One-Dimensional Chain-Type Dicopper Coordination Polymer Linked by 1,4-Di(4-pyridyl)benzene; Synthesis, Crystal Structure, Magnetic Property, and Gas-Adsorption Property


Abstract
:
1. Introduction
2. Results and Discussion
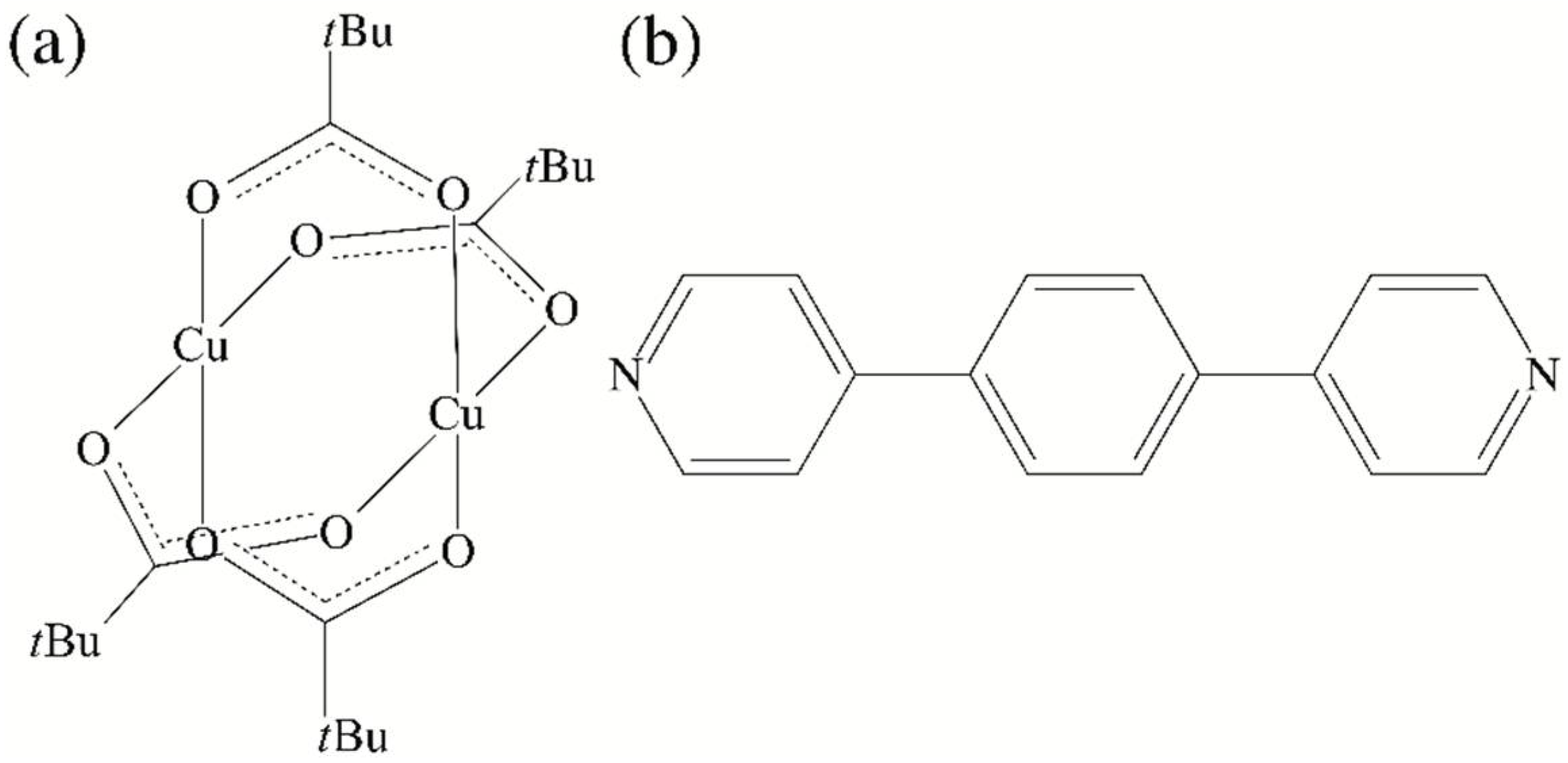
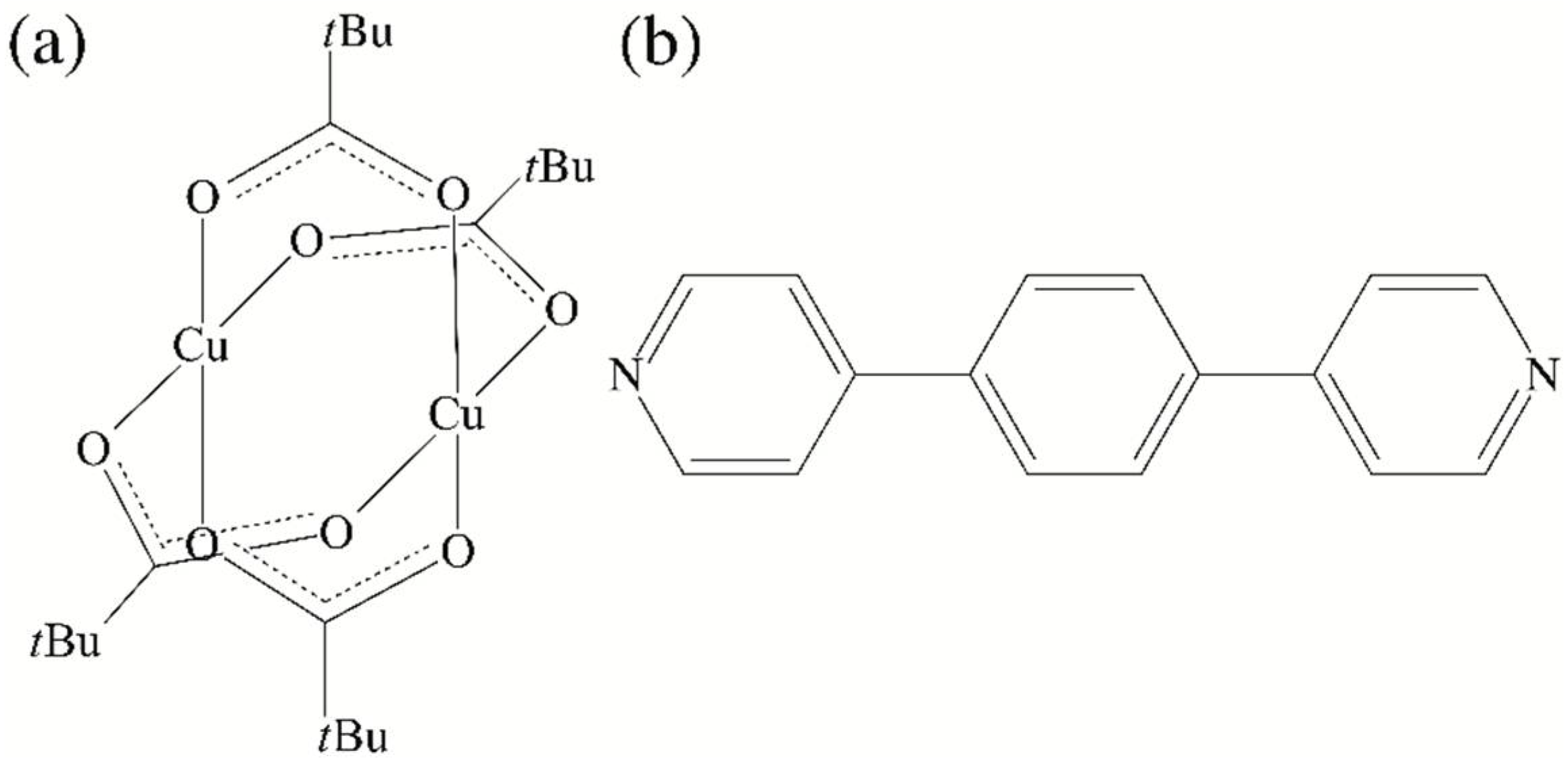
2.1. Synthesis and Characterization of [Cu2(O2C-tBu)4(dpybz)] (1)
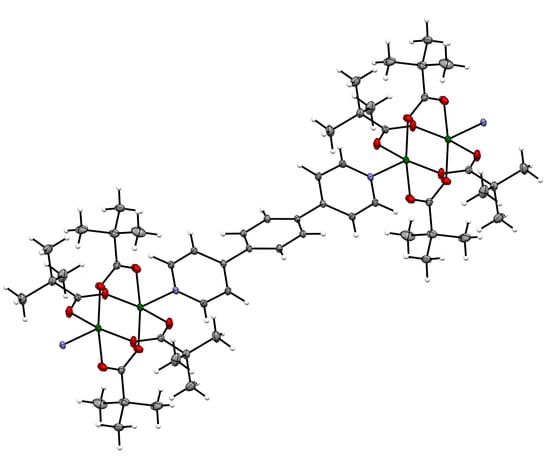
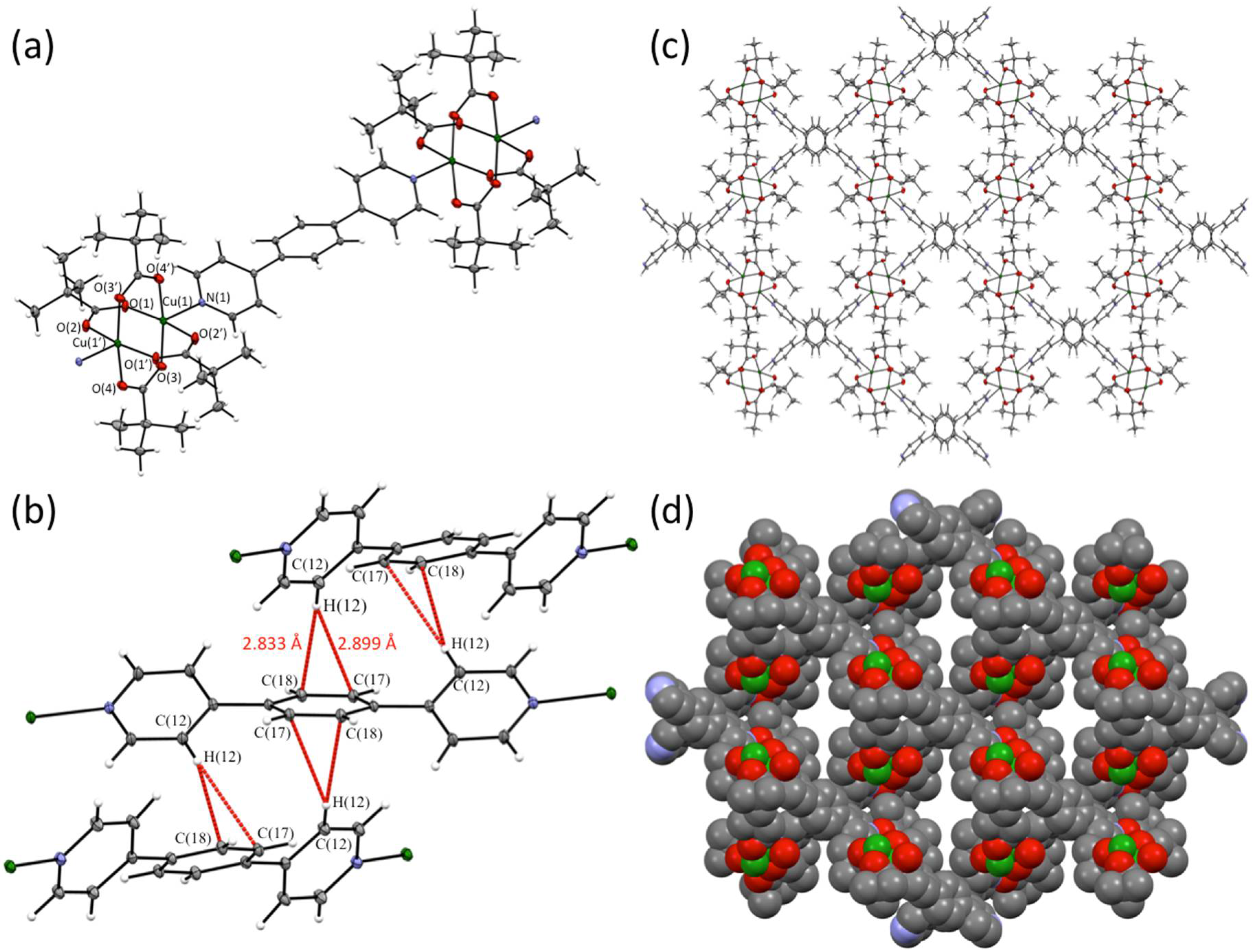
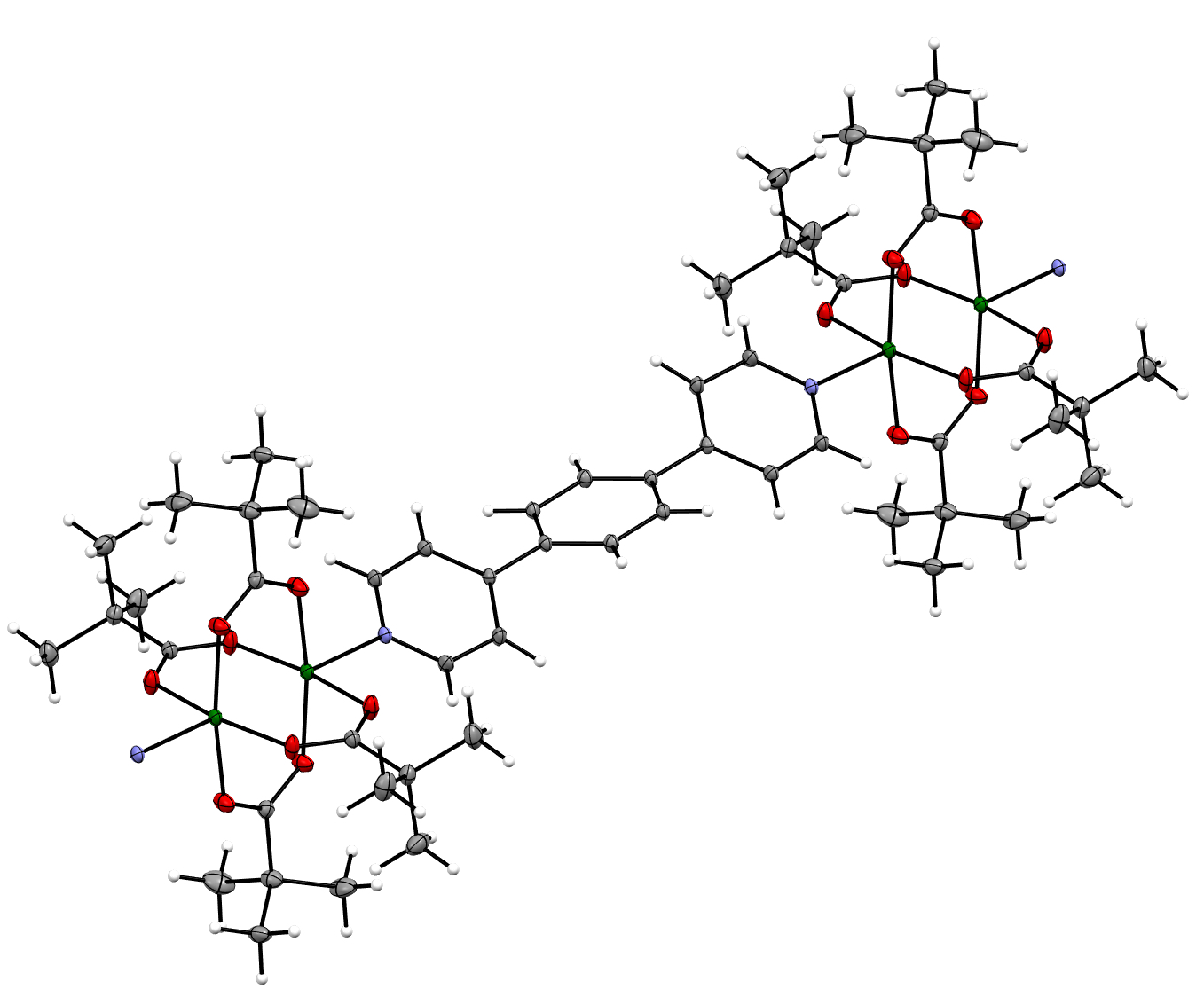
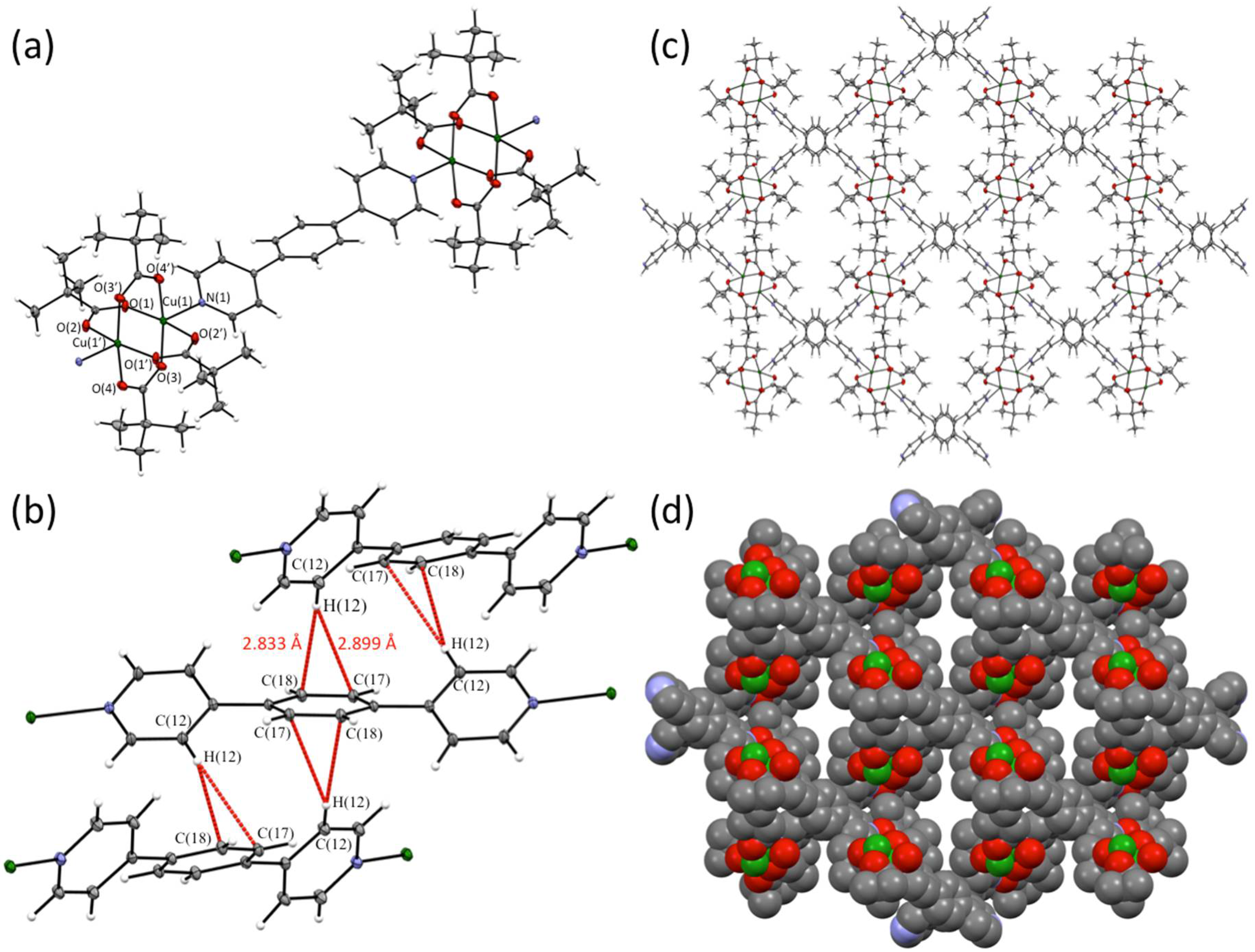
2.2. Single Crystal X-ray Diffraction
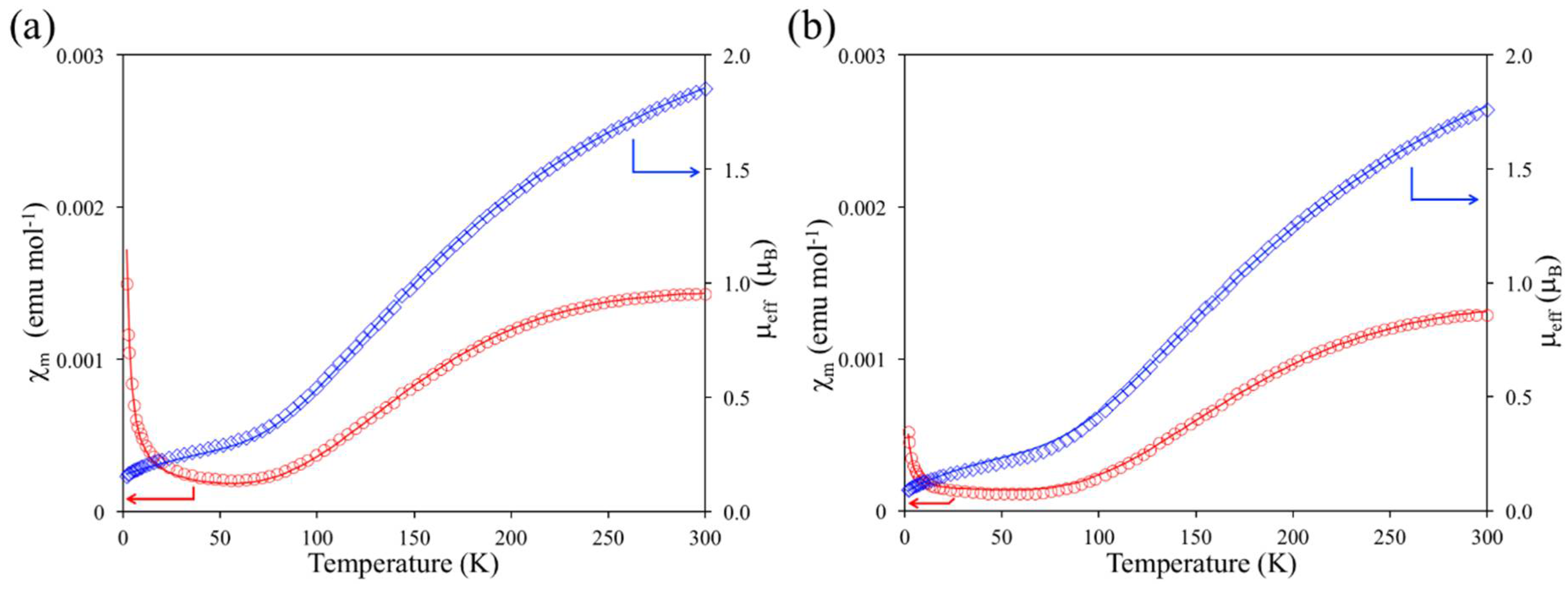
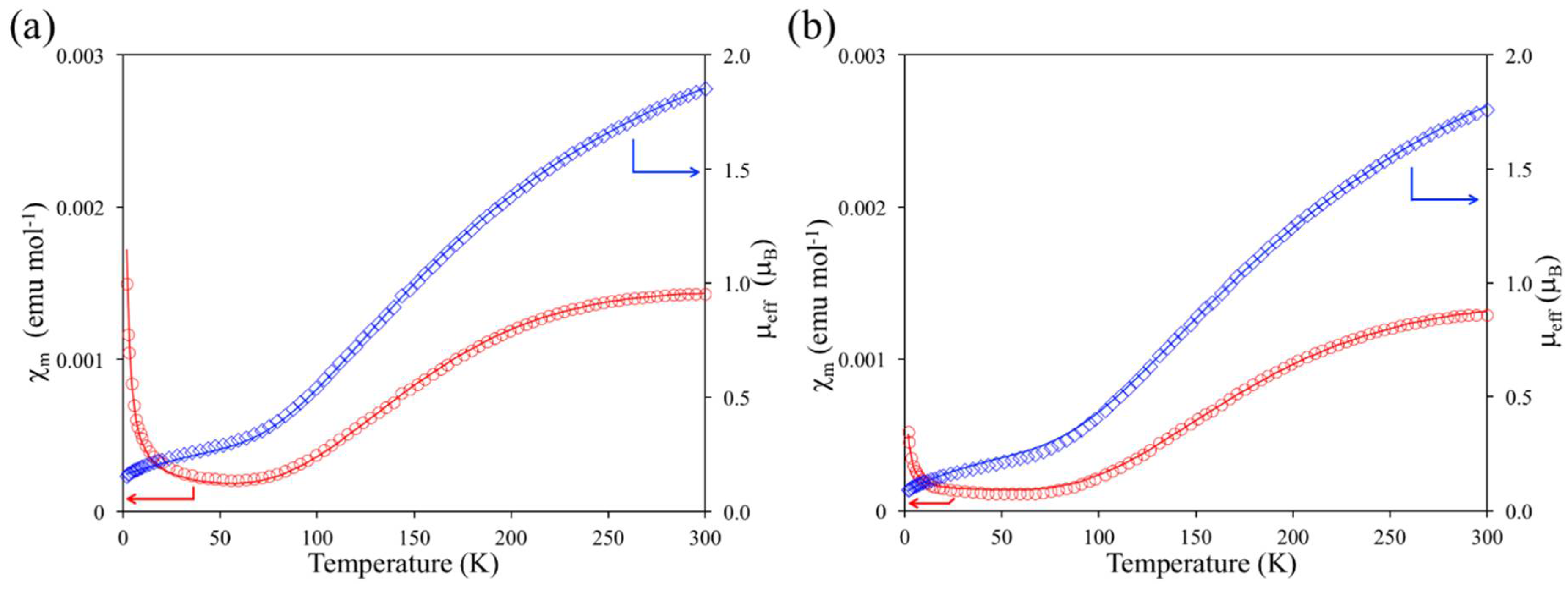
2.3. Magnetic Property of 1
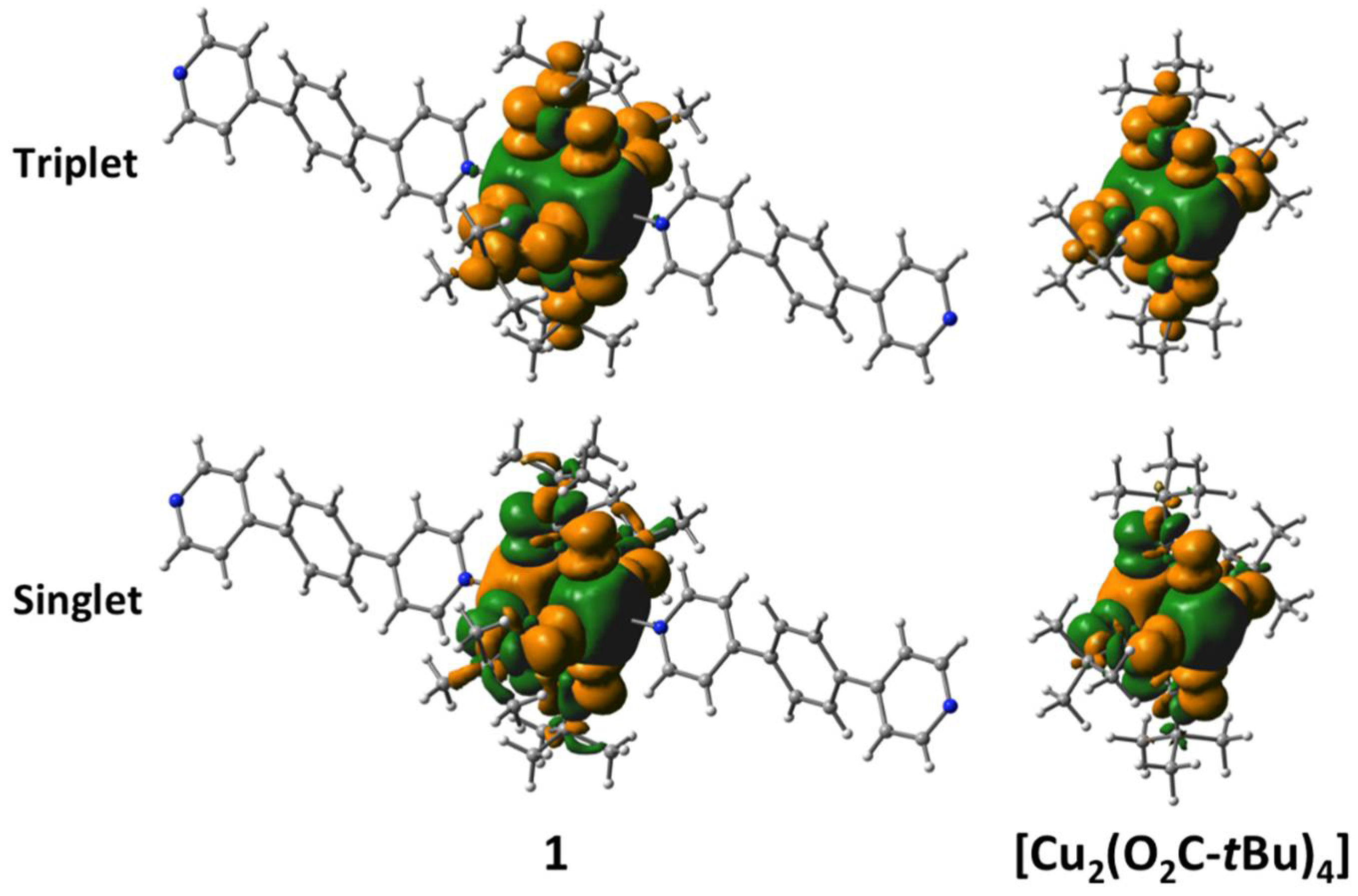
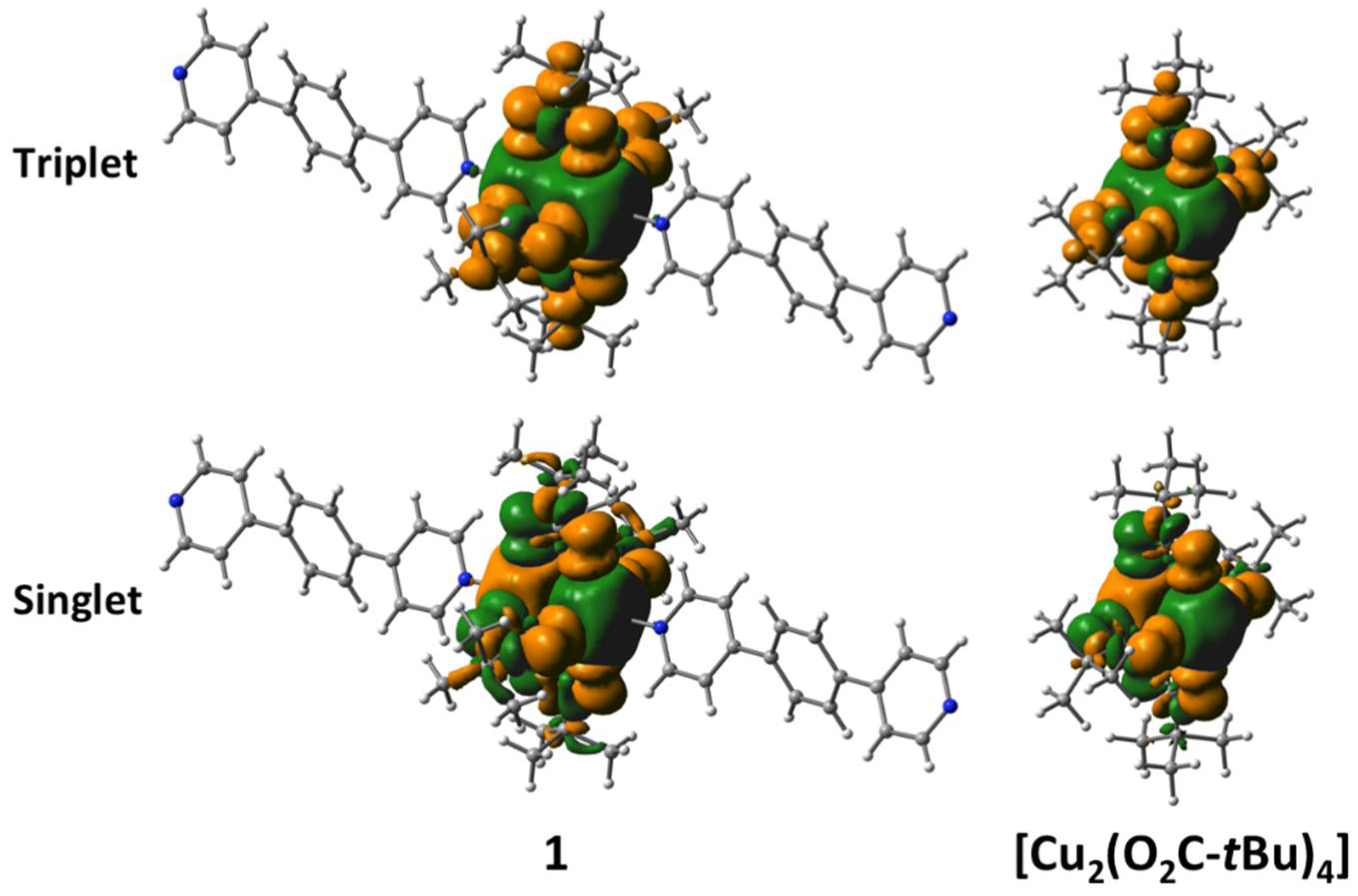
2.4. Results of Theoretical Calculations
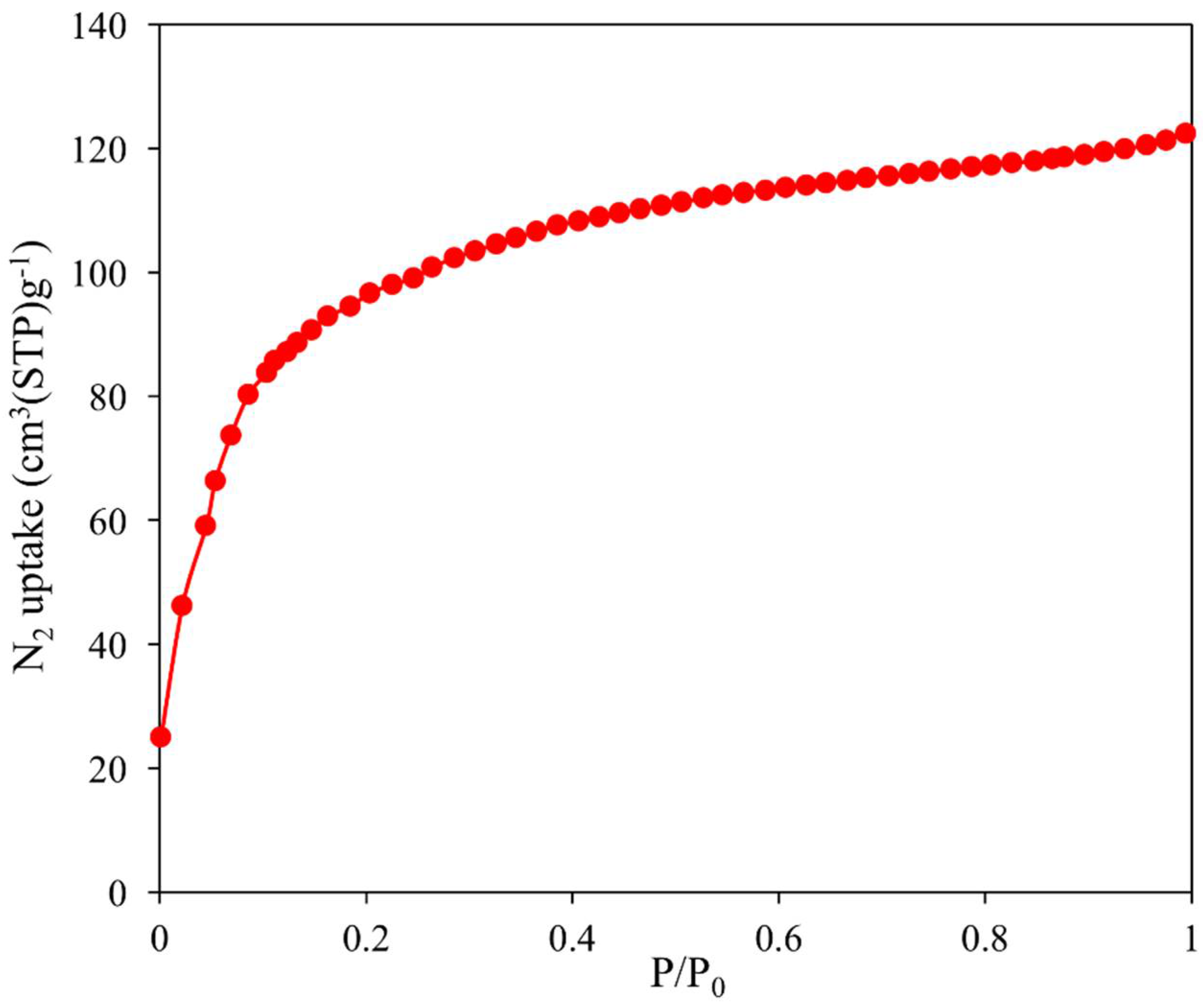
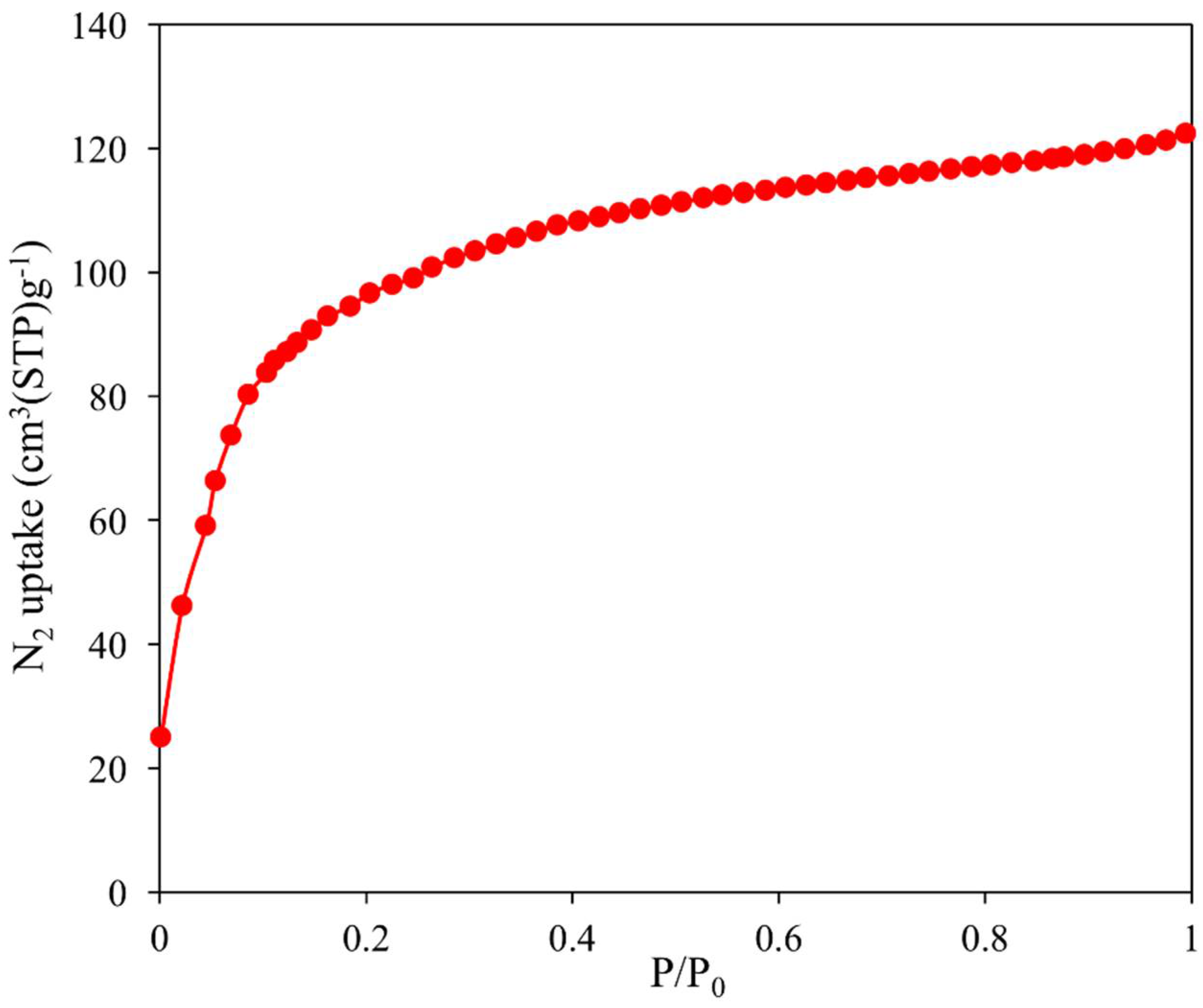
2.5. Nitrogen Adsorption Properties
3. Materials and Methods
3.1. General
3.2. Single Crystal X-ray Diffraction
3.3. Theoretical Calculation
3.4. Synthesis of [Cu2(O2C-tBu)4]
3.5. Synthesis of [Cu2(O2C-tBu)4(dpybz)] (1)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Nierkert, J.N.; Schoening, F.R.L. X-ray Evidence for Metal-toMetal Bonds in Cupric and Chromous Acetate. Nature 1953, 171, 6834–6859. [Google Scholar] [CrossRef]
- Melnik, M. Study of the relation between the structural data and magnetic interaction in oxo-bridged binuclear copper(II) compounds. Coord. Chem. Rev. 1982, 42, 259–293. [Google Scholar] [CrossRef]
- Kato, M.; Muto, Y. Factors affecting the magnetic properties of dimeric copper(II) complexes. Coord. Chem. Rev. 1988, 92, 45–83. [Google Scholar] [CrossRef]
- Köberl, M.; Cokoja, M.; Herrmann, W.A.; Kühn, F.E. From molecules to materials: Molecular paddle-wheel synthons of macromolecules, cage compounds and metal-organic frameworks. Dalton Trans. 2011, 40, 6834–6859. [Google Scholar] [CrossRef] [PubMed]
- Eddaoudi, M.; Kim, J.; Wachter, J.B.; Chae, H.K.; O’Keeffe, M.; Yaghi, O.M. Porous Metal-Organic Polyhedra: 25 Å Cuboctahedron Constructed from 12 Cu2(CO2)4 Paddle-Wheel Building Blocks. J. Am. Chem. Soc. 2001, 123, 4368–4369. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.M.; Sathyanarayana, D.N.; Manohar, H. X-ray crystal structures of somo adducts of dimeric copper(II) acetate. Nature of the copper-copper interaction. J. Chem. Soc. Dalton Trans. 1983, 0, 2167–2173. [Google Scholar] [CrossRef]
- Horikoshi, R.; Mikuriya, M. One-Dimensional Coordination Polymers from the Self-Assembly of Copper(II) Carboxylates and 4,4′-Dithiobis(pyridine). Bull. Chem. Soc. Jpn. 2005, 78, 827–834. [Google Scholar] [CrossRef]
- Mori, W.; Inoue, F.; Yoshida, K.; Nakayama, H.; Takamizawa, S.; Kishita, M. Synthesis of New Adsorbent Copper(II) Terephthalate. Chem. Lett. 1997, 26, 1219–1220. [Google Scholar] [CrossRef]
- Chui, S.S.-Y.; Lo, S.M.-F.; Charmant, J.P.H.; Orpen, A.G.; Williams, I.D. A Chemically Functionalizable Nanoporous Material [Cu3(TMA)2(H2O)3]n. Science 1999, 283, 1148–1150. [Google Scholar] [CrossRef] [PubMed]
- Seki, K.; Takamizawa, S.; Mori, W. Design and Gas Adsorption Property of a Three-Dimensional Coordination Polymer with a Stable and Highly Porous Framework. Chem. Lett. 2001, 30, 332–333. [Google Scholar] [CrossRef]
- Ohmura, T.; Usuki, A.; Fukumori, K.; Ohta, T.; Ito, M.; Tatsumi, K. New-Porphyrin-Based Metal-Organic Framework with High Porosity: 2-D Infinite 22.2 Å Square-Grid Coordination Network. Inorg. Chem. 2006, 45, 7988–7990. [Google Scholar] [CrossRef] [PubMed]
- Mikuriya, M.; Nukada, R.; Morishita, H.; Handa, M. Chain Compounds Formed by the Reaction of Copper(II) Carboxylate [Cu2(O2CR)4] (R = C(CH3)3, CCl3) and Bridging Ligand L (L = Pyrazine, 4,4’-Bipyridine, and 1,4-Diazabicyclo[2,2,2]octane). Chem. Lett. 1995, 24, 617–618. [Google Scholar] [CrossRef]
- Nukada, R.; Mori, W.; Takamizawa, S.; Mikuriya, M.; Handa, M.; Naono, H. Microporous Structure of a Chain Compound of Copper(II) Benzoate Bridged by Pyrazine. Chem. Lett. 1999, 28, 367–368. [Google Scholar] [CrossRef]
- Takahashi, K.; Hishino, N.; Takeda, T.; Noro, S.; Nakamura, T.; Takeda, S.; Akutagawa, T. Structural Flexibilities and Gas Adsorption Properties of One-Dimensional Copper(II) Polymers with Paddle-Wheel Units by Modification of Benzoate Ligands. Inorg. Chem. 2015, 54, 9423–9431. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.J.; Jo, Y.D.; Kim, H.; Kim, K.B.; Jung, K.-D.; Kim, C.; Kim, Y.; Kim, S.-J. Catalytic transesterification reactions of one-dimensional coordination polymers containing paddle-wheel-type units connected by various bridging ligands. Inorg. Chim. Acta 2013, 402, 39–45. [Google Scholar] [CrossRef]
- Liu, C.-S.; Wang, J.-J.; Yan, L.-F.; Chang, Z.; Bu, X.-H.; Sañudo, E.C.; Ribas, J. Copper(II), Cobalt(II), and Nickel(II) Complexes with a Bulky Anthracene-Based Carboxylate Ligand: Syntehsis, Crystal Structures, and Magnetic Properties. Inorg. Chem. 2007, 46, 6299–6310. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, Y.; Yano, N.; Shimodaira, T.; Yan, Y.-N.; Yamasaki, M.; Tanaka, H.; Omata, K.; Kawamoto, T.; Handa, M. Paddlewheel-Type Dirhodium Tetrapivalate Based Coordination Polymer: Synthesis, Characterization, and Self-Assembly and Disassembly Transformation Properties. Eur. J. Inorg. Chem. 2016, 17, 2810–2815. [Google Scholar] [CrossRef]
- Tsuzuki, S.; Fujii, A. Nature and physical origin of CH/π interaction: Significant difference from conventional hydrogen bonds. Phys. Chem. Chem. Phys. 2008, 19, 2584–2594. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Bleaney, B.; Bowers, K.D. Anomalous paramagnetism of copper acetate. Proc. R. Soc. A 1952, 214, 451–465. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Tsunekawa, T.; Yoyoda, Y.; Fueno, T. Ab initio molecular orbital calculations of effective exchange integrals between transition metal ions. Chem. Phys. Lett. 1988, 143, 371–376. [Google Scholar] [CrossRef]
- An, J.; Fiorella, R.P.; Geib, S.J.; Rosi, N.L. Synthesis, Structure, Assembly, and Modulation of the CO2 Adsorption Properties of a Zinc-Adeninate Macrocycle. J. Am. Chem. Soc. 2009, 131, 8401–8403. [Google Scholar] [CrossRef] [PubMed]
- Buria, M.C.; Caliandro, R.; Camalli, M.; Carrozzini, B.; Cascarano, G.L.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polodori, G.; Spagna, R. SIR2011: A new package for crystal structure determination and refinement. J. Appl. Cryst. 2012, 45, 357–361. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, C71, 3–8. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Muto, Y.; Hirashima, N.; Tokii, T.; Kato, M.; Suzuki, I. Magnetic Properties of Dimeric Copper(II) 2,2-Dimethylpropanoate. Bull. Chem. Soc. Jpn. 1986, 589, 3672–3674. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Magnetic Parameters | 1 | [Cu2(O2C-tBu)4] |
|---|---|---|
| J (cm−1) | −175.3 | −201.4 |
| g | 2.09 | 2.17 |
| Nα (cm3 mol−1) | 60 × 10−6 | 60 × 10−6 |
| p | 0.0039 | 0.00088 |
| aR/104 | 4.41 | 3.76 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yano, N.; Handa, M.; Mitsumi, M.; Kataoka, Y. One-Dimensional Chain-Type Dicopper Coordination Polymer Linked by 1,4-Di(4-pyridyl)benzene; Synthesis, Crystal Structure, Magnetic Property, and Gas-Adsorption Property. Magnetochemistry 2018, 4, 26. https://doi.org/10.3390/magnetochemistry4020026
Yano N, Handa M, Mitsumi M, Kataoka Y. One-Dimensional Chain-Type Dicopper Coordination Polymer Linked by 1,4-Di(4-pyridyl)benzene; Synthesis, Crystal Structure, Magnetic Property, and Gas-Adsorption Property. Magnetochemistry. 2018; 4(2):26. https://doi.org/10.3390/magnetochemistry4020026
Chicago/Turabian StyleYano, Natsumi, Makoto Handa, Minoru Mitsumi, and Yusuke Kataoka. 2018. "One-Dimensional Chain-Type Dicopper Coordination Polymer Linked by 1,4-Di(4-pyridyl)benzene; Synthesis, Crystal Structure, Magnetic Property, and Gas-Adsorption Property" Magnetochemistry 4, no. 2: 26. https://doi.org/10.3390/magnetochemistry4020026
APA StyleYano, N., Handa, M., Mitsumi, M., & Kataoka, Y. (2018). One-Dimensional Chain-Type Dicopper Coordination Polymer Linked by 1,4-Di(4-pyridyl)benzene; Synthesis, Crystal Structure, Magnetic Property, and Gas-Adsorption Property. Magnetochemistry, 4(2), 26. https://doi.org/10.3390/magnetochemistry4020026