y.; Miller, D. Hydroxysteroid (17β) dehydrogenase X in human health and disease. Mol. Cell. Endocrinol. 2011, 343, 1–6. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. 1 | 1H NMR 2 | 13C NMR 2 | 1H NMR 3 (Estimated) | Difference 4(ppm) (%) | 13C NMR 3 (Estimated) | Difference 4 (ppm) (%) |
|---|---|---|---|---|---|---|
| CH-1 | 5.62 (dd, 9.5; 21.3) | 52.9 (154.8) 5 | 3.9 | −1.72 (30.6) | 69.0 | 16.1 (30.4) |
| C-2 | — | 126.8 | — | — | 128.6 | 1.8 |
| CH-3 | 7.94 (t, 2.2) | 129.3 (d, 6.0) | 7.78 | −0.16 (2.0) | 129.4 | 0.1 |
| C-4 | — | 113.0 | — | — | 111.7 | −1.3 |
| C-5 | — | 159.5 | — | — | 159.8 | 0.3 |
| CH-6 | 7.01 (d, 8.6) | 117.5 | 6.95 | −0.06 (0.9) | 116.7 | −0.8 |
| CH-7 | 7.65 (dt, 1.8; 8.6) | 135.3 (d, 5.3) | 7.32 | −0.33 (4.0) | 132.6 | −2.7 (2.0) |
| C-8 | — | 168.8 | — | — | 169.7 | 0.9 |
| CH3-9 | 3.91 (s) | 52.5 | 3.95 | 0.04 (1.0) | 51.5 | −1.0 |
| CH3-10/11 | 3.55/3.67 (2d, 10.6; 10.6) | 53.2/53.5 (2d, 6.7/7.1) | 3.66 | 0.11/−0.01 (3.0/0.3) | 53.4 | −0.1/0.2 |
| C-12 | — | 163.6 (d, 10.5) | — | — | 174.5 | 10.9 (6.7) |
| C-13 | — | 131.9 | — | — | 131.9 | 0 |
| C-14 | — | 145.5 | — | — | 145.5 | 0 |
| CH-15 | 7.30 (d, 9.1) | 118.7 | 7.53 | 0.23 (3.2) | 118.2 | −0.5 |
| CH-16 | 6.82 (dd, 2.7; 8.8) | 113.1 | 7.00 | 0.18 (2.6) | 114.6 | 1.5 |
| C-17 | — | 154.6 | — | — | 156.7 | 2.1 |
| CH-18 | 7.31 (d, 3.4) | 105.6 | 7.53 | 0.22 (3.0) | 104.9 | −0.7 |
| CH3-19 | 3.73 (s) | 55.5 | 3.80 | 0.07 (1.8) | 55.8 | 0.3 |
| NH | 8.88 (dd, 3.5; 9.5) | — | 6.79 | −2.09 (23.5) | — | — |
| OH | 10.51 (s) | — | 15.2 | 4.69 (44.6) | — | — |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boutin, S.; Poirier, D. Structure Confirmation and Evaluation of a Nonsteroidal Inhibitor of 17β-Hydroxysteroid Dehydrogenase Type 10. Magnetochemistry 2018, 4, 32. https://doi.org/10.3390/magnetochemistry4030032
Boutin S, Poirier D. Structure Confirmation and Evaluation of a Nonsteroidal Inhibitor of 17β-Hydroxysteroid Dehydrogenase Type 10. Magnetochemistry. 2018; 4(3):32. https://doi.org/10.3390/magnetochemistry4030032
Chicago/Turabian StyleBoutin, Sophie, and Donald Poirier. 2018. "Structure Confirmation and Evaluation of a Nonsteroidal Inhibitor of 17β-Hydroxysteroid Dehydrogenase Type 10" Magnetochemistry 4, no. 3: 32. https://doi.org/10.3390/magnetochemistry4030032
APA StyleBoutin, S., & Poirier, D. (2018). Structure Confirmation and Evaluation of a Nonsteroidal Inhibitor of 17β-Hydroxysteroid Dehydrogenase Type 10. Magnetochemistry, 4(3), 32. https://doi.org/10.3390/magnetochemistry4030032