Local and Average Structural Changes in Zeolite A upon Ion Exchange


Abstract
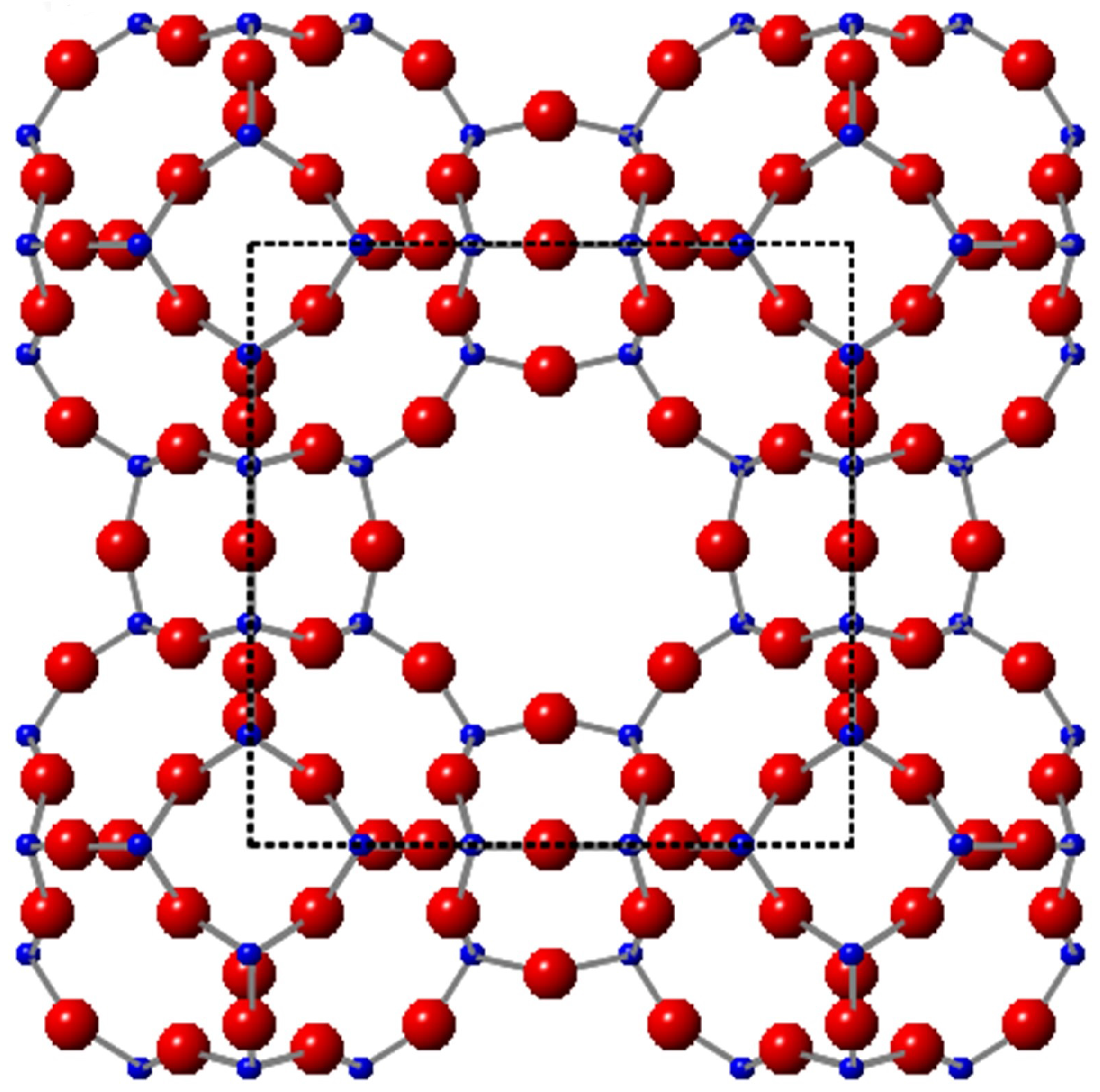
1. Introduction
2. Results and Discussion
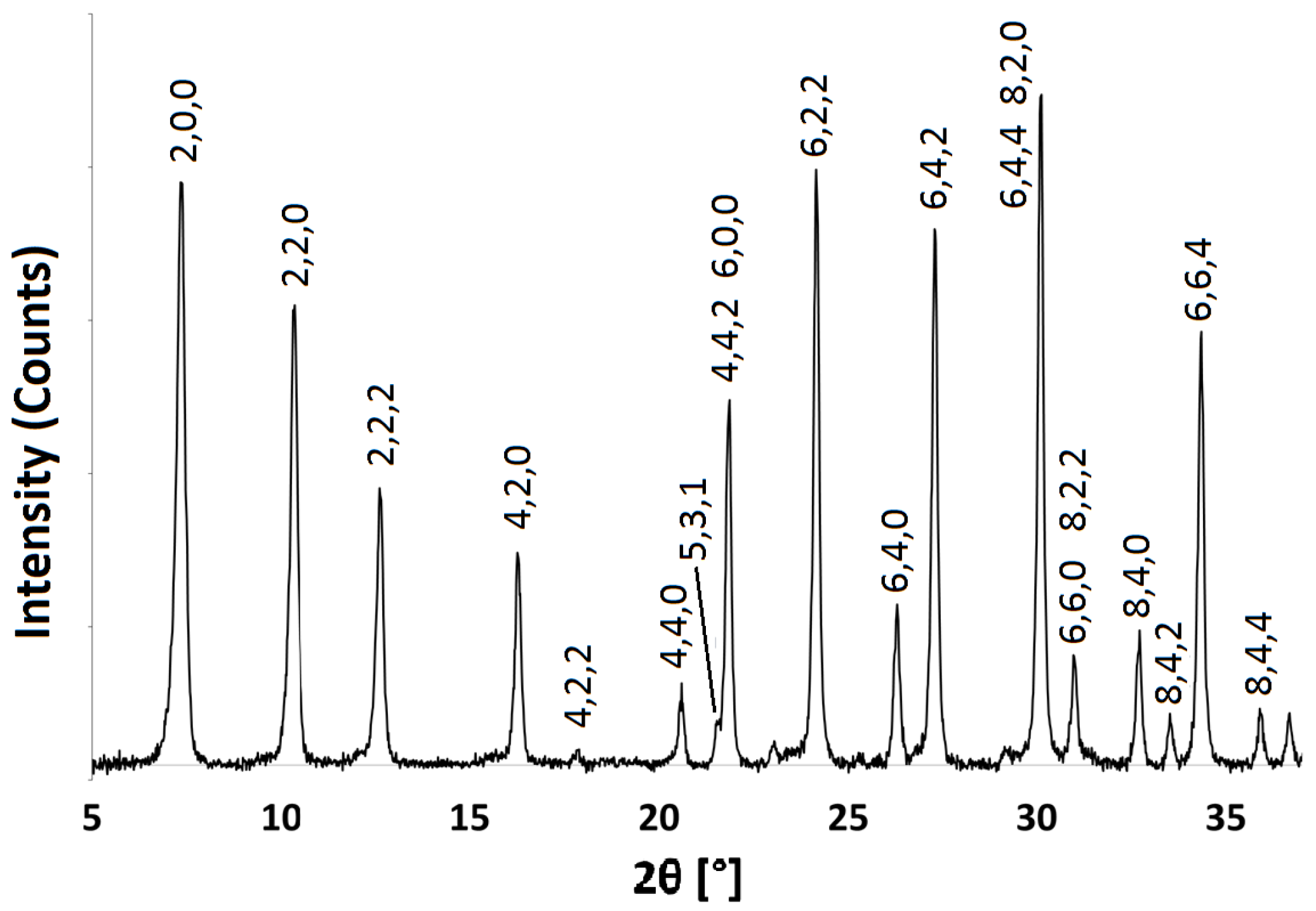
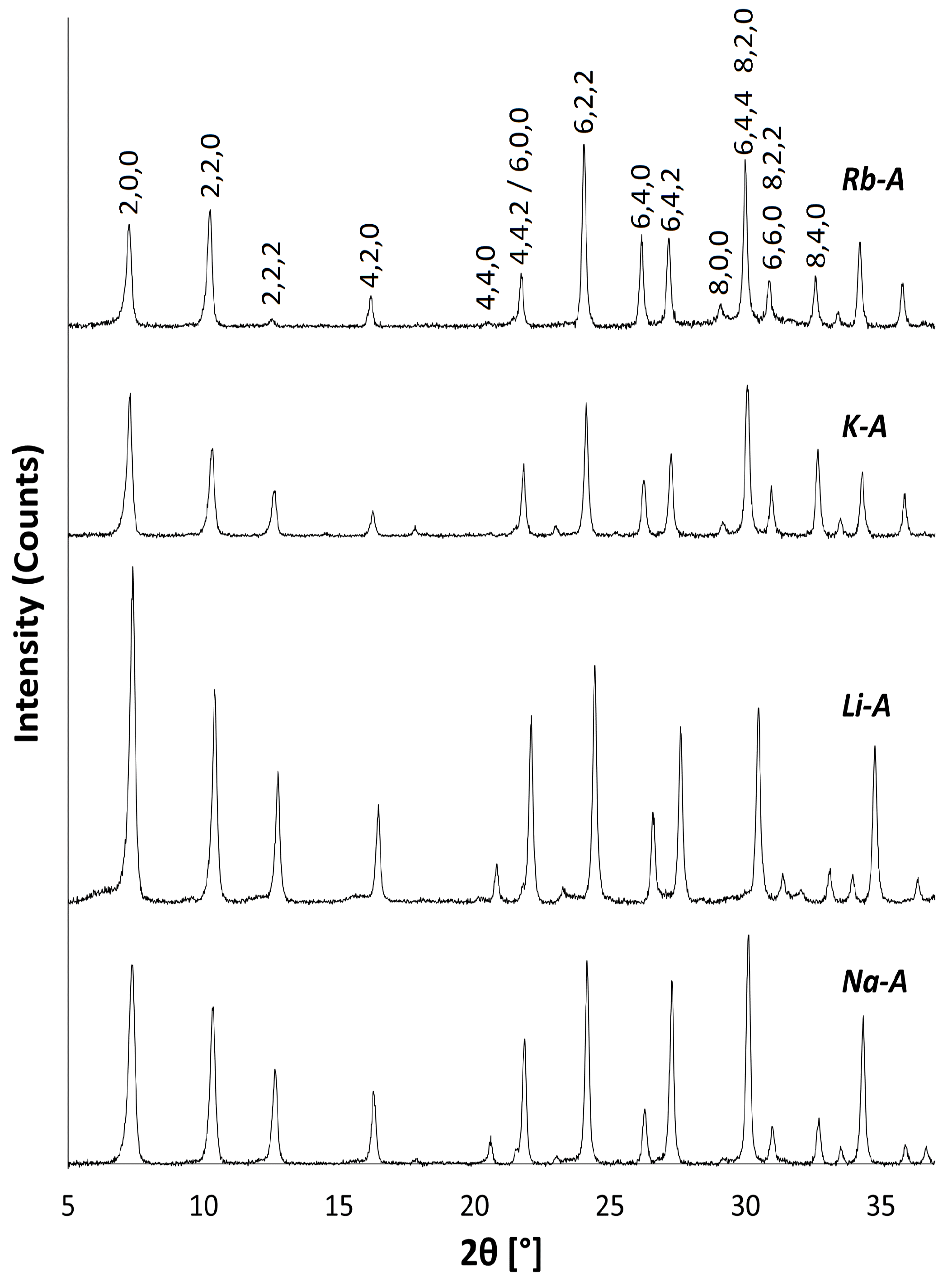
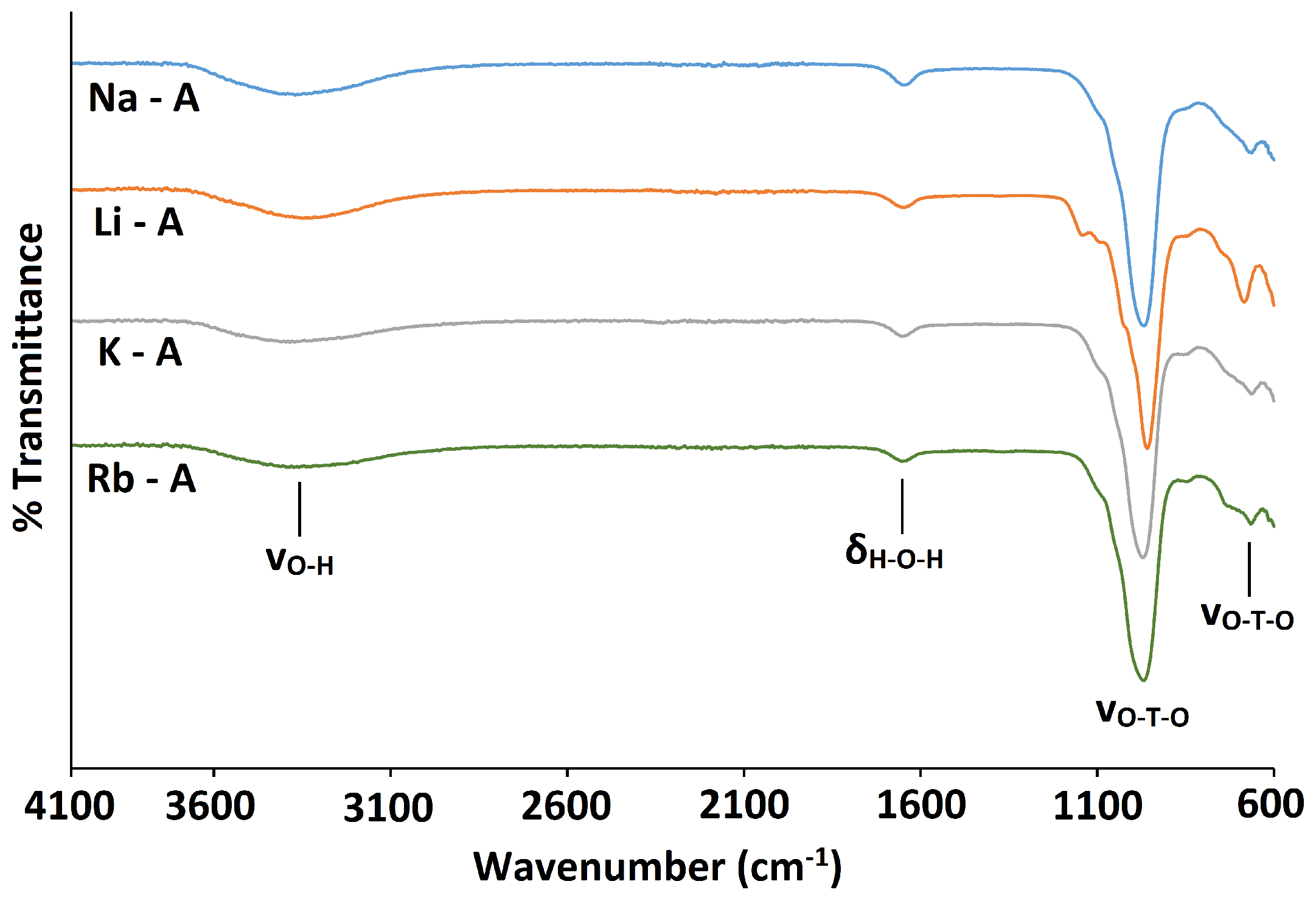
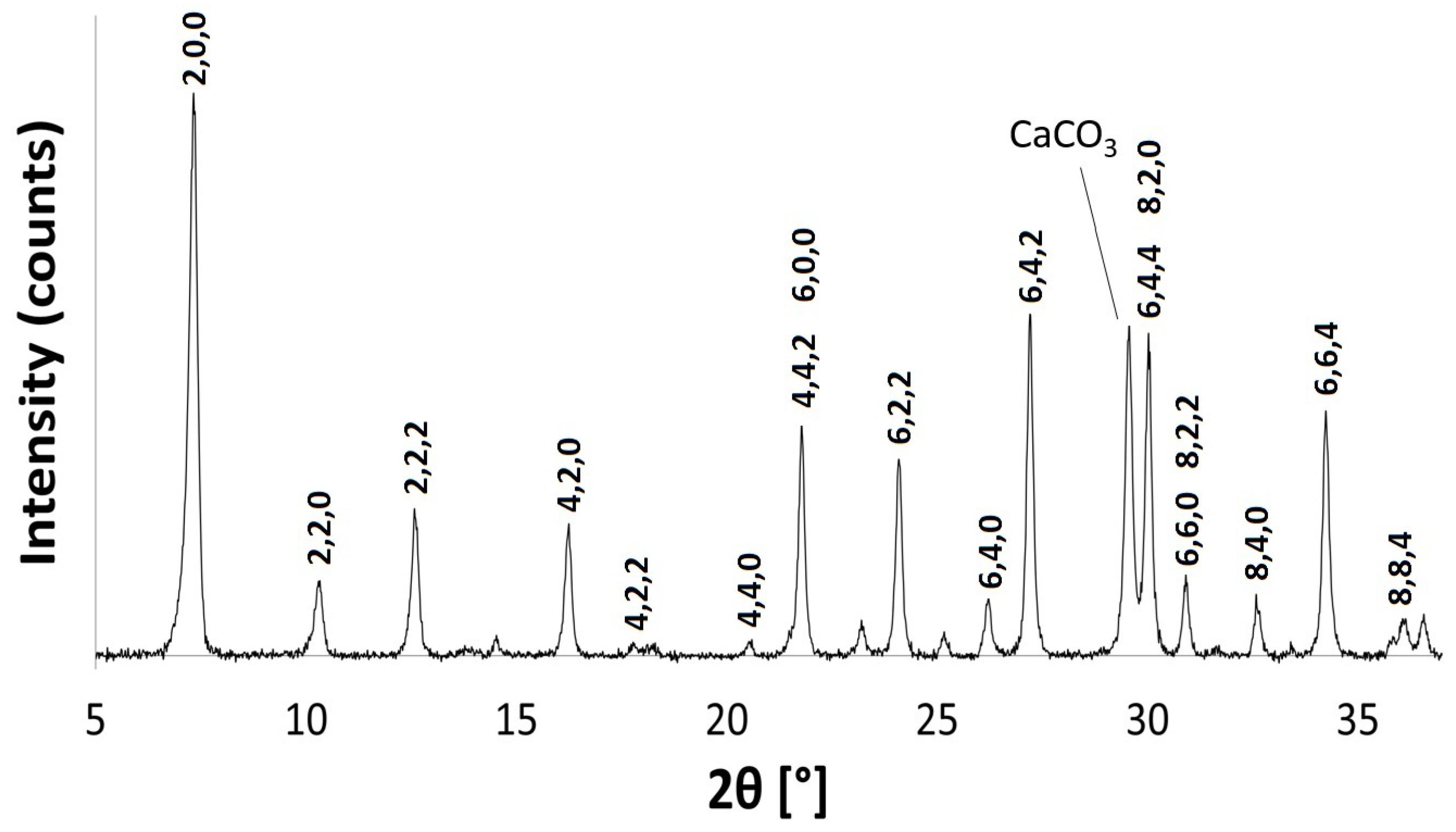
2.1. Na-A, Li-A, K-A and Rb-A
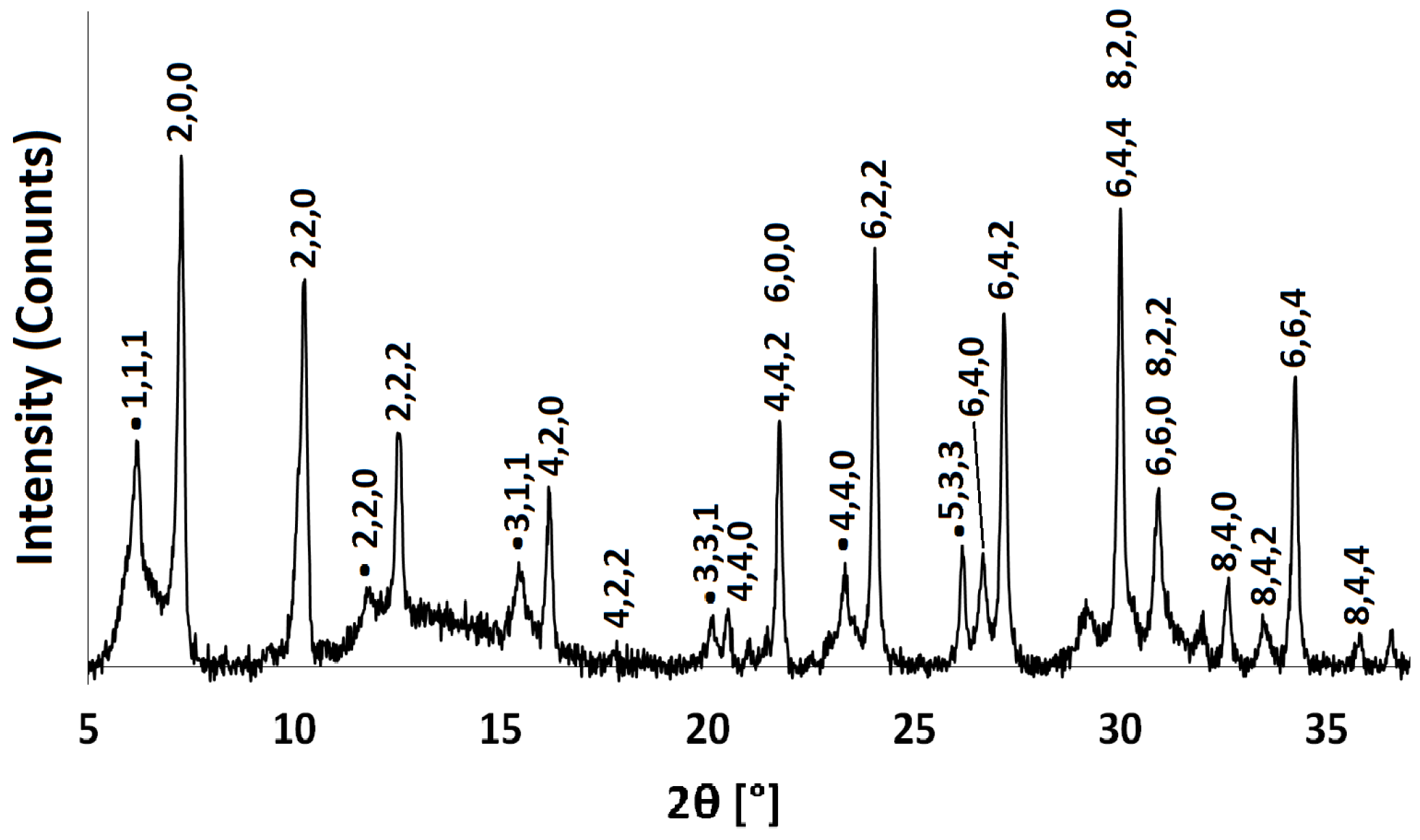
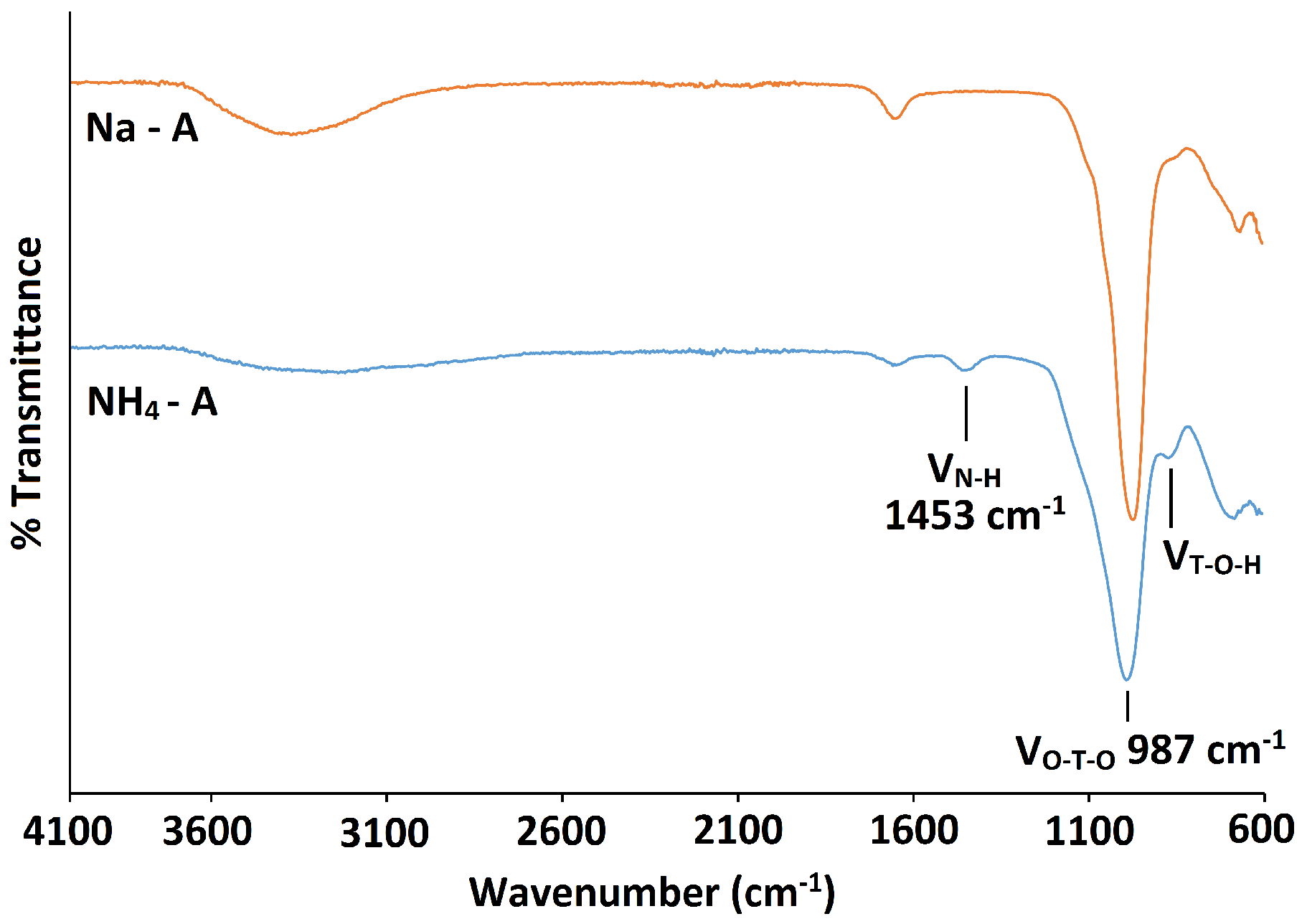
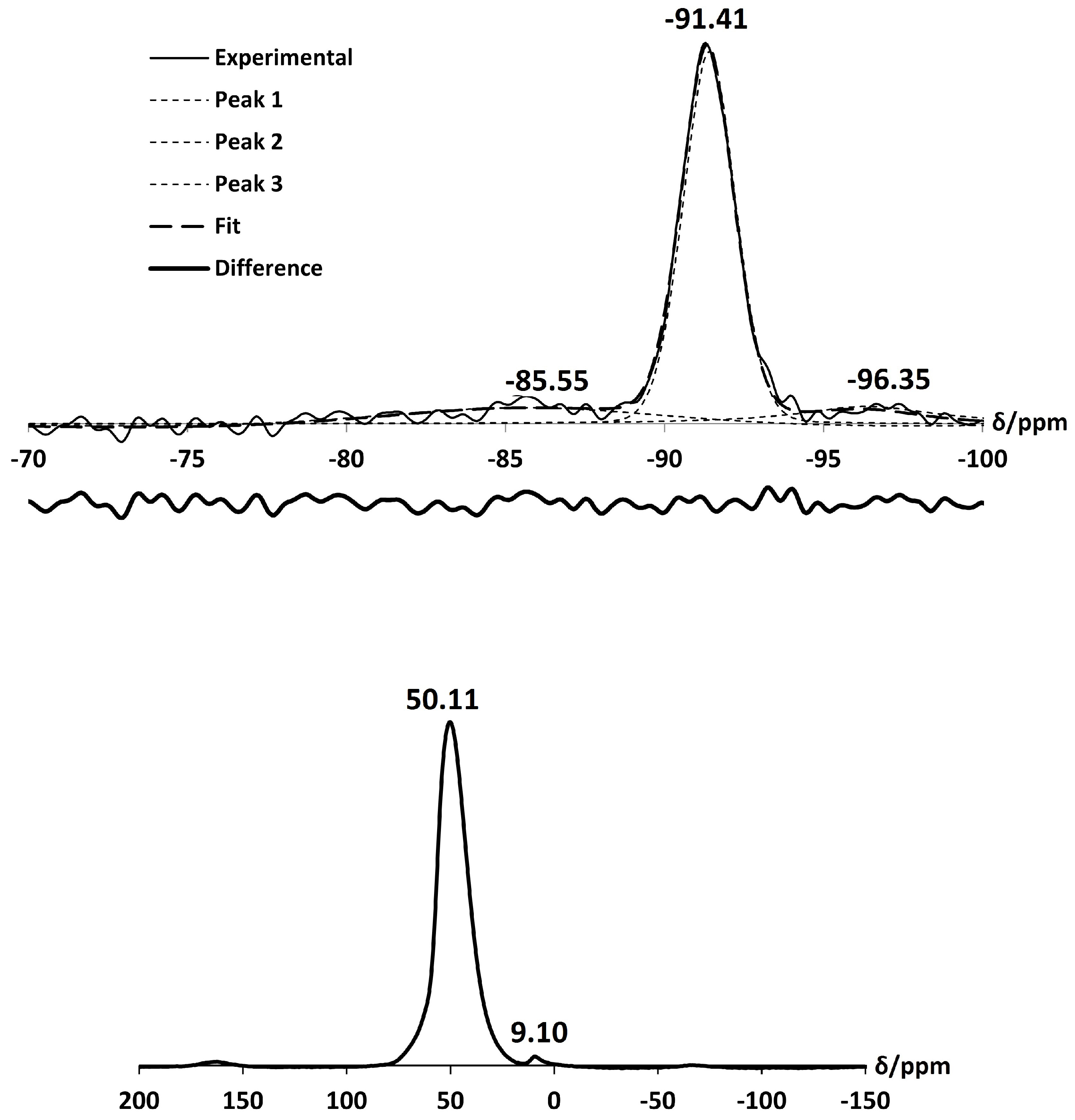
2.2. NH-A
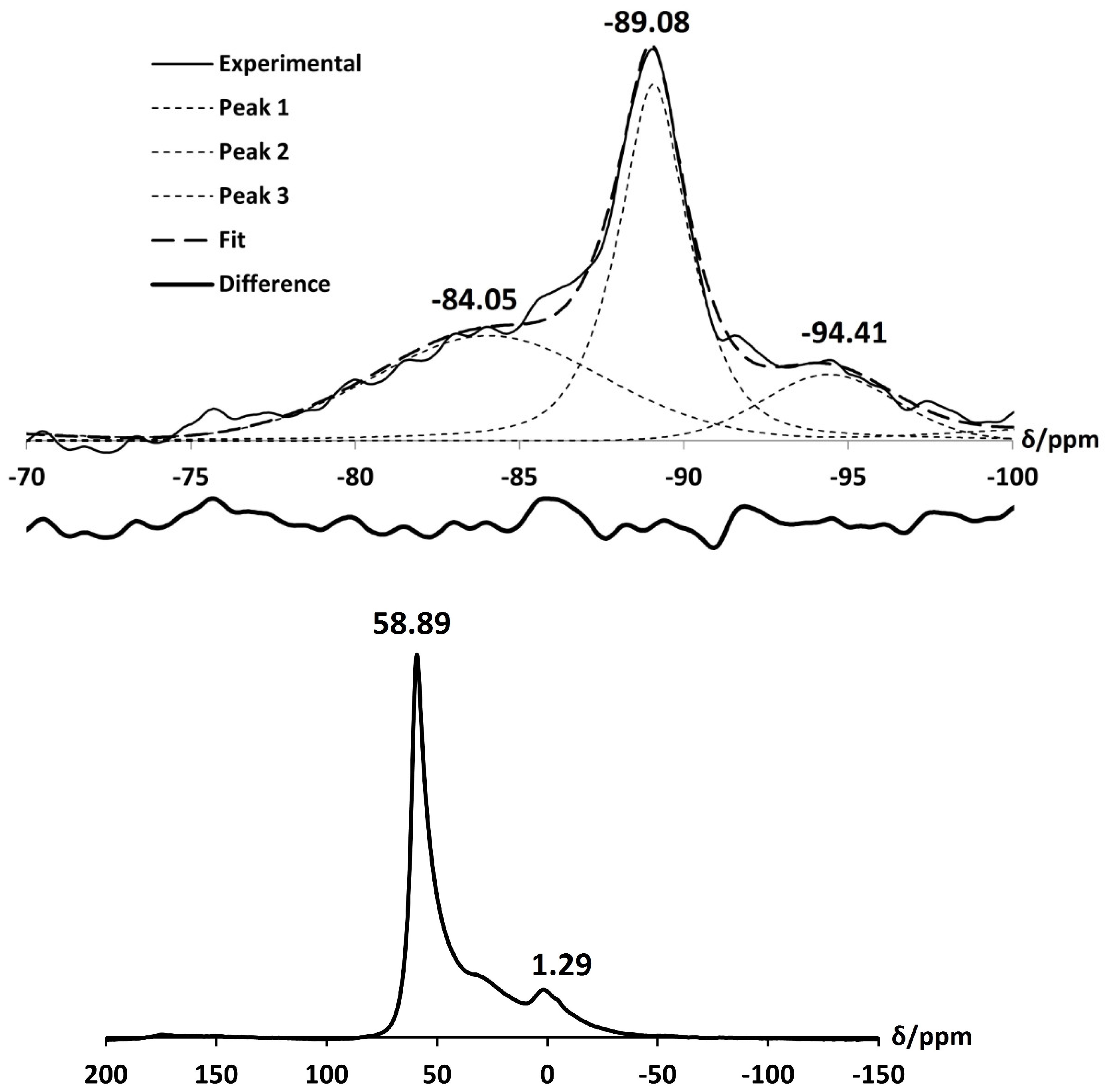
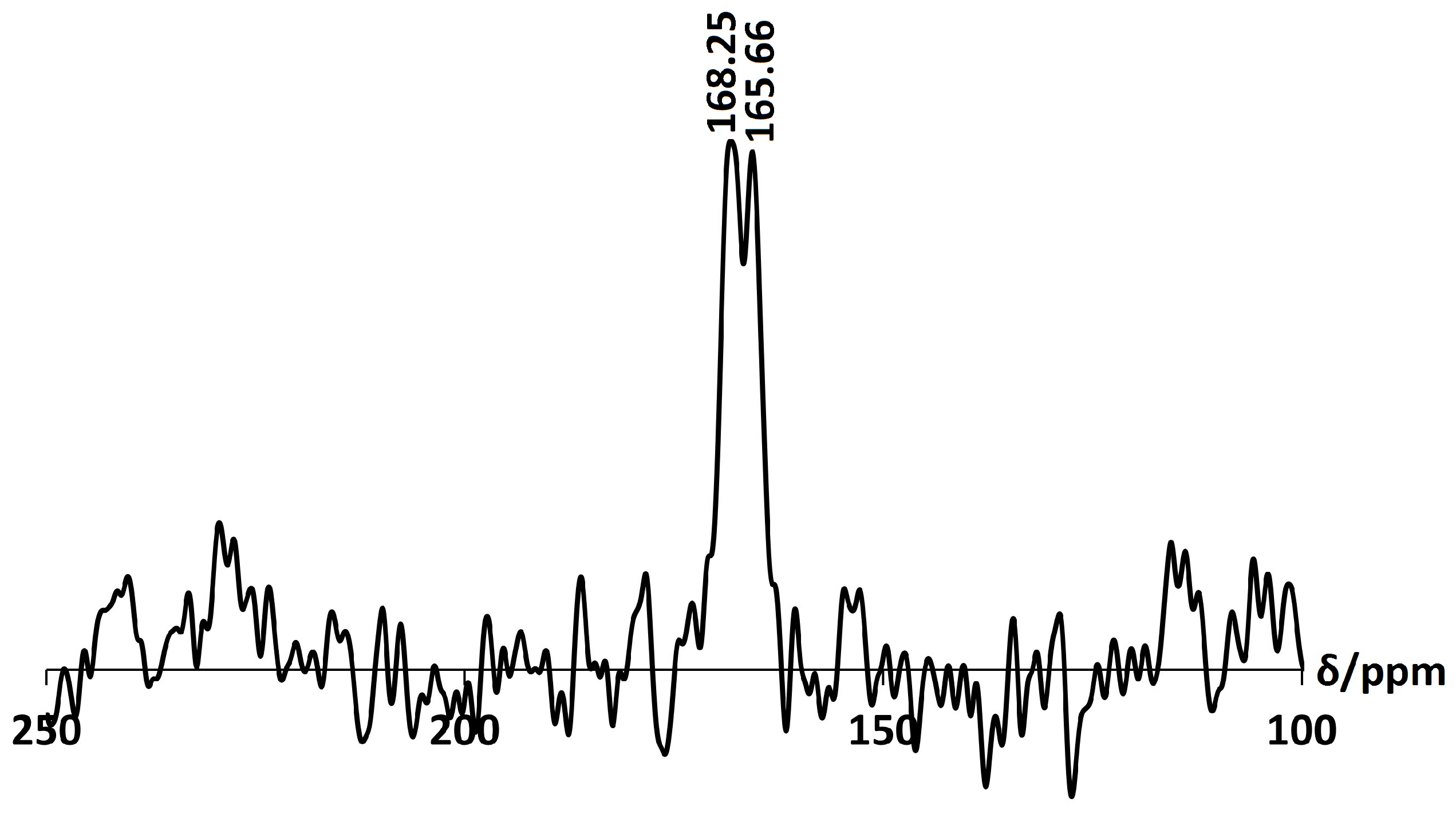
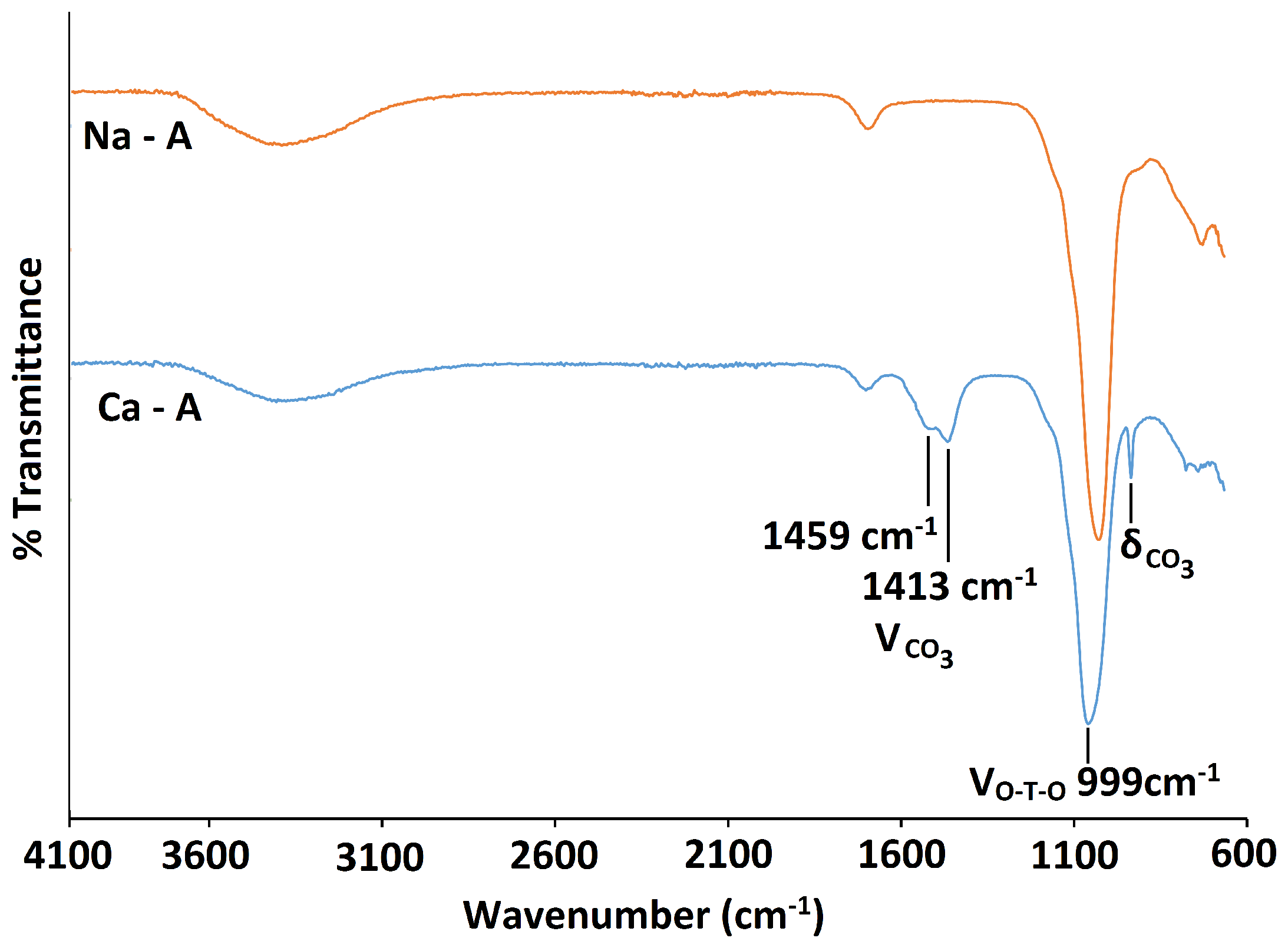
2.3. Ca-A
2.4. SEM
3. Materials and Methods
3.1. Synthesis of Na-A: Low Temperature and Organic Template-Free
3.2. Ion Exchange
3.3. Product Characterisation
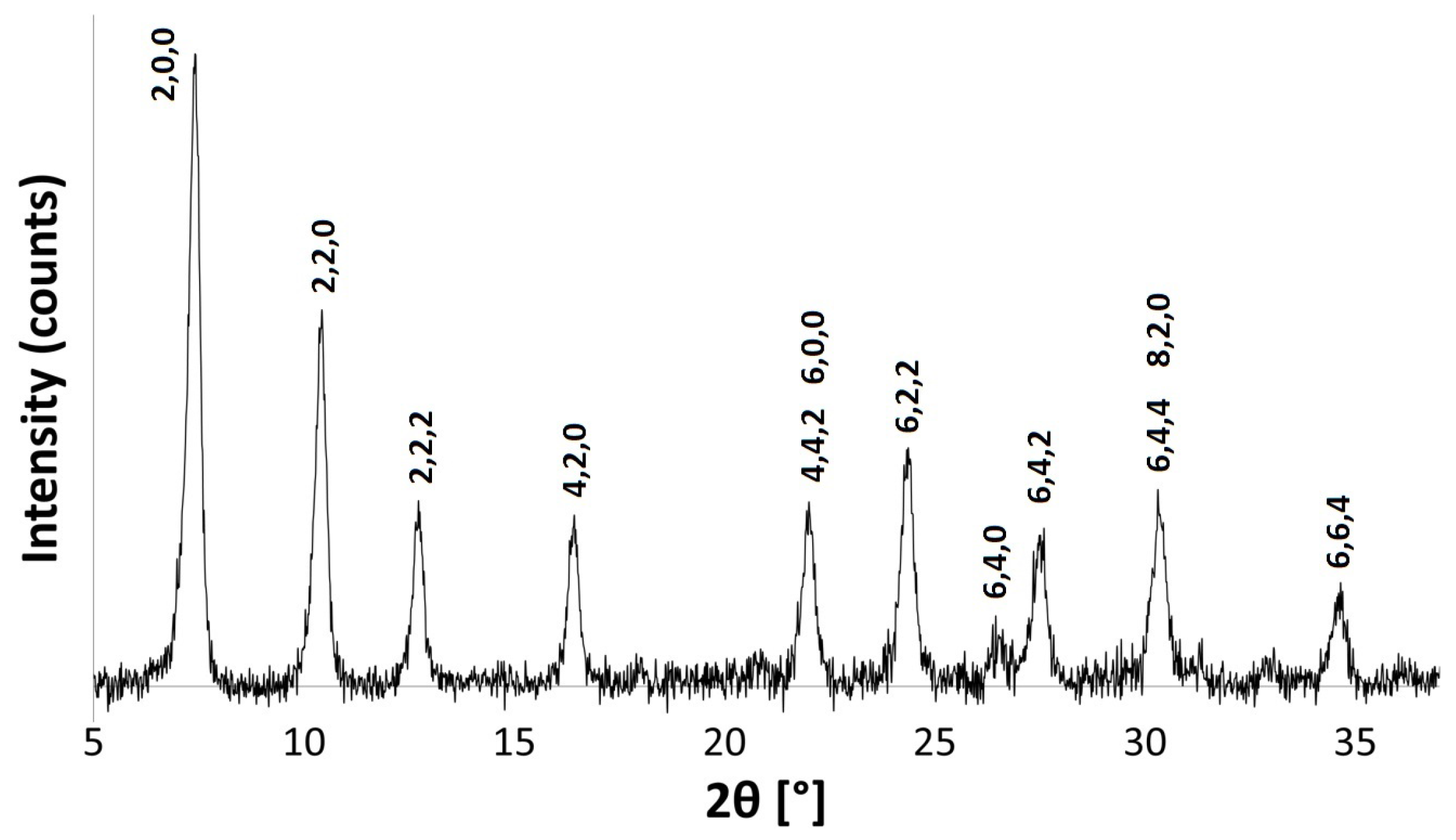
3.3.1. Powder X-ray Diffraction
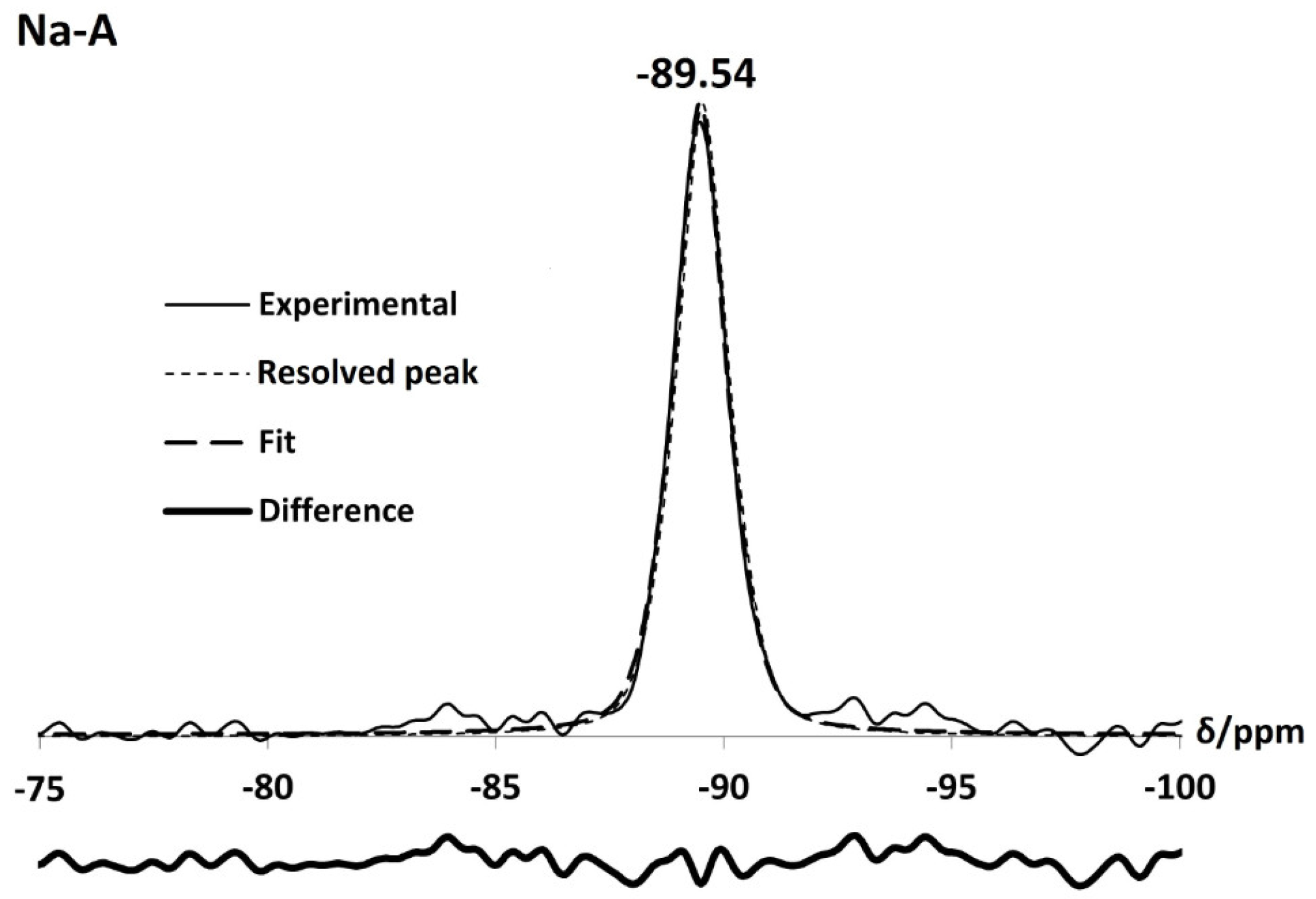
3.3.2. Solid State Nuclear Magnetic Resonance
3.3.3. Scanning Electron Microscopy and Energy Dispersive X-ray Spectroscopy
3.3.4. Fourier Transform Infrared Spectroscopy
3.3.5. Sodium Ion Selective Electrode
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baerlocher, C.; McCusker, L.B.; Olson, D.H. Atlas of Zeolite Framework Types; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Sartbaeva, A.; Wells, S.A.; Treacy, M.M.J.; Thorpe, M.F. The flexibility window in zeolites. Nat. Mat. 2006, 5, 962–965. [Google Scholar] [CrossRef] [PubMed]
- Conato, M.T.; Oleksiak, M.D.; McGrail, B.P.; Motkuri, R.K.; Rimer, J.D. Framework Stabilization of Si-Rich LTA Zeolite Prepared in Organic-Free Media. Chem. Commun. 2014, 51, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.M.; Edwards, P.P.; Jones, E.; Sartbaeva, A. Microwave synthesis of LTN framework zeolite with no organic structure directing agents. RSC Adv. 2015, 5, 35580–35585. [Google Scholar] [CrossRef]
- Nearchou, A.; Raithby, P.; Sartbaeva, A. Systematic approaches towards template-free synthesis of EMT-type zeolites. Microporous Mesoporous Mater. 2018, 255, 261–270. [Google Scholar] [CrossRef]
- Nearchou, A.; Sartbaeva, A. Influence of alkali metal cations on the formation of zeolites under hydrothermal conditions with no organic structure directing agents. CrystEngComm 2008, 17, 2496–2503. [Google Scholar] [CrossRef]
- Maldonado, M.; Oleksiak, M.D.; Chinta, S.; Rimer, J.D. Controlling Crystal Polymorphism in Organic-Free Synthesis of Na-Zeolites. J. Am. Chem. Soc. 2012, 135, 2641–2652. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, C.P. Zeolites in Industrial Separation and Catalysis; Wiley VCH: Weinheim, Germany, 2010; pp. 355–402. [Google Scholar]
- Amorim, R.; Vilaça, N.; Martinho, O.; Reis, R.M.; Sardo, M.; Rocha, J.; Fonseca, A.M.; Baltazar, F.; Neves, I.C. Zeolite Structures Loading with an Anticancer Compound As Drug Delivery Systems. J. Phys. Chem. C 2012, 116, 25642–25650. [Google Scholar] [CrossRef]
- Chaves, T.F.; Soares, F.; Cardoso, D.; Carneiro, R.L. Monitoring of the crystallization of Zeolite LTA using Raman and Chemometric tools. Analyst 2015, 140, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Lalik, E.; Mirek, R.; Rakoczy, J.; Groszek, A. Microcalorimetric study of sorption of water and ethanol in zeolites 3A and 5A. Catal. Today 2006, 114, 242–247. [Google Scholar] [CrossRef]
- Liu, Q.; Mace, A.; Bacsik, Z.; Sun, J.; Laaksonen, A.; Hedin, N. NaKA sorbents with high CO2-over-N2 selectivity and high capacity to adsorb CO2. Chem. Comm. 2010, 46, 4502–4504. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wu, D.; Guo, X.; Shen, B.; Liu, J.; Navrotsky, A. Energetics of Confinement of n-Hexane in Ca-Na Ion Exchanged Zeolite A. J. Phys. Chem. C 2014, 118, 25590–25596. [Google Scholar] [CrossRef]
- Kwakye-Awuah, B.; Labik, L.K.; Nkrumah, I.; Williams, C. Removal of ammonium ions by laboratory synthesized zeolite linde type A adsorption from water samples affected by mining activities in Ghana. J. Water Health 2014, 12, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Sartbaeva, A.; Rees, N.H.; Edwards, P.P.; Ramirez-Cuesta, A.J.; Barney, E. Local probes show that framework modification in zeolites occurs on ammonium exchange without calcination. J. Mater. Chem. A 2013, 1, 7415–7421. [Google Scholar] [CrossRef]
- Seel, A.G.; Sartbaeva, A.; Edwards, P.P.; Rammirez-Cuesta, A.J. Inelastic neutron scattering of Na-zeolite A with in situ ammoniation: An examination of initial coordination. Phys. Chem. Chem. Phys. 2010, 12, 9661–9666. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Yamada, H.; Tanaka, J.; Komatsu, Y.; Moriyoshi, Y. Ammonium Ion Exchange of Synthetic Zeolites: The Effect of Their Open Window Sizes, Pore Structures, and Cation Exchange Capacities. Sep. Sci. Technol. 2005, 39, 2091–2104. [Google Scholar] [CrossRef]
- Navarrete-Casas, R.; Navarrete-Guijosa, A.; Valenzuela-Calahorro, C.; López-González, J.D.; García-Rodríguez, A. Study of lithium ion exchange by two synthetic zeolites: Kinetics and equilibrium. J. Colloid Interface Sci. 2007, 306, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Choi, S.Y.; Lim, W.T. Complete Li+ exchange into zeolite X (FAU, Si\Al = 1.09) from undried methanol solution. J. Porous Mater. 2013, 20, 1449–1456. [Google Scholar] [CrossRef]
- Bignami, G.P.; Dawson, D.M.; Seymour, V.R.; Wheatley, P.S.; Morris, R.E.; Ashbrook, S.E. Synthesis, Isotopic Enrichment, and Solid-State NMR Characterization of Zeolites Derived from the Assembly, Disassembly, Organization, Reassembly Process. J. Am. Chem. Soc. 2017, 139, 5140–5148. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, D.H.; Darton, R.J.; Morris, R.E.; Levitt, M.H. A solid-state NMR method for solution of zeolite crystal structures. J. Am. Chem. Soc. 2005, 127, 10365–10370. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, C.A.; Feng, Y.; Grondey, H.; Kokotailo, G.T.; Gies, H. One- and two-dimensional high-resolution solid-state NMR studies of zeolite lattice structures. Chem. Rev. 1991, 91, 1525–1543. [Google Scholar] [CrossRef]
- Fyfe, C.A.; Gobbi, G.C.; Hartman, J.S.; Klinowski, J.; Thomas, J.M. Solid-state magic-angle spinning. Aluminum-27 nuclear magnetic resonance studies of zeolites using a 400-MHz high-resolution spectrometer. J. Phys. Chem. 1982, 86, 1247–1250. [Google Scholar] [CrossRef]
- Fyfe, C.A.; Thomas, J.M.; Klinowski, J.; Gobbi, G.C. Magic-Angle-Spinning NMR (MAS-NMR) Spectroscopy and the Structure of Zeolites. Angew. Chem. Int. Ed. Engl. 1983, 22, 259–275. [Google Scholar] [CrossRef]
- Lippmaa, A.; Maedi, M.; Samoson, A.; Tarmak, M.; Engelhardt, G. Investigation of the structure of zeolites by solid-state high-resolution silicon-29 NMR spectroscopy. J. Am. Chem. Soc. 1981, 103, 4992–4996. [Google Scholar] [CrossRef]
- Nebel, H.; Neumann, M.; Mayer, C.; Epple, M. On the Structure of Amorphous Calcium Carbonate–A Detailed Study by Solid-State NMR Spectroscopy. Inorg. Chem. 2008, 47, 7874–7879. [Google Scholar] [CrossRef] [PubMed]
- Park, M.B.; Vicente, A.; Fernandez, C.; Hong, S.H. Solid-state NMR study of various mono- and divalent cation forms of the natural zeolite natrolite. Phys. Chem. Chem. Phys. 2013, 15, 7604–7612. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.M.; Klinowsky, J.; Ramadas, S.; Anderson, M.W.; Fyfe, C.A.; Gobbi, G.C. New Approaches to the Structural Characterization of Zeolites: Magic-Angle Spinning NMR (MASNMR). In Intrazeolite Chemistry; Galen, D.S., Francis, G.D., Eds.; ACS Publications: Washington, DC, USA, 1983; pp. 159–180. ISBN 9780841207745. [Google Scholar]
- Gramlich, V.; Meier, W.M. The crystal structure of hydrated NaA: A detailed refinement of a pseudosymmetric zeolite structure. Z. Krist. 1971, 10, 134–149. [Google Scholar] [CrossRef]
- Treacy, M.M.J.; Higgins, J.B. Collection of Simulated XRD Powder Patterns for Zeolites; Elsevier: Amsterdam, The Netherlands, 2007; pp. 252–253. [Google Scholar]
- Alfaro, S.; Rodriguez, C.; Valenzuela, M.A.; Bosch, P. Aging time effect on the synthesis of small crystal LTA zeolites in the absence of organic template. Mater. Lett. 2007, 61, 4655–4658. [Google Scholar] [CrossRef]
- Carey, T.; Tang, C.C.; Hriljac, J.A.; Anderson, P.A. Chemical Control of Thermal Expansion in Cation-Exchanged Zeolite A. Chem. Mater. 2014, 26, 1561–1566. [Google Scholar] [CrossRef]
- Lührs, H.; Derr, J.; Fischer, R.X. K and Ca exchange behavior of zeolite A. Microporous Mesoporous Mater. 2012, 151, 457–465. [Google Scholar] [CrossRef]
- Holland, T.J.B.; Redfern, S.A.T. Unit cell refinement from powder diffraction data–the use of regression diagnostics. Miner. Mag. 1997, 61, 65–77. [Google Scholar] [CrossRef]
- O’Keeffe, M.; Navrotsky, A. Some Aspects of the Ionic Model of Crystals. In Structure and Bonding in Crystals; O’Keeffe, M., Ed.; Academic Press: Cambridge, MA, USA, 1981; pp. 299–322. [Google Scholar]
- Ramdas, S.; Klinowski, J. A simple correlation between isotropic 29Si-NMR chemical shifts and T-O-T angles in zeolite frameworks. Nature 1984, 308, 521–523. [Google Scholar] [CrossRef]
- Edith, M.F.; Hassan, K.; Herman, A.S. Infrared Structural Studies of Zeolite Frameworks. In Molecular Sieve Zeolites-I; Edith, M.F., Leonard, B.S., Eds.; ACS Publications: Washington, DC, USA, 1974; pp. 201–229. ISBN 9780841201149. [Google Scholar]
- Milkey, R.G. Infrared Spectra of some Tectosilicates. Am. Minerol. 1960, 45, 990–1007. [Google Scholar]
- Dyballa, M.; Obenaus, U.; Lang, S.; Gehring, B.; Traa, Y.; Koller, H.; Hunger, M. Brønsted sites and structural stabilization effect of acidic low-silica zeolite A prepared by partial ammonium exchange. Microporous Mesoporous Mater. 2015, 212, 110–116. [Google Scholar] [CrossRef]
- Ucun, F. An Infrared Study of the CaA Zeolite Reacted with CO2. Z. Naturforsch. 2002, 57, 283. [Google Scholar] [CrossRef]
- Holzwarth, U.; Gibson, N. The Scherrer equation versus the ‘Debye-Scherrer equation’. Nat. Nano 2011, 6, 534. [Google Scholar] [CrossRef] [PubMed]
- Cubillas, P.; Gebbie, J.T.; Stevens, S.M.; Blake, N.; Umemura, A.; Terasaki, O.; Anderson, M.W. Atomic Force Microscopy and High Resolution Scanning Electron Microscopy Investigation of Zeolite A Crystal Growth. Part 2: In Presence of Organic Additives. J. Phys. Chem. C 2014, 118, 23092–23099. [Google Scholar] [CrossRef]
- Kliewer, C.E. Electron Microscopy and Imaging. In Zeolite Characterization and Catalysis; Chester, A.W., Derouane, E.G., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 169–196. [Google Scholar]
- Mintova, S.; Olson, N.H.; Valtchev, V.; Bein, T. Mechanism of Zeolite A Nanocrystal Growth from Colloids at Room Temperature. Science 1999, 283, 958–960. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, L.; Valtchev, V.; Nihtianova, D.; Kalvachev, Y. Submicrometer Zeolite A Crystals Formation: Low-Temperature Crystallization Versus Vapor Phase Gel Transformation. Cryst. Growth Des. 2011, 11, 4958–4962. [Google Scholar] [CrossRef]
- Smaihi, M.; Barida, O.; Valtchev, V. Investigation of the Crystallization Stages of LTA-Type Zeolite by Complementary Characterization Techniques. Eur. J. Inorg. Chem. 2003, 2003, 4370–4377. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Li-A | Na-A | K-A | Rb-A | |
|---|---|---|---|---|
| Unit cell parameter, a (Å) | 24.151 (2) | 24.435 (2) | 24.454 (2) | 24.486 (3) |
| Li-A | Na-A | K-A | Rb-A | |
|---|---|---|---|---|
| Si/Al | 1 | 1 | 1 | 1 |
| ∠T-O-T [ ()] | 143.9 | 147.9 | 148.6 | 148.4 |
| Na-A | Ca-A | |
|---|---|---|
| Scherrer Equation (nm) | 242 | 270 |
| FESEM (nm) | 241 | 290 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Price, L.; Leung, K.M.; Sartbaeva, A. Local and Average Structural Changes in Zeolite A upon Ion Exchange. Magnetochemistry 2017, 3, 42. https://doi.org/10.3390/magnetochemistry3040042
Price L, Leung KM, Sartbaeva A. Local and Average Structural Changes in Zeolite A upon Ion Exchange. Magnetochemistry. 2017; 3(4):42. https://doi.org/10.3390/magnetochemistry3040042
Chicago/Turabian StylePrice, Lisa, Ka Ming Leung, and Asel Sartbaeva. 2017. "Local and Average Structural Changes in Zeolite A upon Ion Exchange" Magnetochemistry 3, no. 4: 42. https://doi.org/10.3390/magnetochemistry3040042
APA StylePrice, L., Leung, K. M., & Sartbaeva, A. (2017). Local and Average Structural Changes in Zeolite A upon Ion Exchange. Magnetochemistry, 3(4), 42. https://doi.org/10.3390/magnetochemistry3040042